

Координационная химия, 2022, T. 48, № 8, стр. 458-472
Редокс-активный гермилен на основе 2,4,6,8-тетра-трет-бутил-феноксазин-1-она: синтез, строение и химические свойства
К. В. Арсеньева 1, А. В. Климашевская 1, М. А. Жеребцов 1, М. Г. Чегерев 2, А. В. Черкасов 1, И. А. Якушев 3, А. В. Пискунов 1, *
1 Институт металлоорганической химии им. Г.А. Разуваева РАН
Нижний Новгород, Россия
2 Институт физической и органической химии Южного федерального университета
Ростов-на-Дону, Россия
3 Институт общей и неорганической химии им. Н.С. Курнакова РАН
Москва, Россия
* E-mail: pial@iomc.ras.ru
Поступила в редакцию 02.12.2021
После доработки 20.12.2021
Принята к публикации 24.12.2021
- EDN: NUDJSD
- DOI: 10.31857/S0132344X22070015
Аннотация
Cинтезирован и структурно охарактеризован новый комплекс низковалентного германия PhenoxAPGe (I) на основе редокс-активного лиганда 2,4,6,8-тетра-трет-бутил-феноксазин-1-она. На примере кислотно-основных и окислительно-восстановительных превращений продемонстрирована его разносторонняя реакционная способность. Установлено, что взаимодействие гермилена I с Ni(COD)2 приводит к замещению обеих молекул циклооктадиена, координации четырех гермиленовых фрагментов на нульвалетный никель и формированию соединения (PhenoxAPGe)4Ni (II). Реакция I c [CpNi(CO)]2 в растворе толуола протекает с образованием комплекса (PhenoxAPGe)2(NiCp)2 (III), представляющего собой продукт замещения двух карбонильных групп на два изолобальных гермиленовых фрагмента. Реакция с одноэлектронным окислителем – 3,6-ди-трет-бутил-2-метоксифеноксильным радикалом – приводит к генерации лабильного парамагнитного гермилена IV, охарактеризованного методом спектроскопии ЭПР. Дигермиленоксид V, полученный гидролизом исходного гермилена I, in situ вступает в реакцию с N-гетероциклическим карбеном и KC8 c образованием ионных производных VI и VII, содержащих фрагмент Ge(II)–O–Ge(II). Гермилен I апробирован в качестве катализатора процесса гидроборирования бензальдегида. Молекулярные структуры соединений установлены методом РСА (CCDC № 2117783 (I), 2124277 (II), 2125357 (III), 2118393 (VII)).
В последнее десятилетие в координационной и элементоорганической химии наблюдается интенсивное развитие направления, связанного с низковалентными производными непереходных металлов [1–3]. Несмотря на то, что эти исследования носят прежде всего фундаментальный характер, соединения подобного рода демонстрируют многообещающую реакционную способность в отношении активации малых молекул [4–6]. Данное обстоятельство открывает перспективу использования соединений непереходных элементов в низких степенях окисления в каталитических процессах. В рамках идеи экологически чистого катализа продолжается поиск безметаллических катализаторов. Возрастающее внимание к процессам с меньшим воздействием на окружающую среду стимулирует поиск улучшенных синтетических превращений с минимальным образованием отходов, меньшим потреблением энергии и исключающим образование токсичных веществ. Одним из подходов к достижению этой цели является использование в качестве катализаторов более безопасных соединений главной группы [7].
Для стабилизации низковалентных состояний элементов 12, 13 и 14 групп активно применяются системы на основе α-дииминов [8–10]. Среди большого разнообразия лигандов различной природы также можно обнаружить примеры успешного применения ближайших аналогов дииминов – о-иминохинонов (imQ) в качестве систем, способных стабилизировать непереходные металлы в низких степенях окисления [11–16].
о-Иминохиноны являются яркими представителями редокс-активных органических лигандов. Соединения, содержащие такой тип лигандов, интенсивно изучаются [17–20] и обладают уникальными магнитными и электронными свойствами [21–25], находят применение в качестве спиновых меток [26], а их спектры ЭПР обладают высокой информативностью и могут дать различные сведения о структурах и механизмах превращений [26–30].
Интересным примером редокс-активного лиганда является 2,4,6,8-тетра-трет-бутил-феноксазин-1-он [31]. Несмотря на то, что получен он достаточно давно, количество соединений на основе данного трициклического иминохинона ограничивается лишь небольшим числом соединений, в которых лиганд находится в дианионном [32] или анион-радикальном состояниях [33–39]. Первые работы по координационной химии 2,4,6,8-тетра-трет-бутил-феноксазин-1-она проводились в рамках исследования парамагнитных производных различных металлов в растворах методом спектроскопии ЭПР [31]. И лишь гораздо позже круг металлов был расширен. Появились примеры полностью охарактеризованных соединений металлов 8 [39–41], 9 [34, 42], 10 [39], 11 [43], 12 [36] и 14 [32, 35] групп.
Научные интересы нашей группы лежат в области исследования тяжелых O,N-гетероциклических аналогов карбенов, построенных на основе стерически загруженных о-амидофенолятных лигандов. Ранее нами успешно проводилось изучение разнообразных химических свойств соединений элементов 14 группы в низких степенях окисления – окислительное присоединение, протекающее по металлу или лиганду, восстановление металлоцентра, кислотно-основные взаимодействия, каталитическая активность [11–16, 44, 45]. В настоящем исследовании мы использовали 2,4,6,8-тетра-трет-бутил-феноксазин-1-он для стабилизации низковалентного состояния германия, а также изучили химическое поведение полученного комплекса.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Все операции по синтезу и исследованию химических превращений комплексов германия проводили в условиях отсутствия кислорода и влаги воздуха. Использованные в работе растворители очищали и обезвоживали согласно рекомендациям [46]. Применяли коммерческие реактивы GeCl2 · diox, Ni(COD)2. 2,4,6,8-Tетра-трет-бутил-феноксазин-1-он [32] и 1,3-диизопропил-4,5-диметилимидазол-2-илиден [47] получали согласно известным методикам.
Спектры ЯМР регистрировали на спектрометрах Bruker Avance Neo 300 MГц, Bruker 200 MГц. Спектры ЭПР фиксировали на спектрометре Bruker EMX. В качестве стандарта при определении g-фактора использовали 2,2-дифенил-1-пикрилгидразил (g = 2.0037). Для определения точных параметров спектр ЭПР симулировали с помощью программы WinEPR SimFonia (Bruker). Элементный анализ выполняли на приборе Elementar Vario El cube.
Синтез 3,6-ди-трет-бутил-4-(3,6-ди-трет-бутил-2-метоксифенокси)-2-метоксициклогекса-2,5-диен-1-онa, обратимо диссоциирующего с образованием двух 2-метокси-3,6-ди-трет-бутилфеноксильных радикалов, проводили в рамках опубликованного ранее подхода [48].
3,6-Ди-трет-бутил-2-метоксифенол.
3,6-Ди-трет-бутилпирокатехин (9.75 г, 0.044 моль) растворяли в N,N-диметилформамиде (100 мл) и добавляли иодметан (6.25 г, 0.044 моль), затем карбонат калия (6.07 г, 0.044 моль). Реакционную смесь выдерживали при 60°С в течение 24 ч. После охлаждения смеси добавляли 50 мл воды и 10 мл раствора 30%-ной серной кислоты. Продукт экстрагировали гексаном (150 мл) и экстракт промывали водой (3 × 200 мл). Экстракт сушили над Na2SO4, растворитель удаляли на роторном испарителе и остаток перекристаллизовывали из гексана. Выделен 3,6-ди-трет-бутил-2-метоксифенол в виде белого мелкокристаллического порошка. Выход 9.58 г (72%).
Спектр ЯМР 1Н (CDCl3; 200 МГц; δ, м.д.): 1.42 (с., 9Н, t-Bu), 1.43 (с., 9Н, t-Bu), 3.83 (с., 3Н, ОСН3), 5.77 (с., 1Н, ОН), 6.82 (д., 1Н, Сar–H, J = = 8.5 Гц), 6.99 (д., 1Н, Сar–H, J = 8.5 Гц). Спектр ЯМР 13С (CDCl3; 50 МГц; δ, м.д.): 29.53, 31.15, 34.49, 34.73 (CH3t-Bu); 61.33 (OCH3), 117.39, 121.57 (Car–H); 135.06, 140.18, 147.06, 148.56 (Car).
3,6-Ди-трет-бутил-4-(3,6-ди-трет-бутил-2-метоксифенокси)-2-метоксициклогекса-2,5-диен-1-он.
К раствору 3,6-ди-трет-бутил-2-метоксифенола (5.54 г, 0.024 моль) в диэтиловом эфире при интенсивном перемешивании добавляли водный раствор, содержащий KOH (1.34 г, 0.024 моль) и K3Fe(CN)6 (11.84 г, 0.036 моль). Реакционную массу перемешивали 1 ч при комнатной температуре. Затем продукт экстрагировали эфиром и промывали водой (3 × 100 мл), сушили над сульфатом натрия, растворитель упарили на роторном испарителе. Полученный продукт перекристаллизовали из гексана. Выход желто-зеленых кристаллов 3,6-ди-трет-бутил-4-(3,6-ди-трет-бутил-2-метоксифенокси)-2-метоксициклогекса-2,5-диен-1-онa 1.55 г (28%).
Спектр ЯМР 1Н (CDCl3; 200 МГц; δ, м.д.): 0.92, 1.24, 1.40, 1.50 (с., 9Н, t-Bu); 3.75 (с., 3Н, ОСН3), 3.88 (с., 3Н, ОСН3), 5.94 (д., 1Н, С–H, J = 4.6 Гц), 6.45 (д., 1Н, Сar–H, J = 4.6 Гц), 6.93 (с., 2Н, Сar–Н). Спектр ЯМР 13С (CDCl3; 50 МГц; δ, м.д.): 28.87, 29.85, 30.42, 30.88 (CH3t-Bu); 34.42, 34.63, 34.76, 35.64 (Ct-Bu); 59.39, 61.52 (OCH3); 69.47 (C–H); 120.69, 121.22, 137.30, 141.13, 141.72, 144.02 (Car); 147.16, 149.65, 152.53, 152.97 (Cq); 182.68 (C=O).
Синтез комплекса PhenoxAPGe (I). Навеску иминохинона PhenoximQ (0.4 г, 0.95 ммоль) растворенную в ТГФ (5 мл), добавляли к избытку мелконарезанного металлического лития. Реакцию вели при небольшом нагревании и перемешивании до смены окраски с ярко синей на желтую. Полученный раствор дилитиевой соли PhenoxAPLi2 добавляли к дихлориду диоксаната германия (0.219 г, 0.95 ммоль). Реакционную смесь выдерживали в течение часа на водяной бане, при этом процесс сопровождался выпадением осадка хлорида лития и сменой окраски на интенсивно-оранжевую. ТГФ удалили при пониженном давлении, остаток растворяли в толуоле и отфильтровывали от осадка LiCl при помощи шприцевого фильтра в перчаточном боксе в инертной атмосфере. После концентрирования раствора в три раза, был выделен оранжевый кристаллический продукт I. Выход 0.379 г (0.75 ммоль, 81%).
Спектр ЯМР 1Н (C6D6; 20°С; δ, м.д.): 7.1, 7.09, 6.99 (д., 1H, HАР, JH,H = 2.27); 1.7 (с., 9H, N(t-Bu)); 1.66, 1.62, 1.28 (с., 9H, t-Bu). Спектр ЯМР 13C (C6D6; 20°С; δ, м.д.): 152.3, 144.7 (САР); 139.3, 138.5, 137.5 (С–О); 131.4, 129.9 (C–N); 126.5, 126.0, 119.5, 116.1, 112.7 (САР); 35.1, 34.4, 34.2, 34.0 (Cчетв); 31.06, 30.9, 30.5, 29.4 (Сt-Bu).
Синтез комплекса (PhenoxAPGe)4Ni (II). Навеску комплекса I (0.35 г, 0.711 ммоль), растворенную в толуоле добавляли к замороженному в жидком азоте раствору бис-циклоктадиенила никеля (0.048 г, 0.17 ммоль) в том же растворителе (3 мл). Затем реакционную смесь постепенно нагревали до комнатной температуры. Полученный раствор выдержали в течение 2 сут в темноте для завершения реакции, за это время раствор приобретал красно-коричневый цвет. После концентрирования из раствора выделяли коричневый мелкокристаллический комплекс II и высушивали при нагревании в условиях пониженного давления. Выход 0.86 г (0.43 ммоль, 61%).
Спектр ЯМР 1Н (C6D6; 20°С; δ, м.д.): 7.03, 6.94, 6.81 (д., 1H, HАР); 1.58 (д., 18H, t-Bu); 1.31, 1.27 (с., 9H, t-Bu). Спектр ЯМР 13C (C6D6; 20°С; δ, м.д.): 145.1 (САР); 140.3, 138.6, 136.3 (С–О); 130.8, 130.3 (C–N); 129.1, 128.9, 122.7, 116.3, 115.1, 110.8 (САР); 34.8, 34.4, 33.9, 33.7 (Cчетв); 31.13, 30.8, 30.6, 29.9 (Сt-Bu).
Синтез комплекса (PhenoxAPGe)2(NiCp)2 (III). Навеску комплекса I (0.35 г; 0.711 ммоль), растворенную в толуоле добавляли к раствору димера циклопентадиенилкарбонила никеля (0.215 г, 0.711 ммоль) в том же растворителе (5 мл). Затем реакционную смесь выдержали в течение 2 сут в темноте для завершения реакции, за это время раствор приобретал коричневую окраску. Из концентрированного раствора в гексане выделяли красно-коричневый мелкокристаллический комплекс III. Выход 0.48 г (0.38 ммоль, 54%).
Спектр ЯМР 1Н (C6D6; 20°С; δ, м.д.): 7.5, 7.2, 7.0 (д., 1H, HАР, JH,H = 2.02); 5.15 (с., 5Н, Ср); 1.74, 1.69, 1.67, 1.36. Спектр ЯМР 13C (C6D6; 20°С; δ, м.д.): 151.6, 144.5 (САР); 139.4, 138.1, 137.7 (С–О); 132.3, 130.06 (C–N); 128.9, 126.7, 118.5, 114.9, 111.3 (САР); 88.29 (Ср); 37.1, 35.27, 34.71, 34.36 (Cчетв); 31.19, 31.02, 30.59, 29.6 (Сt-Bu).
Синтез комплекса [(PhenoxAPGe)2O][Im] (VI). К смеси толуол (7 мл) и H2O (0.2 ммоль, 3.64 мкл) (отбирали при помощи микропипетки) при интенсивном перемешивании добавляли раствор гермилена I (0.2 г, 0.4 ммоль) в том же растворителе (10 мл). Реакционная смесь приобретала желтый оттенок, ее выдерживали в течение часа при интенсивном перемешивании и далее использовали без выделения. К реакционной смеси приливали раствор 1,3-диизопропил-4,5-диме-тилимидазол-2-илидена (0.4 ммоль, 0.072 г) в толуоле. После окончания реакции из раствора выпадал бледно-желтый мелкокристаллический комплекс VI. Выход 0.27 г (50%). Вычислено: C 68.48; H 8.91; N 6.14. Найдено: C 68.59; H 9.03; N 6.25.
Спектр ЯМР 1Н (C6D6; 20°С; δ, м.д.): 7.11 (д., 1H, HАР); 6.69 (c., 2H, HАР); 3.8 (CHi-Pr); 1.75, 1.72, 1.69, 1.37 (c., 9H, t-Bu), 1.35 (с., 6Н, С–Me); 1.22 (д., i-Pr). Спектр ЯМР 13C (C6D6; 20°С; δ, м.д.): 145.1 (САР); 140.3, 138.6, 136.3 (С–О); 130.8, 130.3 (C–N); 129.1, 128.9, 122.7, 116.3, 115.1, 110.8 (САР); 34.03, 34.0, 34.4, 34.7 (Cчетв); 31.63, 31.45, 30.9, 30.0 (Сt-Bu); 22.06 (Сi-Pr); 7.55 (СMe Carb).
Синтез комплекса [(PhenoxAPGe)2O][K(THF)3] (VII). Гидролиз гермилена I проводили аналогично описанному выше. Растворитель удаляли при пониженном давлении и остаток растворяли в ТГФ (10 мл). Полученный раствор приливали к KС8 (0.054 г, 0.4 ммоль). Реакционную смесь выдерживали при перемешивании 2 сут. После фильтрации от выделившегося графита и концентрирования из раствора выделяли бесцветные кристаллы комплекса VII. Соединение оказалось крайне неустойчиво и быстро разлагалось после выделения из растворителя, что не позволило получить удовлетворительные аналитические данные. Молекулярная структура установлена методом монокристального РСА.
РСА соединений I, VII проведен на дифрактометрах Bruker APEX II и Bruker D8 Venture соответственно в Центре коллективного пользования ИОНХ РАН. Первичное индицирование, уточнение параметров элементарной ячейки и интегрирование отражений производили с использованием пакета программ Bruker APEX3 [49]. Поправка на поглощение интенсивности отражений произведена по программе SADABS [49]. Сбор дифракционных данных кристаллов соединения II проведен на дифрактометре Rigaku OD Xcalibur E. Экспериментальные наборы интенсивностей для II интегрированы с помощью программы CrysAlisPro [50]. Учет поглощения проведен с использованием алгоритма SCALE3 ABSPACK [50]. Данные рентгеновской дифракции для III получены на рентгеновском пучке станции “Белок” Курчатовского центра синхротронного излучения в Национальном исследовательском центре “Курчатовский институт” (Москва, Российская Федерация) с использованием CCD детектора Rayonix SX165 CCD [51]. Определение параметров элементарной ячейки, их уточнение, интегрирование отражений и учет поглощения интенсивности рефлексов произведены с использованием программного пакета XDS [52].
Структуры расшифрованы прямыми методами и уточнены полноматричным методом наименьших квадратов по F 2 в анизотропном приближении для всех неводородных атомов [53, 54]. Водородные атомы помещены в геометрически рассчитанные положения и уточнены изотропно с фиксированными тепловыми параметрами U(H)изо = 1.2U(C)экв (U(H)изо = 1.5U(C)экв для метильных групп).
В случае сильного разупорядочения в уточнении структуры III применены инструкции SIMU, RIGU, DELU, ISOR, EADP, SADI. Остаточная электронная плотность, относящаяся к разупорядоченному нейтральному растворителю в структуре III удалена при помощи процедуры SQUEEZE в программе PLATON [55].
Кристаллографические данные, параметры рентгеноструктурных экспериментов и уточнения структур приведены в табл. 1.
Таблица 1.
Кристаллографические данные, параметры эксперимента и уточнения структур I, II, III и VII
| Параметр | Значение | |||
|---|---|---|---|---|
| I | II | III | VII | |
| Брутто-формула | C28H39NO2Ge | C112H156N4O8NiGe4, 4C7H8 | C73H96N2O4Ni2Ge2 | C68H100N2O8K2Ge2 |
| М | 494.19 | 2404.00 | 1328.11 | 1296.87 |
| Кристаллическая система | Ромбическая | Триклинная | Моноклинная | Ромбическая |
| Пр. группа | Pnma | $P\bar {1}$ | P21/m | Pbca |
| T, K | 150 | 110 | 100 | 100 |
| λ, Å | 0.71073 (Mo) | 0.71073 (Mo) | 0.74500 (синхротрон) | 1.54178 (Cu) |
| a, Å | 9.2209(7) | 18.1898(3) | 19.690(4) | 18.4255(5) |
| b, Å | 27.822(2) | 18.3046(3) | 9.7250(19) | 26.6762(7) |
| c, Å | 10.0187(9) | 20.5844(4) | 19.904(4) | 27.8650(8) |
| α, град | 90 | 76.862(2) | 90 | 90 |
| β, град | 90 | 82.252(2) | 111.46(3) | 90 |
| γ, град | 90 | 84.212(2) | 90 | 90 |
| V, Å3 | 2570.2(4) | 6595.9(2) | 3547.2(14) | 13696.3(6) |
| Z | 4 | 2 | 2 | 8 |
| ρ(выч.), г/см3 | 1.277 | 1.210 | 1.243 | 1.258 |
| µ, мм–1 | 1.216 | 1.096 | 1.579 | 2.578 |
| Размеры кристалла, мм | 0.30 × 0.30 × 0.28 | 0.42 × 0.24 × 0.11 | 0.240 × 0.080 × 0.040 | 0.11 × 0.05 × 0.02 |
| Область сканирования θ, град | 2.93–30.55 | 2.91–26.02 | 1.165–26.357 | 3.31–66.98 |
| Количество измеренных/ независимых отражений | 29 278/3976 | 91 144/25 958 | 30 756/6622 | 110 103/12032 |
| Rint | 0.0350 | 0.0775 | 0.1275 | 0.0980 |
| Количество независимых отражений с I > 2σ(I) | 3337 | 17 217 | 6622 | 8776 |
| Число уточняемых параметров/ограничений | 184/0 | 1640/441 | 377/575 | 862/101 |
| R (F 2 > 2σ(F 2)) | R1 = 0.0367, wR2 = 0.0956 |
R1 = 0.0596, wR2 = 0.1389 |
R1 = 0.0898, wR2 = 0.2328 |
R1 = 0.0663, wR2 = 0.1551 |
| R (по всем данным) | R1 = 0.0453, wR2 = 0.1004 |
R1 = 0.1049, wR2 = 0.1605 |
R1 = 0.1478, wR2 = 0.2777 |
R1 = 0.0938, wR2 = 0.1667 |
| S (F 2) | 1.031 | 1.036 | 1.027 | 1.076 |
| Остаточная электронная плотность (max/min), e/Å3 | 0.45/–0.25 | 1.48/–0.69 | 1.436/–1.288 | 0.54/–0.45 |
Расчеты выполнены с помощью программного пакета SHELXTL [54] в среде визуализации и обработки структурных данных OLEX2 [54].
Структуры зарегистрированы в Кембриджском банке структурных данных (CCDC № 2117783 (I), 2124277 (II), 2125357 (III), 2118393 (VII); ccdc.cam.ac.uk/structures).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
о-Амидофенолятный комплекс германия(II) PhenoxAPGe (I) синтезировали по двустадийному методу. На первом этапе проводили реакцию восстановления иминохинона избытком щелочного металла, дилитиевое производное использовали далее без выделения. Следующим этапом была реакция обмена дилитиевой соли PhenoxAPLi2 c Ge-Cl2 · diox (cхема 1).
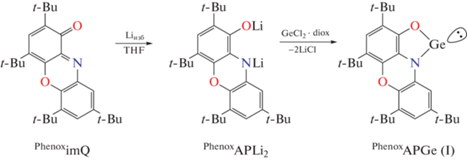
Схема 1 .
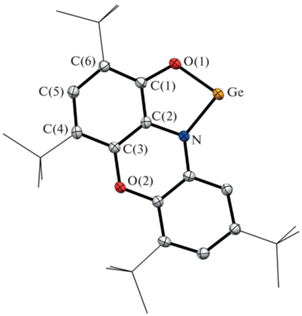
Комплекс I представляет собой ярко-оранжевое кристаллическое вещество, чувствительное к влаге и кислороду воздуха и хорошо растворимое в большинстве органических растворителей. Монокристаллические образцы, пригодные для РСА, были получены из концентрированного раствора в толуоле. Согласно полученным данным, соединение I представляет собой мономерный комплекс, в котором один дианионный органический лиганд бидентатно хелатирует низковалентный атом Ge (pис. 1). Длины связей С–O (1.350(2) Å) и C–N 1.401(2) Å в хелатном фрагменте лежат в области значений, характерных для дианионной формы данного лиганда [32]. Длины связей С–С в шестичленных циклах лежат в области 1.391(2)–1.407(2) Å и типичны для ароматических систем. Расстояния Ge–N (1.879(2) Å) и Ge–O (1.830(2) Å) немногим больше подобных в известных N-гетероциклических [56–60] и алкоксигермиленах [61, 62]. Значение угла NGeO (86.00(5)°) характерно для соединений такого класса. Между соседними мономерными гермиленовыми фрагментами в кристалле реализуется взаимодействие Ge…πарил (3.25 Å), за счет которого в кристаллической упаковке I формируются координационные цепочки. Важным отличием гермилена I от подобных о‑амидофенолятных комплексов Sn [12, 13, 63] и Pb [11] является отсутствие межмолекулярных контактов Ge…O и Ge…N между соседними фрагментами.
Полученный о-амидофенолятный комплекс I, равно как и другие дииминовые, катехолатные [44, 64, 65] и амидофенолятные [11–16, 44, 45] комплексы низковалентных металлов 14 группы, способен демонстрировать разностороннюю реакционную способность. Низковалентный атом металла может вступать в реакции окислительного присоединения с образованием четырехвалентных производных. В то же время в окислительно-восстановительное взаимодействие может быть вовлечен редокс-активный лиганд с сохранением степени окисления металла. Благодаря наличию неподеленной электронной пары и вакантной p-орбитали, металлены проявляют амфотерность Льюиса – они способны выступать в роли как мягких кислот, так и мягких оснований.
Рис. 1.
Молекулярная структура комплекса PhеnoxAPGe (I). Тепловые эллипсоиды избранных атомов приведены с 50%-ной вероятностью. Атомы водорода не показаны для ясности.
В литературе имеются обширные данные о комплексообразовании низковалентных производных 14 группы с переходными металлами путем вовлечения во взаимодействие неподеленной электронной пары металлена [66]. Необходимо отметить, что в данном случае металлены выступают в качестве оснований Льюиса. Реакция I c Ni(COD)2 в растворе толуола завершается при комнатной температуре в течение 2 сут и дает аддукт II в виде мелкокристаллического порошка коричневого цвета (схема 2 ).
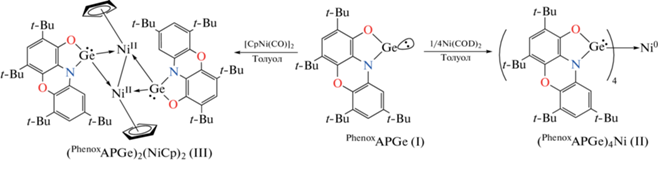
Схема 2 .
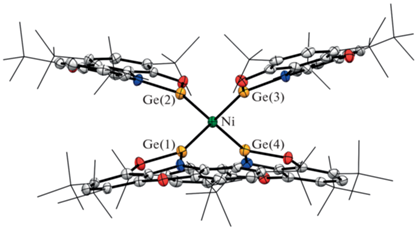
Согласно РСА (рис. 2), комплекс II представляет собой соединение нульвалентного никеля, связанного координационными взаимодействиями с четырьмя нейтральными гермиленами. Сохранение германием его двухвалентного состояния в ходе данной реакции подтверждается распределением длин связей вокруг металлоцентра. Длины связей С–O (1.366(5)–1.380(5) Å) и C–N (1.395(5)–1.406(5) Å) в хелатных фрагментах сопоставимы с аналогичными характеристиками исходного комплекса I (С–O 1.350(2) Å; C–N 1.401(2) Å). Расстояния Ge–O и Ge–N (1.830(2) Å, 1.879(2) Å в I и средние значения в II – 1.807 и 1.842 Å соответственно) несколько сокращаются в ходе координации гермилена I на никель. Тетраэдрическое окружение характерно для Ni0, координированного четырьмя аналогами карбена [67–69], и обеспечивается донорно-акцепторным взаимодействием неподеленных электронных пар германия и вакантных орбиталей никеля. Расстояния Ge–Ni (2.1911(7)–2.2050(7) Å) в гетерометаллическом комплексе II значительно короче аналогичных взаимодействий в ранее опубликованных комплексах тетразамещенных производных никеля [67–69]. Это объясняется снятием стерической нагрузки с атома Ge(II) при переходе от N,N-гетероциклических гермиленов к O,N-хелатному циклу в производном I.
Рис. 2.
Молекулярная структура комплекса (PhenoxAPGe)4Ni (II). Тепловые эллипсоиды избранных атомов приведены с 50%-ной вероятностью. Атомы водорода не показаны для ясности.
Реакция I c димерным соединением никеля [CpNi(CO)]2 в растворе толуола заканчивается в течение 2 сут при комнатной температуре, сопровождаясь выделением СО и изменением цвета с красного на коричневый (схема 2 ). После смены растворителя на гексан и концентрирования примерно в три раза был выделен красно-коричневый комплекс III.
Согласно данным РСА (рис. 3), III представляет собой продукт замещения двух карбонильных групп на два изолобальных гермиленовых фрагмента. В элементарной ячейке кристалла содержится одна молекула сольватированного толуола на одну молекулу комплекса. Оба гермиленовых фрагмента лежат в одной плоскости. Молекула III содержит ядро Ge2Ni2 в конфигурации “бабочка” со связью Ni–Ni 2.581 Å, что несколько больше подобного взаимодействия в исходном димере (2.363 Å) [70]. Расстояние Ge···Ge составляет 3.379 Å и указывает на отсутствие аттрактивного взаимодействия между атомами. Двугранный угол между плоскостями Ni–Ge(1)–Ni и Ni–Ge(2)–Ni, формирующими “крылья бабочки”, составляет 135.8°, что меньше аналогичного угла в [CpNi(CO)]2. Оба атома Ni расположены на одинаковых расстояниях 1.730 Å от центроидов своих Cp-лигандов. Оба циклопентадиенильных лиганда координированы к своим атомам никеля несимметрично псевдо-π-аллильным образом. Такие искажения обычно вызываются неэквивалентностью монодентатных транс-лигандов в полусэндвичевых комплексах [71]. Величины донорно-акцепторных взаимодействий Ge–Ni в комплексе III лежат в интервале 2.223–2.244 Å, что хорошо соотносится с таковыми в комплексе II. Расстояния Ge–O и Ge–N (средние значения в III составляют 1.792 и 1.874 Å соответственно) укорачиваются в ходе координации по сравнению с гермиленом I и сходны с таковыми в II. Длины связей С–O (1.256(15)–1.304(15) Å) и C–N (1.378(14)–1.408(15) Å) в хелатных фрагментах немного короче по сравнению с аналогичными характеристиками соединений I и II, но лежат в области значений характерных для дианионной структуры амидофенолятных лигандов [32]. Раствор комплекса III диамагнитный, обладает хорошо разрешенным спектром ЯМР и не имеет каких-либо сигналов в спектре ЭПР. Это подтверждает, что никель и германий сохраняют свои степени окисления в ходе реакции (схема 2 ).
Рис. 3.
Молекулярная структура комплекса (PhenoxAPGe)2(NiCp)2 (III). Тепловые эллипсоиды избранных атомов приведены с 30%-ной вероятностью. Атомы водорода и трет-бутильные группы не показаны для ясности.
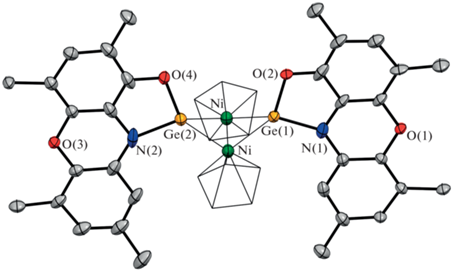
Гетерометаллический комплекс III – это второй пример стабилизации фрагмента СрNi–NiCp в координационном соединении с гермиленами. Единственным известным до настоящего момента соединением с аналогичным по строению кластером является комплекс [(C6F5)2GeNiCp]2, полученный в ходе окислительно-восстановительной реакции между тетракис(пентафторфенил)дигидродигерманом и никелоценом [72]. Авторы [72] предполагали промежуточное образование гермилена (C6F5)2Ge, который при генерации в реакционной смеси координируется на атом никеля. Строение центрального фрагмента опубликованного ранее комплекса [(C6F5)2GeNiCp]2 практически идентично III за исключением значительно меньшего значения двугранного угла между “крыльями бабочки” (117.6 Å).
Металлены, подобные гермилену I, обладают двумя возможными восстановительными центрами: а) низковалентный атом германия, который может участвовать в реакциях окислительного присоединения с последующим образованием производных германия(IV); б) редокс-активная о-амидофенолятная основа, способная претерпевать редокс-процессы без изменения состояния окисления тетрилена. Оба центра реакционноспособны и могут быть индивидуально активированы в зависимости от природы окислителя [11–15, 44, 45, 73]. Известно, что производные тетриленов с различными дианионными редокс-активными лигандами (диамид, амидофенолят, катехолат) реагируют со стабильными радикалами или галогенидами Hg(II) и Ag(I), образуя соответствующие радикальные соединения от детектируемых только спектроскопией ЭПР [64, 65, 74–76] до стабильных [77, 78]. Как правило, такие парамагнитные производные тяжелых аналогов карбена удается наблюдать для соединений олова(II) и свинца(II). Однако в случае германия(II) преимущественно происходит окисление низковалентного центра, поэтому ранее не удавалось наблюдать участие о‑амидофенолятного лиганда в реакциях окисления O,N-гетероциклических гермиленов [14, 15]. Однако недавно сообщалось о первом примере парамагнитного гермилена на основе стерически загруженного N-адамантил-3,5-ди-трет-бутил-о-аминофенола [16], который удалось детектировать в реакционной смеси методом спектроскопии ЭПР, но низкая устойчивость генерируемого соединения не позволила накопить концентрацию, достаточную для наблюдения сверхтонкого взаимодействия (СТВ) неспаренного электрона с магнитным изотопом 73Ge.
Мы провели химическое окисление гермилена I стабильным 3,6-ди-трет-бутил-2-метоксифеноксильным радикалом [48] (схема 3 ). При этом успешно зарегистрировано образование парамагнитного тяжелого аналога карбена IV методом спектроскопии ЭПР (рис. 4). Генерируемое соединение удается наблюдать в растворе при комнатной температуре в течение 15–20 мин, по прошествии которых спектр дополняется целым набором дополнительных сигналов, указывающих на дальнейшую трансформацию IV в растворе.
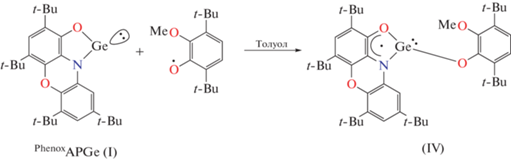
Рис. 4.
Спектр ЭПР парамагнитного соединения IV в толуоле при Т = 300 К. Вкладка A (увеличенная в 10 раз) – область, демонстрирующая СТВ с изотопом 73Ge; вкладка B (экспериментальная и симулированная) – центральная часть спектра.
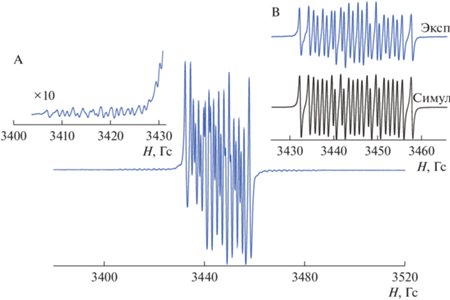
Схема 3 .
Спектр ЭПР соединения IV при Т = 300 К отличает высокое разрешение, обусловленное малой шириной (0.3 Гс) индивидуальных компонент спектра. Их сверхтонкая структура вызвана СТВ неспаренного электрона с магнитными ядрами трех протонов 1H (99.98%, I = 1/2, mN = 2.7928) и одного атома азота 14N (99.63%, I = 1, mN = 0.4037). По краям основного спектра удается наблюдать сателлитное расщепление на магнитном изотопе 73Ge (7.8%, I = 9/2, mN = 0.8795). Параметры спектра: gi = 2.0030, ai(14N) = 8.04 Гс, ai(1H) = 4.16, 3.18, 2.02 Гс, ai(73Ge) = 5.65 Гс. Необходимо отметить, что константа СТВ с магнитным изотопом 73Ge в соединении IV почти в два раза ниже по сравнению с родственными парамагнитными N,N-гетероциклическими гермиленами [75–77]. В то же время ее величина более чем в 5 раз превышает значения, характерные для производных германия(IV) с анион-радикальными лигандами [78]. Данное обстоятельство однозначно указывает на сохранение двухвалентного состояния атома германия в наблюдаемом парамагнитном соединении IV. Причины столь значимого изменения констант СТВ в парамагнитных соединениях элементов 14 группы в степенях окисления 2 и 4 подробно обсуждаются в [79].
В ходе недавних исследований [80, 81] было показано, что гидролиз о-амидофенолятов Ge(II) приводит к образованию соответствующих оксидных производных, атомы германия(II) в которых способны проявлять более сильные нуклеофильные свойства, чем в исходных двухкоординированных гермиленах. Мы получили дигермилен оксид V по реакции I со стехиометрическим количеством воды (схема 4 ). Реакция протекает без дополнительного нагревания и при интенсивном перемешивании в течение часа. Длительная кристаллизация реакционной смеси ведет к разложению комплекса V – медленное упаривание растворителя после окончания реакции приводит к маслянистому остатку, который наряду с целевым дигермиленоксидом V содержит продукты его разложения, в частности соответствующий о-аминофенол. Поэтому мы предприняли попытки стабилизации полученного in situ производного путем депротонирования о-аминофенолятных фрагментов.
Cхема 4.
Обработка реакционной смеси после гидролиза гермилена I N,N-гетероциклическим карбеном приводит к образованию стабильного ионного комплекса VI (схема 5 ). Реакция протекает со скоростью смешения реагентов и заканчивается самопроизвольным выпадением бледно-желтого мелкокристаллического порошка с высоким выходом. Строение комплекса VI было подтверждено методом спектроскопии ЯМР. Нам не удалось получить кристаллы, пригодные для РСА, однако по аналогии с результатами работы [81] можно говорить о том, что VI содержит дианион, в котором два трехкооридинированных атома Ge(II) соединены мостиковым кислородом. В качестве противоионов выступают два имидазолиниевых катиона.
Схема 5 .
При взаимодействии соединения V c калием, интеркалированным в графите, происходит выделение газообразного водорода и образование соединения VII, выделенного с невысоким выходом из реакционной смеси в виде бесцветных кристаллов (схема 5 ). В отличие от соединения VI, комплекс VII весьма чувствителен к следовым количествам влаги и кислорода воздуха и разлагается при удаления маточного растворителя. Все попытки зарегистрировать его спектры ЯМР оказались безуспешными. Перерастворение кристаллического порошка комплекса VII в дейтерированных растворителях вызывает его разложение.
Однако нам удалось определить молекулярную структуру VII методом рентгеноструктурного анализа (рис. 5). Согласно полученным данным, дианион [(PhenoxAPGe)2O]2– в VII координирован двумя катионами калия, которые, в свою очередь, сольватированы тремя молекулами тетрагидрофурана. Германий при этом сохраняет свое двухвалентное состояние. Дианионный фрагмент в VII содержит два трикоординированных гермиленовых центра, связанных между собой μ2-кислородным мостиком. Угол GeO(1)Ge равен 126.3(2)° и значительно меньше, чем в многочисленных известных родственных трикоординированных гермиленовых производных [82, 83]. Расстояния Ge–O(1) составляют 1.863(2), 1.867(2) Å и также превышают значения для подобных производных [83–86]. Суммы углов вокруг атомов Ge(1) (270.8°) и Ge(2) (268.9°) близки к 270° и свидетельствуют о низкой степени вовлечения в гибридизацию неподеленной электронной пары, расположенной на s-орбитали. Распределение длин связей в о-амидофенолятном фрагменте типично для подобных типов лигандов. При этом расстояния Ge–O (1.913(5), 1.920(5) Å) и Ge–N (1.994(4), 1.996(6) Å) в VII заметно длиннее, чем в I (Ge–O 1.830(2) Å, Ge–N 1.879(2) Å). Избранные длины связей для соединений I–III и VII приведены в табл. 2.
Рис. 5.
Молекулярная структура комплекса [(PhenoxAPGe)2O][K2(THF)3] (VII). Тепловые эллипсоиды избранных атомов приведены с 30%-ной вероятностью. Атомы водорода не показаны для ясности.
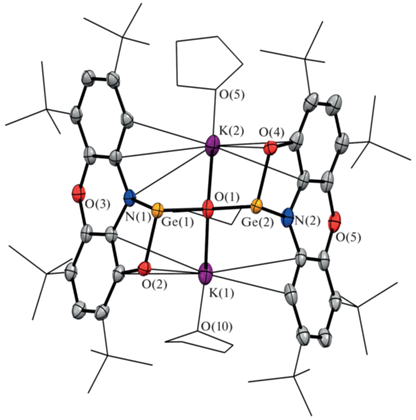
Таблица 2.
Избранные длины связей для комплексов I, II, III и VII
| Связь (Å) | Комплекс | |||
|---|---|---|---|---|
| I | II | III | VII | |
| Ge–N | 1.8297(16) | $\left\langle {{\text{1}}{\text{.842}}} \right\rangle $ | 1.854(8), 1.894(9) | 1.994(4), 1.996(6) |
| Ge–O | 1.8785(13) | $\left\langle {{\text{1}}{\text{.807}}} \right\rangle $ | 1.782(10), 1.802(11) | 1.913(5), 1.920(5) |
| C(1)–O | 1.3501(19) | 1.366(5)–1.380(5) | 1.304(15), 1.256(15) | 1.368(5), 1.396(6) |
| C(2)–N | 1.4011(12) | 1.395(5)–1.406(5) | 1.378(14), 1.408(15) | 1.391(6), 1.384(6) |
| Ge–Ni | 2.1911(7)–2.2050(7) | 2.2233(17)–2.2440(17) | ||
| Ge–O(1) | 1.863(2), 1.867(2) | |||
| Ni–Ni | 2.581 | |||
Существует множество примеров использования гетероциклических соединений низковалентных элементов 14 группы как катализаторов для полимеризации лактидов, цианосилилирования или гидроборирования карбонильных соединений [85–89]. Гидроборирование – важнейшая реакция образования связи элемент–бор в органической химии, используемая в том числе для синтеза боронатных эфиров, представляющих собой класс важнейших органических реагентов в синтетической химии. Борорганические соединения считаются стабильными, простыми в обращении и универсальными реагентами для процессов кросс-сочетания. Гидроборирование ненасыщенных связей предлагает прямой и эффективный путь к борорганическим соединениям [90–93]. Катализируемое Sn и Ge гидроборирование оказалось селективным путем к ценным алкилборонатным эфирам и привлекает внимание исследователей [16, 94–96].
В рамках настоящей работы гермилен I был протестирован как катализатор реакции гидроборирования бензальдегида пинаколбораном (HBpin) (схема 6 ). Были проведены холостые эксперименты, в которых бензальдегид реагировал с одним эквивалентом HBpin в отсутствии катализатора при комнатной температуре, и конверсия наблюдалась лишь на следовом уровне. Соединение I успешно катализирует реакцию гидроборирования бензальдегида с HBpin при комнатной температуре, образуя соответствующий боронатный эфир (схема 6 ) с хорошей конверсией, за контрольный промежуток времени. Условия реакции были оптимизированы и контролировались с помощью спектроскопии ЯМР. Значения конверсии были рассчитаны на основе площади интегрирования продукта и исходного материала в спектрах ЯМР 1Н с использованием мезитилена в качестве внутреннего стандарта. Результаты обобщены в табл. 3. По результатам каталитических тестов установлено, что активность гермилена I как катализатора реакции (схема 6 ) несколько ниже его ближайшего O,N-гетероциклического аналога на основе N-адамантил-3,5-ди-трет-бутил-о-аминофенола [16].
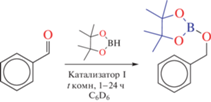
Таблица 3.
Конверсия (%) альдегида в реакции гидроборирования, катализируемая гермиленом I
| Загрузка катализатора I | 1 мол. % | |
|---|---|---|
| субстрат | время, ч | конверсия, % |
 |
1 | 61 |
| 2 | 73 | |
| 3 | 85 | |
| 24 | 91 | |
Схема 6 .
Таким образом, был синтезирован и структурно охарактеризован новый O,N-гетероциклический гермилен на основе редокс-активного 2,4, 6,8-тетра-трет-бутил-феноксазин-1-она. Установлено, что синтезированное соединение способно выступать в качестве нейтрального донорного лиганда за счет неподеленной электронной пары низковалентного атома германия и образовывать комплексы как с нульвалентным, так и с двухвалентным никелем. Окисление о-амидофенолята германия(II) стабильными радикалами приводит к формированию парамагнитного тяжелого аналога карбена, успешно зарегистрированного и охарактеризованного методом спектроскопии ЭПР. Гидролиз гермилена сопровождается формированием дигермиленоксидного производного, который может быть депротонирован действием N-гетероциклического карбена или калия интеркалированного в графите. Исходный гермилен продемонстрировал каталитическую активность в реакции гидроборирования альдегидов.
Авторы заявляют, что у них нет конфликта интересов
Список литературы
Roesky P.W. // Dalton Trans. 2009. V. 11. P. 1887.
Ochiai T., Franz D., Inoue S. // Chem. Soc. Rev. 2016. V. 45. P. 6327.
Jambor R., Dostál L. // Organometallic Pincer Chemistry. Topics in Organometallic Chemistry / Eds van Koten G., Milstein D. Berlin, Heidelberg: Springer, 2013. V. 40. P. 175.
Yadav S., Saha S., Sen S.S. // ChemCatChem. 2015. V. 8. P. 486.
Gendy C., Rautiainen M.J., Mailman A., Tuononen H.M. // Chem. Eur. J. 2021. V. 27. P. 14405.
Inoue S., Weetman C. // ChemCatChem. 2018. V. 10. P. 4213.
Sen N., Khan S. // Chem. Asian. J. 2021. V. 16. P. 705.
Zhang R., Wang Y., Zhao Y. et al. // Dalton Trans. 2021. V. 50. P. 13634.
Majoumo-Mbe F., Lönnecke P., Hey-Hawkins E. // Organometallics. 2005. V. 24. P. 5287.
Ma M., Shen L., Wang H. et al. // Organometallics. 2020. V. 39. P. 1440.
Tsys K.V., Chegerev M.G., Piskunov A.V., Fukin G.K. // Mendeleev Commun. 2018. V. 28. P. 527.
Chegerev M.G., Piskunov A.V., Tsys K.V. et al. // Eur. J. Inorg. Chem. 2019. P. 875.
Пискунов А.В., Цыс К.В., Чегерев М.Г., Черкасов А.В. // Коорд. химия. 2019. Т. 45. С. 527 (Piskunov A.V., Tsys K.V., Cherkasov A.V., Chegerev M.G. // Russ. J. Coord. Chem. 2019. V. 45. № 9. P. 626). https://doi.org/10.1134/S1070328419090069
Tsys K.V., Chegerev M.G., Fukin G.K. et al. // Mendeleev Commun. 2020. V. 30. P. 205.
Arsenyeva K.V., Ershova I.V., Chegerev M.G. et al. // J. Organomet. Chem. 2020. V. 927. 121524.
Arsenyeva K.V., Pashanova K.I., Trofimova O.Yu. et al. // New J. Chem. 2021. V. 45. P. 11758.
Poddel’sky A.I., Cherkasov V.K., Abakumov G.A. // Coord. Chem. Rev. 2009. V. 253. P. 291.
Kaim W. // Inorg. Chem. 2011. V. 50. P. 9752.
Старикова A.A., Минкин В.И. // Успехи химии. 2018. T. 87. C. 1049.
Фоменко И.С., Гущин А.Л. // Успехи химии. 2020. T. 89. C. 966.
Dei A., Gatteschi D., Sangregorio C., Sorace L. // Acc. Chem. Res. 2004. V. 37. P. 827.
Markevtsev I.N., Monakhov M.P., Platonov V.V. et al. // J. Magn. Magn. Mater. 2006. V. 300. P. e407.
Sato O. // J. Photochem. Photobiol. 2004. V. 5. P. 203.
Abakumov G.A., Cherkasov V.K., Nevodchikov V.I. et al. // Inorg. Chem. 2001. V. 40. P. 2434.
Pierpont C.G. // Coord. Chem. Rev. 2001. V. 216/217. P. 99.
Meshcheryakova I.N., Arsenyeva K.V., Fukin G.K. et al. // Mendeleev Commun. 2020. V. 30. P. 592.
Bubnov M.P., Kozhanov K.A., Skorodumova N.A. et al // J. Mol. Struct. 2019. V. 1180. P. 87.
Kozhanov K.A., Bubnov M.P., Teplova I.A. et al. // J. Mol. Struct. 2017. V. 1147. P. 541.
Kozhanov K.A., Bubnov M.P., Abakumov G.A., Cherkasov V.K. // J. Magn. Reson. 2012. V. 225. P. 62.
Bubnov M.P., Teplova I.A., Kozhanov K.A. et al. // J. Magn. Reson. 2011. V. 209. P. 149.
Ивахненко Е.П., Карсанов И.В., Хандкарова В.С. и др. // Изв. АН СССР. Сер. хим. 1986. С. 2755.
Чегерев М.Г., Арсеньева К.В., Черкасов А.В., Пискунов А.В. // Коорд. химия. 2020. Т. 46. С. 672. (Chegerev M.G., Arsenyeva K.V., Cherkasov A.V. et al. // Russ. J. Coord. Chem. 2020. V. 46. P. 746). https://doi.org/10.1134/S1070328420110019
Ивахненко Е.П., Кощиенко Ю.В., Чернышев А.В. и др. // Журн. oбщ. химии. 2016. Т. 86. С. 1188
Ивахненко Е.П., Кощиенко Ю.В., Князев П.А. и др. // Коорд. химия. 2016. Т. 42. С. 221. (Ivakhnenko E.P., Koshchienko Yu.V., Knyazev P.A. et al. // Russ. J. Coord. Chem. 2016. V. 42. P. 509). https://doi.org/10.1134/S1070328416040011
Romanenko G.V., Ivakhnenko E.P., Minkin V.I. et al. // Inorg. Chim. Acta. 2014. V. 417. P. 66.
Ivakhnenko E.P., Starikov A.G., Lyssenko K.A. et al. // Inorg. Chim. Acta. 2014. V. 410. P. 144.
Антипин М.Ю., Ивахненко Е.П., Кощиенко Ю.В. и др. // Изв. АН. Сер. хим. 2013. С. 1744
Speier G., Whalen A.M., Csihony J., Pierpont C.G. // I-norg. Chem. 1995. V. 34. P. 1355.
Bhattacharya S., Pierpont C.G. // Inorg. Chem. 1994. V. 33. P. 6038.
Bhattacharya S., Pierpont C.G. // Inorg. Chem. 1992. V. 31. P. 2020.
Bhattacharya S., Boone S.R., Pierpont C.G. // J. Am. Chem. Soc. 1990. V. 112. P. 4561.
Ивахненко E.П, Кощиенко Ю.В., Князев П.A. и др. // Докл. PАН. 2011. T. 438. С. 485.
DeLearie L.A., Haltiwanger R.C., Pierpont C.G. // I-norg. Chem. 1989. V. 28. P. 644.
Tsys K.V., Chegerev M.G., Pashanova K.I. et al. // In-org. Chim. Acta. 2019. V. 490. P. 220.
Aysin R.R., Bukalov S.S., Leites L.A. et al. // Organometallics. 2019. V. 38. P. 3174.
Гордон А., Форд Р. // Спутник химика. М.: Мир, 1976. P. 543.
Ryan S.J., Schimler S.D., Bland D.C., Sanford M.S. // Org. Lett. 2015. V. 17. P. 1866.
Abakumov G.A., Cherkasov V.K., Nevodchikov V.I. et al. // Tetrahedron Lett. 2005. V. 46. P. 4095.
APEX3, SAINT and SADABS. Madison (WI, USA): Bruker AXS Inc., 2016.
Rigaku Oxford Diffraction. CrysAlisPro Software System. Version 1.171.38.46. Wroclaw (Poland): Rigaku Corporation, 2015.
Svetogorov R.D., Dorovatovskii P.V., Lazarenko V.A. // Cryst. Res. Tech. 2020. V. 55. 1900184.
Kabsch W. // Acta Crystallogr. D. 2010. V. 66. P. 125.
Sheldrick G.M. // Acta Crystallogr. A. 2015. V. 71. P. 3.
Sheldrick G.M. // Acta Crystallogr. C. 2015. V. 71. P. 3
Spek A.L. // Acta Crystallogr. C. 2015. V. 71. P. 9.
Matson E.M., Opperwall S.R., Fanwick P.E., Bart S.C. // Inorg. Chem. 2013. V. 52. P. 7295.
Chegerev M.G., Piskunov A.V., Starikova A.A. et al. // Eur. J. Inorg. Chem. 2018. P. 1087.
Hill M.S. // Science. 2006. V. 311. P. 1904.
Aysin R.R., Leites L.A., Bukalov S.S. et al. // Inorg. Chem. 2016. V. 55. P. 4698.
Zabula A.V., Hahn F.E., Pape T., Hepp A. // Organometallics. 2007. V. 26. P. 1972.
Zemlyansky N.N., Borisova I.V., Khrustalev V.N. et al. // Organometallics. 2003. V. 22. P. 5441.
Kitschke P., Mertens L., Rüffer T. et al. // Eur. J. Inorg. Chem. 2015. P. 4996.
Aivaz’yan I.A., Piskunov A.V., Fukin G.K. et al. // Inorg. Chem. Commun. 2006. V. 9. P. 612.
Chegerev M.G., Piskunov A.V., Maleeva A.V. et al. // Eur. J. Inorg. Chem. 2016. P. 3813.
Piskunov A.V., Aivaz’yan I.A., Cherkasov V.K., Abakumov G.A. // J. Organomet. Chem. 2006. V. 691. P. 1531.
Baumgartner J., Marschner C. // Rev. Inorg. Chem. 2014. V. 34. P. 119.
Gendy C., Mansikkamäki A., Valjus J. et al. // Angew. Chem. Int. Ed. 2019. V. 58. P. 154.
Ullah F., Kühl O., Bajor G. et al. // Eur. J. Inorg. Chem. 2009. V. 2. P. 221.
Bazinet P., Yap G.P.A., Richeson D.S. // J. Am. Chem. Soc. 2001. V. 123. P. 11162.
Byers L.R., Dahl L.F. // Inorg. Chem. 1980. V. 19. P. 680.
Hermann W.A., Herdtcheck E., Floel M. et al. // Polyhedron. 1987. V. 6. P. 1165.
Pankratov L.V., Nevodchikov V.I., Zakharov L.N. et al. // J. Organomet. Chem. 1992. V. 429. P. 13.
Piskunov A.V., Aivaz’ya, I.A., Poddel’sky A.I. et al. // Eur. J. Inorg. Chem. 2008. P. 1435.
Tumanskii B., Pine P., Apeloig Y. et al. // J. Am. Chem. Soc. 2004. V. 126. P. 7786.
Tumanskii B., Pine P., Apeloig Y. // J. Am. Chem. Soc. 2005. V. 127. P. 8248.
Абакумов Г.А., Черкасов В.К., Пискунов А.В. и др. // Докл. РАН. 2005. Т. 404. С. 496.
Fedushkin I.L., Khvoinova N.M., Baurin A.Y. // Inorg. Chem. 2004. V. 43. P. 7807.
Абакумов Г.А., Черкасов В.К., Ермолаев Н.И. и др. // Изв. АН. Сер. хим. 1995. С. 1568.
Абакумов Г.А., Черкасов В.К., Пискунов А.В. и др. // Изв. АН. Сер. хим. 2006. С. 1103.
Janes T., Zatsepin P., Song D.T. // Chem. Commun. 2017. V. 53. P. 3090.
Arsenyeva K.V., Chegerev M.G., Cherkasov A.V. et al. // Mendeleev Commun. 2021. V. 31. P. 330.
Пискунов А.В., Арсеньева К.В., Климашевская А.В., Черкасов А.В. // Коорд. химия. 2022. Т. 48. № 5. С. 277 (Piskunov A.V., Arsenyeva K.V., Klimashev-skaya A.V., Cherkasov A. V.K. // Russ. J. Coord. Chem. 2022. V. 48. P. 278).
Siwatch R.K., Yadav D., Mukherjee G. et al. // Inorg. Chem. 2013. V. 52. P. 13384.
Hadlington T.J., Kefalidis C.E., Maron L., Jones C. // ACS Catal. 2017. V. 7. P. 1853.
Kelly J.A., Juckel M., Hadlington T.J. et al. // Chem. Eur. J. 2019. V. 25. P. 2773.
Pal S., Dasgupta R., Khan S. // Organometallics. 2016. V. 35. P. 3635.
Rittinghaus R.D., Tremmel J., Růžička A. et al. // Chem. Eur. J. 2020. V. 26. P. 212.
Praban S., Yimthachote S., Kiriratnikom J. et al. // J. Polym. Sci. A. 2019. V. 57. P. 2104.
Karmakar A., Hazra S., Rúbio, G.M.D.M. et al. // New J. Chem. 2018. V. 42. P. 17513.
Goswami B., Feuerstein T.J., Yadav R. et al. // Chem. Eur. J. 2021. V. 27. P. 4401.
Yadav S., Dixit R., Bisai M.K. et al. // Organometallics. 2018. V. 37. P. 4576.
Eedugurala N., Wang Z., Chaudhary U. et al. // ACS Catal. 2015. V. 5. P. 7399.
Garhwal S., Kroeger A.A., Thenarukandiyil R. et al. // Inorg Chem. 2021. V. 60. P. 494.
Dasgupta R., Das S., Hiwase S. et al. // Organometallics. 2019. V. 38. P. 1429.
Dasgupta R., Khan S. // Adv. Organomet. Chem. 2020. V. 74. P. 105.
Schneider J., Sindlinger C.P., Freitag S.M. et al. // Ange-w. Chem. Int. Ed. 2017. V. 56. P. 333.
Дополнительные материалы отсутствуют.
Инструменты
Координационная химия