

Кристаллография, 2019, T. 64, № 6, стр. 979-986
Новые кристаллы в системе CsHSO4–CsH2PO4–NH4H2PO4–H2O
В. А. Коморников 1, *, И. С. Тимаков 1, О. Б. Зайнуллин 1, В. В. Гребенев 1, И. П. Макарова 1, Е. В. Селезнева 1, Д. А. Ксенофонтов 2
1 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
2 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
* E-mail: v.a.kom@mail.ru
Поступила в редакцию 09.04.2019
После доработки 25.04.2019
Принята к публикации 27.05.2019
Аннотация
Изучены фазовые равновесия в четырехкомпонентной водно-солевой системе CsHSO4–CsH2PO4–NH4H2PO4–H2O. Определены области кристаллизации и растворимости следующих фаз: CsHSO4, NH4H2PO4, (Cs,NH4)H2PO4 и впервые полученных (Cs,NH4)4(HSO4)3(H2PO4) и (Cs,NH4)2(HSO4)(H2PO4). Впервые получены также кристаллы стехиометрических составов Cs3NH4(HSO4)3(H2PO4) и CsNH4(HSO4)(H2PO4).
ВВЕДЕНИЕ
Изучение кристаллов-суперпротоников является актуальной задачей как для фундаментальных исследований, так и с точки зрения создания материалов для практического применения. Кристаллы семейства MmHn(AO4)(m + n)/2 · yH2O (M = = K, Rb, Cs, NH4, AO4 = SO4, SeO4, HРO4), названные суперпротониками, способны проявлять аномально высокую протонную проводимость при относительно невысоких температурах. Высокая протонная проводимость в этих соединениях обусловлена наличием разупорядоченной сетки водородных связей, что и обеспечивает условия для диффузии протонов. Проводимость связана со структурными особенностями данных соединений, является собственной и поэтому не зависит от влажности, а также не является следствием дефектов реальной структуры или влияния легирующих добавок [1]. Отметим, что именно в ИК РАН впервые была экспериментально обнаружена и исследована аномально высокая протонная проводимость кристаллов CsHSO4 и CsHSeO4 [2].
Практическая значимость упомянутых соединений обусловлена перспективами их применения в качестве функциональных материалов, включая мембраны водородных топливных элементов, о чем упоминалось в ведущих научных журналах, в том числе “Nature” [3], “Physics Today” [4].
Технологически важным является вопрос о получении и исследовании новых кристаллов, структурной обусловленности их свойств, возможности расширения температурного диапазона существования суперпротонной фазы. Такие исследования активно ведутся как в России [1, 5–8], так и в других странах [9–13].
Помимо поиска новых соединений, обладающих суперпротонной проводимостью, немаловажной задачей является модификация свойств уже известных соединений. Одним из способов существенного влияния на свойства кристаллов, принадлежащих упомянутому семейству, является модификация сеток водородных связей за счет внедрения ионов аммония в катионные позиции структуры. Ранее [14–16] наглядно были продемонстрированы возможности такого подхода.
Для получения новых соединений семейства MmHn(AO4)(m + n)/2 · yH2O были изучены фазовые равновесия в тройной водно-солевой системе CsHSO4–NH4H2PO4–H2O и первично исследованы сечения в четырехкомпонентной системе CsHSO4–CsH2PO4–NH4H2PO4–H2O при содержании CsH2PO4 25, 50 и 75 мол. %.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В настоящей работе использовали следующие реактивы: гидросульфат цезия (CsHSO4, CSH) марки Ч, ТУ 6-09-04-198-83; дигидроортофосфат цезия (CsH2PO4, CDP) марки ХЧ, ТУ 6-09-04-201-82; дигидроортофосфат аммония (NH4H2PO4, ADP) марки ХЧ, ГОСТ 3771-74.
Фазовые равновесия изучали методом одновременных параллельных процессов кристаллизации. Данное исследование проводилось в специальном лабораторном термостате-шейкере с подвижным держателем сосудов лоткового типа (для обеспечения перемешивания в термостатируемых сосудах) и программируемым ПИД-регулятором температуры. Упомянутый термостат является установкой самостоятельной сборки. В качестве основы использован термостат водяной лабораторный LR-20 (ГДР), на который смонтирован привод подвижного лотка, а стандартное реле контроля температуры заменено на программный ПИД-регулятор ОВЕН ТРМ151 (Россия). Объем рабочей емкости термостата 20 л; теплоноситель – вода дистиллированная; рассогласование температур по объему термостата – ±0.2°С; точность поддержания/регулирования температуры ±0.1°С; тип движения лотка-держателя возвратно-поступательный. Для минимизации тепловых потерь и испарения теплоноситель укомплектован крышкой с резиновыми уплотнителями. Общий вид установки с обозначением основных узлов представлен на рис. 1.
Рис. 1.
Общий вид термостата-шейкера: 1 – корпус, 2 – блок управления температурой (ПИД-регулятор ТРМ151) и перемешиванием, 3 – привод подвижного лотка-держателя, 4 – лоток-держатель, 5 – крышка.

Готовили серию маточных растворов в одинаковых герметичных сосудах (кристаллизаторах) с переменным соотношением исходных сухих компонентов и минимальным количеством дистиллированной воды (5 мл). Затем при температуре, незначительно превышающей комнатную, определяли растворимость навесок компонентов посредством повторного добавления малых порций (1–5 мл) воды до получения насыщенных растворов с минимальным (не более 3–5 кристалликов соли) содержанием осадка на дне сосуда.
Дополнительно контроль количества растворенной в насыщенном растворе соли анализировали по содержанию в насыщенном растворе суммы аммония и цезия весовым методом в виде тетрафенилборатов [17]. В качестве реактива для определения суммы аммония и цезия применяли 0.6%-ный водный раствор (C6H5)4BNa марки ЧДА. Для приготовления реактива 1.5 г сухого (C6H5)4BNa и 0.5 г сухого Al(OH)3 перемешивали в течение 10 мин с 250 мл воды. После перемешивания раствор фильтровали через воронку со стеклянным пористым фильтром № 4. В качестве промывной жидкости использовали воду, к которой на каждые 100 мл добавляли 3 мл указанного реактива и 0.5 мл ледяной уксусной кислоты.
Перед добавлением реактива в пробу анализируемого раствора приливали соляную кислоту для улучшения коагуляции образующегося осадка (0.5 мл концентрированной соляной кислоты на пробу объемом 10 мл). Осаждение проводили, постепенно добавляя реактив к анализируемому раствору. Выпавший осадок рекомендуется, для простоты фильтрации, отстаивать в течение 0.5–1 ч для укрупнения частиц осадка, затем фильтровать через пористый фильтр-тигель № 4, промывать специальной жидкостью и затем водой (2 мл). Фильтр с осадком высушивали в сушильном шкафу при температуре 100–110°С в течение 1 ч и взвешивали. Ошибка определения в данном случае не превышает 0.5%.
После определения растворимости маточные растворы дополнительно выдерживали в течение двух суток при установленной температуре. Эта процедура необходима для установления в кристаллизаторе динамического межфазного равновесия между насыщенным раствором и осадком. После выдерживания температуру кристаллизаторов снижали по одинаковой программе до требуемого значения в течение нескольких суток. Образовавшиеся кристаллы имели размеры 2–5 мм и легко декантировались от маточного раствора, поверхность образцов отмывали 96%-ным этиловым спиртом.
Полученные таким образом монокристаллы использовали для определения фазового состава равновесных с маточным раствором твердых фаз методом порошкового рентгенофазового анализа (РФА). Он выполнен при комнатной температуре на рентгеновском дифрактометре Rigaku Miniflex 600 (Япония) (Cu-излучение без фильтра, непрерывный режим съемки со скоростью 2 град/мин, шаг 0.02° в интервале углов 2θ 5°–70°, без вращения образца, в воздушной атмосфере).
РЕЗУЛЬТАТЫ
Для изучения фазовых равновесий в системе CsHSO4–NH4H2PO4–H2O была приготовлена серия из девяти исходных маточных растворов с соотношением от 10CSH : 90ADP до 90CSH : 10ADP с шагом 10 мол. %. Точки 0CSH : 100ADP и 100CSH : 0ADP соответствуют справочным величинам растворимости исходных компонентов, и их решено было не синтезировать.
На основе данных о псевдотройной системе CsHSO4–CsH2PO4–H2O, хорошо изученной авторами [18, 19], для первичного анализа фазовых равновесий в четырехкомпонентной системе CsHSO4–CsH2PO4–NH4H2PO4–H2O было решено синтезировать дополнительные исходные растворы xCSH : yCDP : zADP (при x + y + z = = 100 мол. %) с шагом 10 мол. %. при y = 25, 50 и 75 мол. %. Составы синтезированных растворов представлены в табл. 1.
Таблица 1.
Составы исходных растворов
| Номер процесса кристаллизации (мол. % NH4H2PO4 без учета воды) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 (10) | 2 (20) | 3 (30) | 4 (40) | 5 (50) | 6 (60) | 7 (70) | 8 (80) | 9 (90) | |||
| Номер ряда (мол. % CsН2PO4 без учета воды) | 1 (0) | NH4H2PO4, г | 1.6 | 3.3 | 5.3 | 7.5 | 10.0 | 12.9 | 16.2 | 20.0 | 24.5 |
| CsHSO4, г | 28.4 | 26.7 | 24.7 | 22.5 | 20.0 | 17.1 | 13.8 | 10.0 | 5.5 | ||
| H2O, г | 6.0 | 5.5 | 5.0 | 5.0 | 6.0 | 6.5 | 7.0 | 12.0 | 12.0 | ||
| 2 (25) | NH4H2PO4, г | 1.6 | 3.3 | 5.3 | 7.5 | 10.0 | 12.9 | 16.2 | |||
| CsHSO4, г | 20.5 | 18.3 | 15.9 | 13.1 | 10.0 | 6.4 | 2.3 | ||||
| CsH2PO4, г | 7.9 | 8.3 | 8.8 | 9.3 | 10.0 | 10.7 | 11.5 | ||||
| H2O, г | 5.5 | 5.5 | 5.5 | 5.5 | 8.0 | 11.0 | 13.0 | ||||
| 3 (50) | NH4H2PO4, г | 1.6 | 3.3 | 5.3 | 7.5 | ||||||
| CsHSO4, г | 12.6 | 10.0 | 7.1 | 3.8 | |||||||
| CsH2PO4, г | 15.8 | 16.7 | 17.6 | 18.7 | |||||||
| H2O, г | 6.5 | 7.5 | 9.5 | 11.0 | |||||||
| 4 (75) | NH4H2PO4, г | 1.6 | 3.3 | 4.3 | |||||||
| CsHSO4, г | 4.8 | 1.7 | 0 | ||||||||
| CsH2PO4, г | 23.7 | 25.0 | 25.7 | ||||||||
| H2O, г | 10.0 | 10.5 | 11.0 | ||||||||
Была задана температура определения растворимости – 50°С, что обусловлено сочетанием малого времени установления динамического равновесия в растворе (менее суток) и значительным температурным запасом для получения кристаллических образцов большого размера методом управляемого снижения температуры раствора. Использование более высокой температуры осложнено конструкционными особенностями экспериментальной установки, а использование более низкой температуры значительно увеличивает время эксперимента и осложняет получение приемлемых по размеру и качеству кристаллических образцов. Во всех процессах кристаллизации температуру снижали одинаково – до 40°С, что позволило получить достаточное количество кристаллических образцов для исследований в каждом процессе кристаллизации без значительного изменения состава маточного раствора. При учете того, что во всех 23 процессах кристаллизации количество кристаллов на выходе не превышало 1 г, а суммарное количество исходных реактивов (NH4H2PO4, CsHSO4 и CsH2PO4) составляло 30 г, максимальное суммарное отклонение от расчетного значения не превышало ±3 мас. %, или ±0.8 мол. % для NH4H2PO4, или ±0.4 мол. % для CsHSO4 и CsH2PO4.
Распределение растворимостей и составов, равновесных маточным растворам, твердых фаз, определенных по данным РФА, представлено в табл. 2. В указанных условиях в системе выявлены пять областей кристаллизации. Области кристаллизации исходных компонентов – CsHSO4, NH4H2PO4 и CsH2PO4, область кристаллизации исходного компонента CsH2PO4 представлена не в виде чистого дигидроортофосфата цезия, а в виде предельного состава твердого раствора Cs1 ‒ x(NH4)xH2PO4, который подробно рассмотрен в [20]. Отметим, что чистый дигидроортофосфат цезия и твердый раствор Cs1 – x(NH4)xH2PO4 со значениями x меньше предельного также кристаллизуются в данной системе, но область кристаллизации этих фаз ограничена содержанием NH4H2PO4 в исходных растворах – концентрацией меньше 10 мол. %, поэтому матрицей синтеза не была охвачена.
Таблица 2.
Растворимость исходных растворов и составы равновесных твердых фаз
| Номер процесса кристаллизации (мол. % NH4H2PO4 (ADP) без учета воды) | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 (10) | 2 (20) | 3 (30) | 4 (40) | 5 (50) | 6 (60) | 7 (70) | 8 (80) | 9 (90) | |||
| Номер ряда (мол. % CsH2PO4 (CDP) без учета воды) | 1 (0) | Раствор, мас. % | 83.3 | 84.5 | 85.7 | 85.7 | 83.3 | 82.2 | 81.1 | 71.4 | 71.4 |
| Раствор, мол. % | 29.2 | 34.3 | 35.6 | 37.0 | 34.3 | 32.4 | 34.0 | 24.6 | 26.2 | ||
| Твердая фаза | CsHSO4 | Cs3NH4(HSO4)3 (H2PO4) + +CsHSO4 | Cs3NH4(HSO4)3 (H2PO4) | CsNH4(HSO4) (H2PO4)+ Cs3NH4 (HSO4)3(H2PO4) | CsNH4(HSO4)(H2PO4) | NH4H2PO4 | |||||
| 2 (25) | Раствор, мас. % | 84.5 | 84.5 | 84.5 | 84.5 | 78.9 | 73.2 | 69.8 | |||
| Раствор, мол. % | 31.0 | 32.2 | 33.4 | 34.8 | 28.1 | 23.4 | 21.8 | ||||
| Твердая фаза | (Cs,NH4)4(HSO4)3(H2PO4) | (Cs,NH4)4(HSO4)3 (H2PO4) + (Cs,NH4)2 (HSO4)(H2PO4) + + NH4H2PO4 | NH4H2PO4 | ||||||||
| 3 (50) | Раствор, мас. % | 82.2 | 80.0 | 75.9 | 73.2 | ||||||
| Раствор, мол. % | 27.6 | 25.8 | 22.5 | 21.1 | |||||||
| Твердая фаза | (Cs,NH4)H2PO4 + + (Cs,NH4)4(HSO4)3 (H2PO4) | (Cs,NH4)H2PO4 + + (Cs,NH4)4(HSO4)3 (H2PO4) + NH4H2PO4 | NH4H2PO4 | ||||||||
| 4 (75) | Раствор, мас. % | 78.9 | 75.0 | 71.4 | |||||||
| Раствор, мол. % | 23.6 | 20.7 | 18.7 | ||||||||
| Твердая фаза | (Cs,NH4)H2PO4 | ||||||||||
По этой же причине (ограниченности матрицы синтеза) отображение результатов исследования фазовых равновесий в виде соответствующей диаграммы, пусть и схематической, не будет корректным. Построение фазовой диаграммы данной системы требует отдельного исследования с матрицей синтеза, включающей в себя не менее 45 точек, а также дополнительные дифракционные эксперименты для установления концентрационных координат точек нонвариантного равновесия.
Помимо исходных компонентов в данной системе образуются кислые сульфатофосфаты – твердые растворы (Cs,NH4)4(HSO4)3(H2PO4) и (Cs,NH4)2(HSO4)(H2PO4). Микрофотографии полученных кристаллических образцов представлены на рис. 2, а дифрактограммы полученных фаз – на рис. 3.


ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ И ВЫВОДЫ
В результате проведенных исследований впервые получены кристаллы Cs3NH4(HSO4)3(H2PO4) и твердый раствор (Cs,NH4)4(HSO4)3(H2PO4), а также CsNH4(HSO4)(H2PO4) и твердый раствор (Cs,NH4)(HSO4)(H2PO4) (рис. 4, 5).
Рис. 4.
Рентгенофазовый анализ образцов Cs3NH4(HSO4)3(H2PO4) (1), твердого раствора (Cs,NH4)4(HSO4)3(H2PO4) (2) и кристалла Cs4(HSO4)3(H2PO4) (3).
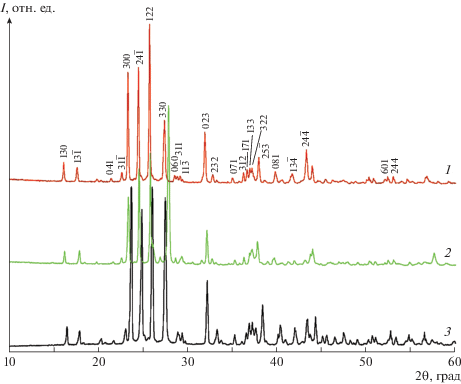
Рис. 5.
Рентгенофазовый анализ образцов CsNH4(HSO4)(H2PO4) (1) и твердого раствора (Cs,NH4)(HSO4)(H2PO4) (2).

Составы кристаллов определены в соответствии с составами маточных растворов. В пользу этого соответствия (пусть и косвенно) свидетельствует и конгруэнтный характер растворимости соединений (способность к перекристаллизации из водного раствора без изменения фазового состава), и выявленное структурное сходство новых соединений с кристаллами, состав которых доподлинно установлен [18, 20].
Фактически твердый раствор (Cs,NH4)4(HSO4)3(H2PO4) ограничен составами: ранее изученным [18, 19] Cs4(HSO4)3(H2PO4) и Cs3NH4(HSO4)3(H2PO4). Оказалось, что положения основных рефлексов на дифрактограмме Cs3NH4(HSO4)3(H2PO4) практически совпадают с позициями рефлексов для Cs4(HSO4)3(H2PO4) (рис. 3), и соответственно по данным РФА были определены параметры моноклинной элементарной ячейки: a = 12.311(4), b = 18.744(1), c = = 9.349(3) Å, β = 109.488(31)°, V = 2004.11 Å3. Эти результаты, по-видимому, указывают на существенное сходство структур соединений Cs4(HSO4)3(H2PO4) и Cs3NH4(HSO4)3(H2PO4). Более подробное изучение как самого соединения Cs3NH4(HSO4)3(H2PO4), так и твердого раствора (Cs,NH4)4(HSO4)3(H2PO4), несомненно, является предметом отдельного исследования.
Твердый раствор (Cs,NH4)(HSO4)(H2PO4) также ограничен составом CsNH4(HSO4)(H2PO4) с одной стороны и, по-видимому, составом Cs2(HSO4)(H2PO4), с другой стороны, с той лишь разницей, что соединение Cs2(HSO4)(H2PO4) в условиях проведения исследования (t ~ 40°C) не кристаллизуется из водных растворов [18, 21]. Параметры ячейки соединения CsNH4(HSO4)(H2PO4) по данным РФА составляют: a = 7.786(2), b = = 7.612(2), c = 7.654(4) Å, β = 101.07(3)°, V = = 445.1(7) Å3. Они весьма близки к параметрам ячейки Cs2(HSO4)(H2PO4): a = 7.856(8), b = = 7.732(7), c = 7.827(7) Å, β = 99.92(4)°, V = = 468.3(8) Å3 [20]. Как и в случае соединения Cs3NH4(HSO4)3(H2PO4), более подробное изучение соединения CsNH4(HSO4)(H2PO4) и твердого раствора (Cs,NH4)(HSO4)(H2PO4) является предметом отдельного исследования.
В работе использовано оборудование ЦКП ИК РАН (Минобрнауки, проект RFMEFI62114X0005). Работа выполнена при поддержке Министерства науки и высшего образования РФ в рамках Государственного задания ФНИЦ “Кристаллография и фотоника” РАН в части получения кристаллов сложных сульфатофосфатов цезия-аммония. Рентгеновские исследования выполнены при финансовой поддержке Российского фонда фундаментальных исследований (проект № 18-32-20050).
Список литературы
Баранов А.И. // Кристаллография. 2003. Т. 48. № 6. С. 1081.
Баранов А.И., Шувалов Л.А., Щагина Н.М. // Письма в ЖЭТФ. 1982. Т. 36. № 11. С. 381.
Norby T. // Nature. 2001. V. 410. P. 877.
Fitzgerald R. // Physics Today. 2001. V. 54. № 7. P. 22.
Иванов-Шиц А.К., Мурин И.В. Ионика твердого тела. Т. 2. СПб.: Изд-во СПбГУ, 2010. 1000 с.
Коморников В.А., Гребенев В.В., Макарова И.П. и др. // Кристаллография. 2016. Т. 61. № 4. С. 665.
Коморников В.А., Гребенев В.В., Андреев П.В., Дмитричева Е.В. // Кристаллография. 2015. Т. 60. № 3. С. 496.
Makarova I., Grebenev V., Dmitricheva E. et al. // Acta Cryst. B. 2016. V. 72. P. 133.
Haile S.M., Chisholm C.R.I., Sasaki K. et al. // Faraday Discussions. 2007. V. 134. P. 17.
Varga A., Brunelli N.A., Louie M.W. et al. // J. Mater. Chem. 2010. V. 20. P. 2190.
Qing G., Kikuchi R., Takagaki A. et al. // Electrochim. Acta. 2015. V. 169. P. 219.
Lim A.R., Hyung M. // Chem. Phys. 2015. V. 459. P. 59.
Ostrowski A., Bednarski W. // Solid State Ionics. 2017. V. 301. P. 152.
Dmitricheva E.V., Makarova I.P., Grebenev V.V. et al. // Solid State Ionics. 2014. V. 268. P. 68.
Селезнева Е.В., Макарова И.П., Гребенев В.В., Коморников В.А. // Кристаллография. 2018. Т. 63. № 4. С. 548.
Selezneva E.V., Makarova I.P., Malyshkina I.A. et al. // Acta Cryst. B. 2017. V. 73. P. 1105.
Geilmann W., Gebauhr W. // Anal. Chem. 1953. V. 139. P. 161.
Коморников В.А., Гребенев В.В., Макарова И.П. и др. // Кристаллография. 2016. Т. 61. № 4. С. 645.
Makarova I., Grebenev V., Dmitricheva E. et al. // Acta Cryst. B. 2016. V. 72. P. 133.
Baranov A.I., Grebenev V.V., Khodan A.N. et al. // Solid State Ionics. 2005. V. 176. P. 2871.
Chisholm C.R.I., Haile S.M. // Acta Cryst. B.1999. V. 55. P. 937.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография