

Кристаллография, 2019, T. 64, № 6, стр. 884-890
Синтез и строение 1,3-дикарбонильных соединений на основе 9,10-фенантренхинона. кристаллическая и молекулярная структура биядерного комплекса-фонарика меди(II) Cu2[μ2-OOCCH2(C14H8)(CO)2OC2H5]4(NCCH3)2
Р. В. Линко 1, *, М. А. Рябов 1, Н. А. Полянская 1, П. В. Дороватовский 2, В. Н. Хрусталев 1, 2
1 Российский университет дружбы народов
Москва, Россия
2 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
* E-mail: rlinko@mail.ru
Поступила в редакцию 26.06.2018
После доработки 26.06.2018
Принята к публикации 11.08.2018
Аннотация
Взаимодействием 9,10-фенантренхинона с ацетилацетоном и ацетоуксусным эфиром и последующим восстановлением получены 1,3-дикарбонильные соединения, которые охарактеризованы совокупностью спектральных и квантово-химических методов. Установлено, что полученные соединения в кристаллическом состоянии и в растворах находятся в кетонной форме. Методом рентгеноструктурного анализа определена кристаллическая и молекулярная структура биядерного карбоксилатного комплекса меди(II) с геометрией “китайского фонарика” Cu2[μ2-OOCCH2(C14H8)(CO)2OC2H5]4(NCCH3)2, образовавшегося в результате окисления исходного 1,3-дикарбонильного соединения при комплексообразовании. Координационным полиэдром каждого атома меди является квадратная пирамида (4 + 1) с четырьмя атомами О четырех карбоксильных групп в основании и атомом N ацетонитрила в апикальной позиции. Формирование димера-фонарика обусловлено бидентатно-мостиковым μ2-O,O' характером координации карбоксильных групп.
ВВЕДЕНИЕ
В последнее время 9,10-фенантренхинон (ФХ) и его производные являются объектами активных исследований с целью построения на их 1,2-дикарбонильной основе конденсированных гетероциклических соединений ряда имидазола [1–4], пиразина [5–8] и 1,2,4-триазина [9, 10] с достаточно широким спектром полезных свойств.
Интермедиатами в синтезе других конденсированных гетероциклов, содержащих фенантреновую систему, а также лигандами для получения металлокомплексов могут являться продукты нуклеофильного присоединения СН-кислот (нитрометан [11], ацетон [12–14], малонодинитрил [15], 1-(1-циклопентенил)пирролидин [16] и др.) по карбонильным группам ФХ и его производных. При взаимодействии ФХ и его нитропроизводных с ацетоном [12, 14] образуются 1,4-дикарбонильные соединения, из которых реакцией с гидразином или его замещенными могут быть получены производные пиридазина. Для синтеза пиримидиновых производных может быть использована реакция бинуклеофилов (мочевина, тиомочевина, гуанидин, бензамидин и др.) с 1,3-дикарбонильными соединениями, полученными взаимодействием ФХ с ацетоуксусным эфиром [17, 18].
Цель настоящей работы – установление строения и изучение свойств продуктов конденсации, полученных взаимодействием ФХ с ацетилацетоном и ацетоуксусным эфиром, их восстановления и комплексообразования.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез. 3-Ацетил-11b-гидрокси-1H-циклопента[l]фенантрен-2(11bH)-он (I). 2.0 г (9.6 ммоля) ФХ растворяли в 70 мл DMSO, добавляли 9 мл ацетилацетона, 6 капель 20%-ного раствора NaOH и перемешивали при комнатной температуре до исчезновения ФХ (2 дня, контроль методом тонкослойной хроматографии). Реакционную массу выливали в 400 мл воды, подкисленной HCl. Осадок отфильтровывали, промывали водой и сушили; после перекристаллизации из этанола получено 1.4 г соединения I (выход 50%), Тпл = = 177–178°С. Найдено, %: С 78.32, Н 4.72. Для С19Н14О3 вычислено, %: С 78.61, Н 4.86. Спектр ЯМР 1Н (δ, м.д.; J, Гц): 2.46 (с, 3Н, СОМе); 2.62 (с, 1Н, ОН); 3.15, 3.33 (оба д, по 1Н, С(1)Н, спектр АВ, JАВ = 19.0); 7.36–7.98 (м, 8Н, Ar).
Этил-11b-гидрокси-2,11b-дигидро-2-оксо-1H-циклопента[l]фенантрен-3-карбоксилат (II). Суспензию 5.0 г (24 ммоля) ФХ, 25 мл CH3CN, 20 мл DMSO, 5 мл ацетоуксусного эфира и четырех капель 20%-ного NaOH перемешивали при комнатной температуре до исчезновения ФХ (2 дня, контроль методом тонкослойной хроматографии). Реакционную массу выливали в 600 мл воды, подкисленной HCl. Полученный осадок отфильтровывали, промывали водой и сушили. После перекристаллизации из смеси этанола с ацетонитрилом получено 4.2 г соединения II (выход 54%), Тпл = 188–189°С (из этанола); по данным [17] Тпл = 190–191.6°С (из CH3CN).
1-Ацетил-1H-циклопента[l]фенантрен-2(3H)-он (III). Суспензию 9.5 г (32.7 ммоля) I и 25 г KI в 90 мл концентрированного раствора HCl перемешивали при 70°С в течение 7 мин. Реакционную массу выливали в 500 мл воды, осадок отфильтровывали на стеклянном фильтре, отжимали на фильтре, промывали 20%-ным раствором KI, водой и сушили. После перекристаллизации из 70 мл этанола и 30 мл хлороформа получено 3.1 г соединения III (выход 34%), Тпл = 184–186°С. Найдено, %: С 83.42, Н 5.26. Для С19Н14О2 вычислено, %: С 83.19, Н 5.14. Спектр ЯМР 1Н (δ, м.д.; J, Гц): 2.31 (с, 3Н, СОМе); 3.91, 4.09 (оба д, по 1Н, С(3)Н, спектр АВ, JАВ = 23.0); 4.98 (с, 1Н, С(1)Н); 7.35–7.85, 8.55–8.85 (оба м, по 6Н и 2Н, Ar).
Этил-2,3-дигидро-2-оксо-1H-циклопента[l]фенантрен-1-карбоксилат (IV). Суспензию 2.8 г (8.7 ммоля) II в 8.5 мл 55–58%-ного раствора HI перемешивали при 70°С в течение 3 мин. Реакционную массу выливали в 200 мл воды, осадок отфильтровывали на стеклянном фильтре, отжимали на фильтре, промывали 20%-ным раствором KI, водой и сушили. После перекристаллизации из смеси 12 мл этанола и 8 мл хлороформа получено 1.1 г соединения IV (выход 40%), Тпл = 123–124°С (из этанола), по данным [18] Тпл = 122–124°С (из этанола).
Cu2[μ2-OOCCH2(C14H8)(CO)2OC2H5]4 (NCCH3)2 (V). Смешивали горячие растворы 0.2 г (0.66 ммоль) IV в 5 мл CH3CN и 0.112 г (0.66 ммоль) CuCl2 · 2H2O в 10 мл CH3CN. Полученный раствор коричневого цвета выдерживали 2 дня на водяной бане при 60–65°С, фильтровали и оставляли на трое суток при комнатной температуре. Кристаллы зеленого цвета отфильтровывали на стеклянном фильтре, промывали CH3CN и сушили на воздухе; получено 0.034 г соединения V. Монокристаллы для рентгеноструктурного анализа (РСА) были отобраны из маточного раствора после нагревания и хранились в запаянном капилляре.
ИК-спектры регистрировали в диапазоне 4000–400 см–1 на спектрометрах Nicolet 6700 FT-IR в кристаллическом состоянии методом нарушенного полного внутреннего отражения (приставка НПВО с алмазным кристаллом) и Specord 75-IR в кристаллическом состоянии (таблетки с KBr) и в растворах (C2H4Cl2).
Электронные спектры поглощения (ЭСП) соединений I–IV в этаноле получены на спектрометрах Specord M-40 и Varian Cary 50 Scan в кварцевых кюветах толщиной 1.0 см при концентрациях веществ 4.0 × 10–5 моль/л.
Спектры ЯМР 1Н регистрировали на ЯМР-спектрометре Bruker WP-80 (рабочая частота 80 МГц) в CDCl3, внутренний стандарт – тетраметилсилан.
Рентгеноструктурное исследование V проводили в НИЦ “Курчатовский институт”. Параметры элементарной ячейки и интенсивности отражений измерены на синхротронной станции ”БЕЛОК”.
Структура определена прямым методом и уточнена в анизотропном приближении для неводородных атомов. Атомы водорода включены в уточнение с фиксированными позиционными параметрами (модель “наездника”) и изотропными параметрами смещения (Uизо(H) = 1.5Uэкв(О), 1.2Uэкв(N) и 1.2Uэкв(C)). Кристаллографические данные, параметры эксперимента и уточнения структуры V приведены в табл. 1.
Таблица 1.
Кристаллографические характеристики, данные эксперимента и уточнения структуры C84H66Cu2N2O20 (V)
| Сингония, пр. гр., Z | Тетрагональная, P4nc, 2 |
| a, c, Å | 16.189(3), 16.895(3) |
| V, Å3 | 4427.9(13) |
| Dx, г/см3 | 1.163 |
| Излучение; λ, Å | Синхротрон, 0.96990 |
| μ, мм–1 | 1.246 |
| Т, K | 100(2) |
| Размер образца, мм | 0.20× 0.15 × 0.10 |
| Дифрактометр | Rayonix SX165 CCD |
| Тип сканирования | φ |
| Учет поглощения, Tmin/Tmax | Полуэмпирический, 0.790/0.880 |
| θmax, град | 19.21 |
| Пределы h, k, l | –20 ≤ h ≤ 20, –20 ≤ k ≤ 20, ‒21 ≤ l ≤ 21 |
| Число отражений: измеренных/независимых (N1), Rint/с I > 2σ(I) (N2) | 46972/4819, 0.103/4326 |
| Метод уточнения | Полноматричный МНК по F 2 |
| Число параметров | 244 |
| Коэффициент экстинкции | 0.0045(4) |
| R1/wR2 по N1 | 0.0742/0.1646 |
| R1/wR2 по N2 | 0.0696/0.1612 |
| S | 1.022 |
| Δρmin/Δρmax | –1.974/1.757 |
| Программы | iMosflm, CCP4 [19], Scala [20], SHELXTL [21] |
Полные данные РСА для соединения V депонированы в Кембриджский банк структурных данных (CCDC № 1849832).
Квантово-химическое моделирование электронной структуры молекул проводили в рамках приближения теории функционала плотности (DFT) с использованием гибридного трехпараметрического обменного функционала Беке [22] с корреляционным функционалом Ли–Янга–Парра [23] (B3LYP) [24] и базисного набора def2-SV(P) [25]. Для анализа электронной структуры основного состояния молекул лигандов и комплексов применяли подход естественных связывающих орбиталей [26]. Все вычисления проводились средствами программного комплекса Firefly 7.1.G [27].
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Взаимодействием ФХ с ацетилацетоном и ацетоуксусным эфиром в присутствии каталитических количеств NaOH получены соединения I и II, при восстановлении которых образуются соединения III и IV соответственно (схема 1). При нагревании IV с хлоридом меди(II) в ацетонитриле получено комплексное соединение V. Соединения I, III и V получены впервые.
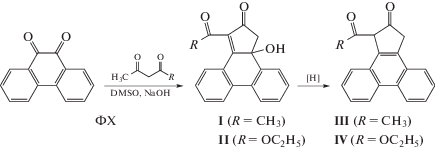
Для соединений I–IV потенциально возможна кето-енольная таутомерия, поскольку в α-положении к карбонильным группам находятся подвижные атомы водорода. Формулы соответствующих кетонных и енольных форм приведены на схеме 2.

Расчеты, выполненные методом DFT, показывают, что для молекул I–IV более устойчивы кетонные формы (рис. 1).

Енольные формы, в которых атом водорода переходит от атома С15 на атомы O3 (I и II) или О1 (III и IV) (нумерация атомов по рис. 1), менее устойчивы на 99 (Ie), 73 (IIe), 71 (IIIe) и 58 (IVe) кДж/моль, чем соответствующие кетонные формы. Енольные формы Ie’ и IIIe''', в которых атом водорода переходит на атом О2 от атома С19, менее устойчивы на 62 и 71 кДж/моль соответственно. Енольные формы молекул IIIe’, IIIe'' и IVe’, в которых атомы водорода переходят на атомы О1 или О2 не от атома С15, а от атома С17, менее выгодны на 19, 8 и 13 кДж/моль, чем соответствующие кетоны. В ходе расчета енольной формы IVe'' атом водорода переходит от атома О2 на атом О1 с образованием енольной формы IVe’.
ИК-спектры соединений I–IV также указывают на существование этих соединений в 1,3-дикарбонильных формах. Так, в спектрах каждого соединения в кристаллическом состоянии и в растворах имеется по две полосы поглощения карбонильных групп (табл. 2). Согласно расчетам эти полосы относятся к валентным колебаниям связей С16–О и С18–О (рис. 1). Рассчитанные с понижающим коэффициентом 0.945 значения волновых чисел этих полос равны 1718 и 1691 см–1 (I), 1735 и 1711 см–1 (II), 1776 и 1728 см–1 (III), 1759 и 1733 см–1 (IV) и хорошо согласуются с экспериментальными данными, полученными в растворах.
Таблица 2.
Данные ИК и электронной спектроскопии соединений I–IV
| Соединение | ИК-спектры, ν, см–1 | ЭСП | |||
|---|---|---|---|---|---|
| Условия | ν(O–H) | ν(C=O) | Условия | λ, нм (lg ε) | |
| I | НПВО | 3453 | 1705, 1683 | C2H5OH | 301 (4.08), 263 (4.59) |
| KBr | 3450 | 1710, 1683 | C2H5OH + KOH | 309 (4.23), 284 (4.22), 263 (4.39) | |
| C2H4Cl2 | 3550 | 1710, 1694 | |||
| II | НПВО | 3443 | 1726, 1698 | C2H5OH | 299 (4.07), 264 (4.60) |
| KBr | 3445 | 1730, 1700 | |||
| C2H4Cl2 | 3555 | 1733, 1709 | C2H5OH [8] | 300 (4.06), 264 (4.58) | |
| нуйол [8] | 3350 | 1715, 1680 | |||
| III | НПВО | 1744, 1698 | C2H5OH | 301 (4.20), 287 (4.19), 278 (4.30), 253 (4.94) | |
| KBr | 1748, 1698 | ||||
| C2H4Cl2 | 1755, 1712 | C2H5OH + KOH | 345 (4.16), 301 (4.06), 278 (4.23), 250 (4.80) | ||
| IV | НПВО | 1763, 1713 | C2H5OH | 301 (4.14), 287 (4.12), 278 (4.23), 256 (4.94) | |
| KBr | 1762, 1709 | ||||
| C2H4Cl2 | 1765, 1729 | C2H5OH + KOH | 368 (4.01), 321 (4.04), 278 (4.44), 250 (4.74) | ||
В ИК-спектрах I и II (в KBr) в области 3450 см–1 наблюдаются интенсивные полосы валентных колебаний гидроксильных групп. При переходе от кристаллического состояния к растворам значение ν(О–Н) смещается в высокочастотную область на ~110 см–1, что объясняется разрывом межмолекулярных Н-связей. В ИК-спектрах III и IV полосы в этой области отсутствуют, что также указывает на отсутствие енольных форм.
Расчеты методом DFT указывают, что моноанион I, образованный при депротонировании атома О1, на 67 кДж/моль стабильнее, чем моноанион, образованный при депротонировании атома С15. Дианион молекулы I может быть образован при депротонировнии обоих этих атомов.
Электронный спектр поглощения I характеризуется наличием интенсивных полос 301 и 263 нм (табл. 2). При добавлении раствора КОН к этанольному раствору I длинноволновая полоса (ДП) 301 нм батохромно смещается до 309 нм, слегка возрастая по интенсивности, а полоса 263 нм сохраняет свое положение, кроме того, появляется дополнительная полоса 284 нм. Батохромные сдвиги интенсивных ДП наблюдаются при подщелачивании этанольных растворов и для III, IV.
Расчеты ЭСП молекул I и II указывают, что батохромный сдвиг ДП наблюдается при депротонировании атома С15, в данном случае при образовании дианиона, поскольку депротонирование атома О1 не приводит к батохромному сдвигу ДП. Так, рассчитанные для нейтральной молекулы I полосы 304 и 259 нм сдвигаются аналогично экспериментальному ЭСП I до значений 315, 285 и 262 нм в рассчитанном спектре дианиона молекулы I.
Для молекул III и IV депротонирование атома С17 несколько выгоднее, чем С15: на 1 и 16 кДж/моль соответственно. При этом каждое депротонирование приводит к батохромному сдвигу ДП. Таким образом, наблюдаемый в эксперименте значительный (44 и 67 нм) батохромный сдвиг ДП может быть обусловлен образованием дианионов III и IV, образующихся при депротонировании атомов С17 и C15.
При взаимодействии 1,3-дикарбонильного соединения IV с хлоридом меди(II) в ацетонитриле при нагревании получен комплекс V, строение которого, отличное от ожидаемого, установлено методом РСА (схема 3).
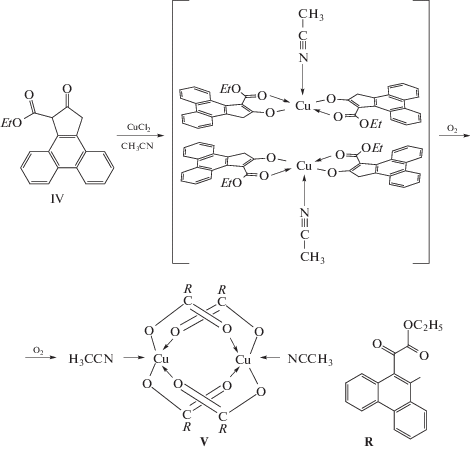
Оказалось, что комплекс V (рис. 2) относится к достаточно распространенной группе биядерных карбоксилатных комплексов меди(II) с геометрией “китайского фонарика” и общей формулой Cu2(μ2-OOCR)4L2 [28–31]. Существенной особенностью V является наличие в нем связанного с карбоксильной группой объемного радикала (R = = CH2(C14H8)(CO)2OC2H5), содержащего фенантреновую систему. Каждый атом меди в биядерном комплексе имеет квадратно-пирамидальное (КП 4 + 1) окружение с четырьмя атомами О четырех карбоксильных групп в основании и атомом N ацетонитрила в апикальной позиции (КП1 и КП2 для пирамид Cu1 и Cu2 соответственно). Формирование димера-фонарика обусловлено бидентатно-мостиковым μ2-O,O'-характером координации каждой из четырех карбоксильных групп.

Молекула V обладает собственной симметрией С4 и в кристалле занимает частное положение на оси четвертого порядка, проходящей через молекулы CH3CN и атомы Cu: C22–C21–N1–Cu1–Cu2–N2–C23–C24, поэтому основаниями пирамид являются правильные квадраты. Атомы Cu1 и Cu2 выходят из их плоскостей к вершинам пирамид на 0.239 и 0.194 Å, что существенно ниже аналогичного значения 0.264 Å для Cu2(μ2-OOCCF3)4(NCCH3)2 [30]. В КП2, в которой наблюдается наименьший выход атома Cu2 из плоскости основания, расстояние Cu2–N2 2.173(11) Å увеличено по сравнению с соответствующим расстоянием 2.141(11) Å в КП1 и 2.114 Å в [30].
Вследствие внутренней симметрии расстояния Cu–O в основании каждой из пирамид равны, что наблюдалось для комплекса Cu2(μ2-OOCCF3)4(DBE)2 (где DBE – дибензиловый эфир) [28] и достаточно редко встречается для подобных комплексов с небольшими заместителями у карбоксильной группы. Межатомные расстояния Cu–O (1.978(4) и 2.002(4) Å, табл. 3) близки между собой и немногим больше соответствующих расстояний в аналогичных комплексах: 1.960(5)–1.976(5) Å в Cu2(μ2-OOCCF3)4(DBE)2 и 1.929(3)–1.974(3) Å в Cu2(μ2-OOCBut)4(DBE)2 [28], 1.964(3)–1.972(2) Å в Cu2(μ2-OOCCF3)4(NCCH3)2 [30] и 1.9626(17)–1.9655(16) Å в Cu2(μ2-OOCBut)4(THF)2 (где THF – тетрагидрофуран) [31]. Такое увеличение расстояний Cu–O в V может быть обусловлено пространственными затруднениями, связанными с большим размером заместителя у карбоксильных групп. Длины связей C–O в карбоксильных группах одинаковы (С1–О1 1.261(8) и С1–О2 1.263(8) Å), что указывает на практически полную делокализацию π-электронной плотности карбонильных групп и бидентатно-мостиковый характер координации.
Таблица 3.
Основные длины связей d (Å) и валентные углы ω (град) в молекуле V по данным РСА
| Связь | d | Угол | ω |
|---|---|---|---|
| Cu1–Cu2 | 2.6460(17) | O1–Cu1–O1A | 166.3(3) |
| Cu1–O1 | 2.002(4) | O1–Cu1–O1B | 89.18(3) |
| Cu2–O2 | 1.978(4) | O2–Cu2–O2A | 168.7(3) |
| Cu1–N1 | 2.141(11) | O2–Cu2–O2B | 89.45(3) |
| Cu2–N2 | 2.173(11) | O1–Cu1–N1 | 96.87(13) |
| O1–C1 | 1.261(8) | O2–Cu2–N2 | 95.63(13) |
| O2–C1 | 1.263(8) | C1–O1–Cu1 | 121.6(4) |
| N1–C21 | 1.155(8) | C1–O2–Cu2 | 120.9(4) |
| N2–C23 | 1.156(8) | O1–C1–O2 | 126.7(5) |
| С21–С22 | 1.477(9) | O1–C1–C2 | 116.2(5) |
| С23–С24 | 1.477(9) | O2–C1–C2 | 117.0(5) |
Расстояние Cu1···Cu2 2.6460(17) Å в V имеет промежуточное значение относительно соответствующих величин в аналогичных комплексах: 2.5692(7) Å в Cu2(μ2-OOCBut)4(DBE)2 [28] и 2.5747(10) Å в Cu2(μ2-OOCBut)4(THF)2 [31], 2.6882(18) Å в Cu2(μ2-OOCCF3)4(DBE)2 [28] и 2.766(1) Å в Cu2(μ2-OOCCF3)4(NCCH3)2 [30]. Согласно [29, 30] расстояния Cu···Cu существенным образом зависят от природы углеводородного радикала в RCOO, а также донорных свойств и размера аксиального лиганда, что согласуется с полученными данными для комплекса V.
Таким образом, в комплексе V лигандом является не соединение IV, а продукт его окисления. Столь неожиданный результат можно объяснить каталитическим окислением IV, содержащего в α-положении к карбонильным группам активированную связь CH, кислородом воздуха в присутствии комплексов меди(II) [28, 32]. Можно предположить, что первоначально образуется квадратно-пирамидальный комплекс меди(II) с IV состава 1 : 2 и молекулой ацетонитрила в апикальном положении (схема 3), в котором происходит окисление лиганда по связи С16–С17 (рис. 1) с дальнейшим преобразованием в биядерный карбоксилатный комплекс V.
Рентгеновские измерения проведены на оборудовании уникальной научной установки Курчатовский источник синхротронного излучения НИЦ “Курчатовский институт” при финансовой поддержке Министерства образования и науки РФ (идентификатор проекта RFMEFI61917X0007). Публикация подготовлена при поддержке Программы РУДН “5-100”.
Список литературы
Zhong-Yi W., Bin L., Jue-Wen Z. et al. // Organic Electronics. 2018. V. 52. P. 89.
Anupama E., Si Hyun H., Thaksen J. et al. // J. Mater. Chem. C. 2018.V. 6. № 8. P. 2077.
Ma-Yong G., Jian-Quan L., Xiang-Shan W. // Tetrahedron. 2017. V. 73. № 31. P. 4698.
Javid A., Khojastehnezhad A., Pombeiro A.J.L. // Russian J. General Chem. 2017. V. 87. № 12. P. 3000.
Bahadur Sk, Khodia S., Patra A. // Chem. Commun. 2018. V. 54. P.1786.
Ткаченко Ю.Н., Попов Л.Д., Пожарский А.Ф. и др. // Журн. орган. химии. 2017. Т. 53. № 10. С. 1536.
Oliveira P.F.M., Haruta N., Chamayou A. et al. // Tetrahedron. 2017. V. 73. № 16. P. 2305.
Fukushima T., Drisdell W., Yano J. at al. // J. Am. Chem. Soc. 2015. V. 137. № 34. P. 10926.
Bachollet S.P.J.T., Volz D., Fiser B. et al. // Chemistry - A European Journal. 2016. V. 22. № 35. P. 12430.
Kopchuk D.S., Chepchugov N.V., Khasanov A.F. et al. // Tetrahedron Lett. 2016. V. 57. № 34. P. 3862.
Гридунова Г.В., Стручков Ю.Т., Линко Р.В. и др. // Изв. РАН. Сер. Хим. 1992. № 7. С. 1575.
Гридунова Г.В., Шкловер В.Е., Стручков Ю.Т. и др. // Кристаллография. 1988. Т. 33. Вып. 2. С. 390.
Smith-Verdier P., Florencio F., Garcia-Blanco S. // Crystal Structure Commun. 1980. V. 9. P. 578.
Линко Р.В., Бельский В.К., Варламов А.В. и др. // Изв. РАН. Сер. Хим. 2001. № 9. С. 1548.
Junek H., Hamböck H., Hornischer B. // Monatshefte Chemie. 1967. B. 98. № 2. S. 315.
Kandile N.G., Abdel-Latif T.M. // Acta Chim. Hungarica. 1989. V. 126. P. 271.
Cope A.C., Field L., MacDowell D.W.H., Wright M.E. // J. Am. Chem. Soc. 1956. V. 78. P. 2547.
Cope A.C., MacDowell D.W.H. // J. Am. Chem. Soc. 1958. V. 80. P. 5513.
Battye T.G.G., Kontogiannis L., Johnson O. at al. // Acta Cryst. D. 2011. V. 67. P. 271.
Evans P.R. // Acta Cryst. D. 2006. V. 62. P. 72.
Sheldrick G.M. // Acta Cryst. C. 2015. V. 71. P. 3.
Becke A.D. // J. Chem. Phys. 1993. V. 98. P. 5648.
Lee C., Yang W., Parr R.G. // Phys. Rev. B. 1988. V. 37. P. 785.
Stephens P.J., Devlin F.J., Chabalowski C.F., Frisch M.J. // J. Phys. Chem. 1994. V. 98. P. 11623.
Schaefer A., Huber C., Ahlrichs R. // J. Chem. Phys. 1994. V. 100. P. 5829.
NBO 5.G. Glendening E.D., Badenhoop J.K., Reed A.E. et al. Theoretical Chemistry Institute, University of Wisconsin, Madison, WI. 2004. http://www.chem.wisc.edu/~nbo5.
Granovsky A., Firefly version 7.1.G. http://classic.chem.msu.su/gran/firefly/index.html.
Нефедов С.Е., Кушан Е.В., Яковлева М.А. и др. // Координац. химия. 2012. Т. 38. № 3. С. 233.
Rao V.M., Sathyanarayana D.N., Manohar H. // J. Chem. Soc., Dalton Trans. 1983. № 10. P. 2167.
Karpova E.V., Boltalin A.I., Zakharov M.A. et al. // Z. Anorg. Allg. Chem. 1998. V. 624. P. 741.
Денисова Т.О., Амельченкова Э.В., Прусс И.В. и др. // Журн. неорган. химии. 2006. Т. 51. № 7. С. 1098.
Гончарук В.В., Камалов Г.Л., Ковтун Г.А. и др. Катализ. Кластерные подходы, механизмы гетерогенного и гомогенного катализа. Киев: Наук. думка, 2002. 541 с.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография