

Кристаллография, 2020, T. 65, № 3, стр. 477-483
Синтез и термическая устойчивость кремнийсодержащих фосфатов кальция
О. А. Голованова 1, *, В. С. Чиркова 1
1 Омский государственный университет им. Ф.М. Достоевского
Омск, Россия
* E-mail: golovanoa2000@mail.ru
Поступила в редакцию 09.01.2019
После доработки 22.07.2019
Принята к публикации 17.09.2019
Аннотация
Выполнен синтез кремнийсодержащих фосфатов кальция. Синтезированные твердые фазы исследованы методами рентгенофазового анализа, ИК-спектроскопии, оптической микроскопии, определены удельные поверхности образцов методом БЭТ (Брунауэра–Эммета–Теллера). Показано, что с возрастанием концентрации силикат-ионов в исходном реакционном растворе уменьшается содержание фосфат-ионов в составе синтезированных образцов, что говорит о возможном изоморфном замещении на силикат-ионы. Проведено динамическое растворение и сравнение скоростей растворения твердой фазы с разным содержанием силикат-ионов до и после термической обработки. Установлено, что после термообработки образцов образуется смесь кремнийсодержащего гидроксилапатита и β-трикальций фосфата.
ВВЕДЕНИЕ
Разработка биоматериалов, необходимых для восстановления и замены костных тканей, является актуальным направлением медицинского материаловедения.
Биогенный материал для замены костной ткани на основе карбонатсодержащего нестехиометрическго гидроксилапатита (ГА) Ca10 – x – y/2(HPO4)x(CO3)y(PO4)6 – x – y(OH)2 – x и белка коллагена уникален по свойствам и составу. Он характеризуется высокой минерализацией межклеточного матрикса и содержит 50 мас. % неорганических соединений, 25 мас. % органических и 25 мас. % воды. Минеральные вещества придают ему твердость, органические – эластичность и упругость [1, 2].
Результаты клинических исследований [3–5] показали, что наряду с преимуществами материалы на основе ГА обладают такими недостатками, как небольшая скорость биорезорбции и слабое стимулирующее действие на рост новой костной ткани [6, 7].
Модифицирование ГА различными биологически активными ионами за счет изоморфных замещений дает возможность целенаправленно изменять свойства ГА и получать на его основе материалы с элементным составом, близким к костной ткани человека [8–13].
Одним из таких ионов-допантов является кремний – жизненно важный микроэлемент для формирования кости и поддержания ее структуры в норме [14–17]. Установлено, что в состав межтканевой жидкости человека входят силикат-ионы. При повреждении костной ткани кремний локализуется в области формирования нового костного каркаса и расходуется на образование основного вещества кости и хряща. Синтетические кальций-фосфатные биоматериалы, в структуру которых входит кремний, обладают повышенной биологической активностью по сравнению с незамещенным ГА, способствуют росту внеклеточного матрикса и ускорению минерализации костной ткани [18–25]. Si-ГА является биосовместимым материалом и не вызывает отторжения при введении в ткани живого организма [26]. Таким образом, силикат-ионы являются весьма перспективными для улучшения биологической активности ГА.
Цель работы – синтез и исследование физико-химических характеристик модифицированного кремнийсодержащего гидроксилапатита и керамики на его основе.
ОБЪЕКТЫ И МЕТОДЫ ИССЛЕДОВАНИЯ
Синтез ГА проводили путем осаждения при сливании растворов нитрата кальция Ca(NO3)2 · · 4H2O (ч.д.а), двузамещенного фосфата аммония (NH4)2HPO4 (ч. д. а) и водного раствора аммиака NH4ОН. Силикат-ионы вводили в виде Na2SiO3 · · 9H2O (ч. д. а). Концентрации исходных реагентов рассчитывали таким образом, чтобы nисх = = Са(NO3)2/(NH4)2HPO4 = 1.70. Концентрацию силикат-ионов изменяли в диапазоне от 1 до 30%. Время синтеза составляло 24 и 48 ч.
При проведении синтеза к 250 мл раствора, содержащего Са(NO3)2 и NH4OH, с помощью делительной воронки при постоянном перемешивании реакционной смеси приливали 250 мл раствора (NH4)2HPO4 и Na2SiO3 со скоростью 4.5–5.0 мл/мин. рН раствора корректировали до значения pH = 9.00 ± 0.05 с растворами НNO3 (50%) и/или NаОН (20%) и оставляли для кристаллизации на 24 и 48 ч. После отстаивания раствор фильтровали, осадок не менее трех раз промывали водой (${{V}_{{{{{\text{H}}}_{{\text{2}}}}{\text{O}}}}}$ = 50 мл) и сушили в сушильном шкафу при температуре ∼80°С до полного удаления воды. Затем осадок переносили в маркированную емкость, взвешивали на аналитических весах и исследовали с помощью группы физико-химических методов.
Концентрацию Са2+ определяли титрометрически стандартным раствором этилендиаминтетрауксусной кислоты (ЭДТА) с индикатором мурексидом. Погрешность составила 1 отн. %. Титрование проводили с предварительным отделением фосфатов путем осаждения солями висмута в слабокислой среде [27].
Определение фосфат-ионов проводили фотометрически по методу молибденовой сини [27] на приборе КФК-2 с красным светофильтром (λэфф = 690 нм) и кюветами с толщиной светопоглощающего слоя 2 см. Погрешность составила 2–4 отн. %.
Определение содержания кремния фотометрическим методом основано на реакции взаимодействия мономерно-димерной формы кремниевой кислоты и силикатов с молибдатом аммония в кислой среде с образованием молибдокремниевой гетерополикислоты желтого цвета. Максимум в спектре поглощения образовавшегося соединения наблюдался при λ = 410 нм. Погрешность составила 2–4 отн. %.
Фазовый состав синтезированных твердых фаз проводили методом рентгенофазового анализа (РФА). Порошковые дифрактограммы получали на рентгеновском дифрактометре ДРОН-3. Идентификацию пиков проводили с использованием международной картотеки ASTM и программных пакетов Crystallographica Search-Match и DifWin4.0. Погрешность измерений составила 3–5 отн. %.
Для получения дополнительной информации о составе образцов снимали ИК-спектры на спектрофотометре ФСМ 2202. Пробы готовили прессованием образцов в таблетки с KBr. Погрешность измерений <5%.
Изучение морфологии, определение формы частиц твердых фаз проводили методом оптической микроскопии с помощью микроскопа серии “XSP-104”.
Удельную площадь поверхности образцов по методу БЭТ (SБЭТ-N2) определяли методом одноточечной адсорбции стандартного газа при определенном равновесном давлении на адсорбционном анализаторе Cорбтомeтр, ИК CО РAН (ОНЦ CО РAН, г. Омск). Данный метод БЭТ позволяет определить поверхность в диапазоне от 0.1 до 2000 м2/г с относительной погрешностью 2–5%.
Для термогравиметрического анализа образцы массой 0.1000 ± 0.0002 г нагревали в муфельной печи LF-7/13-G1 LOIPLF при температуре от 600 до 1000°C с интервалом 100°C в течение 1 ч (после выхода печи на рабочий режим). После прокаливания образцы охлаждали на воздухе до комнатной температуры, взвешивали на аналитических весах и переносили в маркированные емкости. По разнице масс до и после прокаливания рассчитывали убыль массы вещества в результате термообработки. Для каждого образца проводили по три эксперимента.
Для характеризации биоактивности образцы растворяли в модельных растворах, для моделирования активной фазы резорбции материала – в ацетатном буфере (рН = 5.5), для моделирования пассивной фазы резорбции материала – в растворе 0.9% NaCl (рН = 7.4). Измерения проводили при комнатной температуре в течение 15 мин при постоянном объеме жидкой фазы в условиях постоянного перемешивания. В ходе эксперимента контролировали концентрацию ионов кальция, переходящих в жидкую фазу в зависимости от времени. Концентрацию ионов кальция определяли методом прямой потенциометрии на иономере И-160МИ с использованием Ca-селективного электрода (поливинилхлоридный пленочный электрод с твердым внутренним контактом ЭЛИC-121Ca), предварительно откалиброванного по серии стандартных растворов CaCl2, совместно с хлорсеребряным электродом сравнения и температурным датчиком. Обработка данных кинетических измерений выполнена в программе Origin 8.0.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Согласно результатам РФА все твердые фазы, полученные при варьировании концентрации силикат-ионов, идентичны и представлены фазой карбонатгидроксилапатита (КГА). С повышением исходной концентрации силикат-ионов в экспериментальных системах наблюдалось снижение степени разделения рефлексов ГА в области 30°–33° брегговских углов отражения. Слабое разрешение рефлексов обусловлено малой степенью кристалличности и высокодисперсным состоянием образцов при замещении фосфат-ионов силикат-ионами в структуре КГА (рис. 1). Более длительное отстаивание осадка под маточным раствором приводило к улучшению степени кристалличности твердых фаз, вследствие чего формировались кристаллиты меньшего размера (табл. 1).
Рис. 1.
Дифрактограмма осадков, время синтеза 48 ч: КГА (1), Si-КГА (C(SiO$_{3}^{{2 - }}$) = 15 мас. % (2), Si-КГА (C(SiO$_{3}^{{2 - }}$) = 30 мас. % (3).
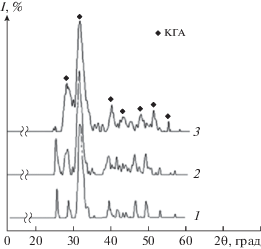
Таблица 1.
Характеристики твердых фаз образцов (время синтеза 48 ч)
| Концентрация SiO$_{3}^{{2 - }}$, мас. % | Средние размеры кристаллитов d, нм | Соотношение концентраций | Масса образцов m, г | Удельная площадь поверхности образцов Sуд, м2/г | |
|---|---|---|---|---|---|
| Ca/P | Ca/(P + Si) | ||||
| 0 | 6.31 | 1.65 | 5.2442 | 86 | |
| 15 | 6.10 | 1.63 | 1.47 | 5.4435 | 138 |
| 30 | 6.18 | 1.61 | 1.30 | 5.9014 | 151 |
Методом ИК фурье-спектроскопии было установлено, что полученные образцы содержат набор полос, характерных для КГА (рис. 2). Структуру ГА определяют полосы, характерные для деформационных (620 и 589 см–1) и валентных (1040–1080 и 960 см–1) колебаний О–Р–О в РО$_{4}^{{3 - }}$. Полоса поглощения на 3570 см–1 отнесена к валентным колебаниям связи О–Н гидроксильных ионов, локализующихся в структуре ГА. Развитая поверхность порошков ГА способствует адсорбции воды, которой соответствует появление интенсивных полос поглощения в области деформационных (1640 см–1) и валентных (3500−3000 см–1) колебаний воды. Поскольку ГА синтезируется в воздушной среде, то в процессе его образования из атмосферы воздуха сорбируется углекислый газ, и в решетке локализуются ионы карбоната в положении гидроксид-иона, которым на ИК-спектрах соответствуют полосы валентных колебаний О–С–О в СО$_{3}^{{2 - }}$ на 1422–1457 см–1, что соответствует замещению А-типа. По сравнению со спектром КГА в ИК-спектре Si-КГА начиная с концентрации 15% появляются полосы поглощения с частотами от 450–550 см–1, характерные для колебания связей силикатной группы в структуре ГА, обусловленные деформационными колебаниями связей Si–O. Валентные колебания этих связей могут перекрываться с интенсивной полосой поглощения фосфатной группы в области частот 900–1100 см–1. Различие ИК-спектров КГА и Si-КГА также наблюдается в интервале частот, отвечающих за деформационные колебания групп РО$_{4}^{{3 - }}$. В ИК-спектре КГА имеется характерный триплет с частотами 653, 620 и 589 см–1, а в спектрах Si-КГА в этой области присутствуют только две полосы на 651 и 589 см–1, соответствующие деформационным колебаниям групп РО$_{4}^{{3 - }}$. Кроме этого, в ИК-спектре Si-КГА при увеличении концентрации силикат-ионов наблюдается уменьшение интенсивности полосы валентных колебаний О–Н в решетке КГА при 3570 см–1, что говорит о потере групп ОН и появлении вакансий, обеспечивающих компенсацию заряда при замещении фосфатных групп на силикатные. Кроме этого, с возрастанием концентрации вводимых силикат-ионов изменяются пики, соответствующие карбонатным группам: увеличивается интенсивность пиков 873–880 см–1 и расширяется пик 1383 см–1. Это указывает на рост концентрации карбонат-ионов в синтезированных образцах с возрастанием концентрации силикат-ионов, что способствует формированию материала, приближенного по составу к естественной кости [22].
Рис. 2.
ИК-спектры образцов, время синтеза 48 ч: КГА (1), Si-КГА (C(SiO$_{3}^{{2 - }}$) = 15 мас. % (2), Si-КГА (C(SiO$_{3}^{{2 - }}$) = 30 мас. % (3).
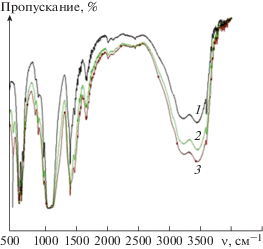
Рис. 3.
Вид частиц КГА и Si-КГА при разном времени синтеза: КГА 24 ч (а), Si-КГА (C(SiO$_{3}^{{2 - }}$) = 30 мас. %, 24 ч (б), КГА 48 ч (в), Si-КГА (C(SiO$_{3}^{{2 - }}$) = 30 мас. %, 48 ч (г) (увеличение – ×10).

При изучении морфологии твердой фазы выявлено, что при увеличении концентрации силикат-ионов происходит образование более мелких и пористых агрегатов, доказательством чего являются данные метода БЭТ (табл. 1). Видно, что удельная поверхность увеличивается с ростом содержания силикат-ионов в исходном растворе.
Мольные соотношения Са/Р оценивали по разнице начальных и конечных концентраций ионов в системе. Коэффициент Са/Р в полученных образцах изменяется (табл. 1). С увеличением концентрации силикат-ионов происходит уменьшение соотношения Са/(Р + Si). Отклонение коэффицента Са/Р синтезированных образцов от стехиометрического значения (1.67) при больших концентрациях силикат-ионов в реакционной среде может быть связано с тем, что часть SiO$_{3}^{{2 - }}$ не встраивается в позиции РО$_{4}^{{3 - }}$-тетраэдров, а адсорбируется на поверхности образующейся твердой фазы [22].
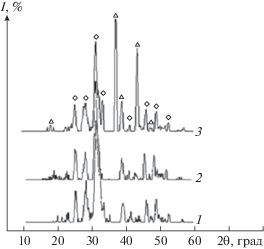
Биокерамика, в производстве которой используют ГА, широко применяется в медицине для лечения зубных и костных дефектов. Также керамику применяют совместно с полимерами для того, чтобы заполнить костные полости при реставрации [28]. Поэтому была предпринята попытка создания керамики путем воздействия на синтезированные кремнийсодержащие образцы температур в диапазоне 600–1000°С. После термообработки для всех образцов наблюдалось увеличение общей потери массы с ростом температуры прокаливания. В интервале температур 25–230°C происходит удаление из состава образцов несвязанной и адсорбционной воды. При t > 600°C из структуры КГА выходят СО$_{3}^{{2 - }}$. Для полученных осадков термодеструкция начинается при температуре около 800°C. В результате выхода СО$_{3}^{{2 - }}$ из структуры происходит разложение КГА и формирование двух фаз: β-ТКФ (трикальций фосфат) и Si-ГА. Результаты РФА подтверждают наличие двух фаз в осадках после прокаливания. По мере повышения исходной концентрации силикат-ионов интенсивность дифракционных максимумов увеличивается, а их ширина уменьшается, что свидетельствует о большей степени закристаллизованности твердой фазы по сравнению с беспримесным образцом (рис. 4).
Рис. 4.
Дифрактограммы осадков Si-КГА после прокаливания при t = 800°С, 48 ч: C(SiO$_{3}^{{2 - }}$) 5% (1), C(SiO$_{3}^{{2 - }}$) 15% (2), C(SiO$_{3}^{{2 - }}$) 30% (3).
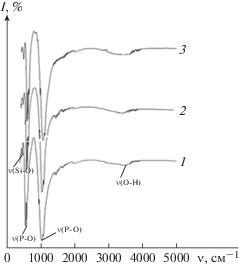
Результаты РФА согласуются с данными ИК-спектроскопии. На спектрах образцов, полученных после прокаливания при t = 800°C, наблюдаются полосы поглощения, характерные для колебания связи Р–О в тетраэдре РО$_{4}^{{3 - }}$. Полосы поглощения на 450–550 см–1 указывают на присутствие силикат-ионов в структуре синтезированных фосфатов кальция. После термообработки исчезают пики на 1383 и 873–880 см–1, что соответствуют валентным и деформационным колебаниям связи С=О. С увеличением концентрации силикат-ионов в образцах происходит возрастание интенсивности и расширение пика при 1096 см–1, характерного для колебаний связи Р–О и валентных колебаний Si–O (рис. 5).
Рис. 5.
ИК-спектры осадков Si-КГА после прокаливания при t = 800°С, 48 ч: C(SiO$_{3}^{{2 - }}$) 5% (1), C(SiO$_{3}^{{2 - }}$) 15% (2),C(SiO$_{3}^{{2 - }}$) 30% (3).
Наличие в составе осадков более растворимых фаз, чем ГА, подтверждают результаты, полученные при изучении их биоактивности. Биорезорбируемость синтезированных образцов Si-КГА показала, что максимальная скорость перехода Са2+ наблюдается на начальном этапе резорбции. На основании кинетических зависимостей логарифма концентрации ионов кальция от времени рСа = f(t) рассчитаны значения начальных скоростей выделения ионов кальция в растворе V как тангенс угла наклона линейного участка прямой
Затем с течением времени происходит уменьшение перехода ионов кальция из твердой фазы в раствор Са2+, что характерно для всех образцов, и кинетика растворения твердой фазы подчиняется экспоненциальной зависимости:
где C0 – условная начальная концентрация, Cm – концентрация насыщения, b – коэффициент, t – время.Данная зависимость соответствует кинетике реакции первого порядка, так как скорость изменения “активных участков растворения” пропорциональна их количеству в растворяемом материале в данный момент [1].
В табл. 2 приведены результаты начальных скоростей растворения для твердых фаз. Выявлено, что скорость растворения кремнийсодержащих образцов увеличивается с возрастанием концентрации силикат-ионов в твердой фазе. Такое поведение скорости растворения твердой фазы обусловлено достижением высокой степени дефектности кристаллической структуры, в связи с чем существенно возрастает реакционная способность вещества. Следовательно, введение силикат-ионов в структуру ГА эффективно повышает его взаимодействие с биологическими жидкостями.
Таблица 2.
Значения начальных скоростей растворения образцов ${v}$, 48 ч
| Образец | Этапы раство-рения | t, мин | Ацетатный буферный раствор | 0.9%-ный раствор NaCl | ||
|---|---|---|---|---|---|---|
| Кинетическое уравнение | ${v}$ × 104, мин–1 | Кинетическое уравнение | ${v}$ × 105, мин–1 | |||
| КГА | 1 | 0–5 | С(Са2+) = 5.79 × 10–4t + + 5.81 × 10–4 | 5.79 | C(Ca2+ ) = 5.88 × 10–5t + + 1.20 × 10–4 | 5.88 |
| 2 | 6–15 | С(Са2+) = 3.04 × 10–4 + + 2.90 × 10–3e2.10E–02t | 30.4 | C(Ca2+) = 2.75 × 10–4 + + 2.63E–04e5.47E–03t | 27.5 | |
| КГА+ C(SiO$_{3}^{{2 - }}$) 1% | 1 | 0–5 | С(Са2+) = 5.43 × 10–4t + + 1.07 × 10–3 | 5.43 | C(Ca2+) = 2.20 × 10–5t + + 1.99 × 10–4 | 2.20 |
| 2 | 6–15 | С(Са2+) = 3.16 × 10–3 + + 3.04E–03e1.50E–02t | 31.6 | C(Ca2+) = 2.32 × 10–4 + + 2.24E–04e4.91E–03t | 23.2 | |
| КГА+ C(SiO$_{3}^{{2 - }}$) 5% | 1 | 0–5 | С(Са2+) = 4.48 × 10–4t + + 8.34 × 10–4 | 4.48 | C(Ca2+) = 3.70 × 10–5t + + 2.85 × 10–4 | 3.70 |
| 2 | 6–15 | С(Са2+) = 2.94 × 10–3 + + 2.73 × 10–3e1.69E–02t | 29.4 | C(Ca2+) = 3.06 × 10–4 + + 2.92 × 10–4e7.92E–03t | 30.6 | |
| КГА+ C(SiO$_{3}^{{2 - }}$) 10% | 1 | 0–5 | С(Са2+) = 5.35 × 10–4t + + 8.21 × 10–4 | 5.35 | C(Ca2+) = 1.58 × 10–5t + + 1.85 × 10–4 | 1.58 |
| 2 | 6–15 | С(Са2+) = 3.37 × 10–3 + + 3.10 × 10–4e1.84E–02t | 33.7 | C(Ca2+) = 2.41 × 10–4 + + 2.36E–04e3.96E–03t | 24.1 | |
| КГА+ C(SiO$_{3}^{{2 - }}$) 15% | 1 | 0–5 | С(Са2+) = 7.30 × 10–4t + + 1.20 × 10–3 | 7.30 | C(Ca2+) = 7.96 × 10–5t + + 2.02 × 10–4 | 7.96 |
| 2 | 6–15 | С(Са2+) = 3.61 × 10–3 + + 3.43 × 10–3e1.34E–02t | 36.1 | C(Ca2+) = 4.56 × 10–4 + + 4.48 × 10–4e3.35E–03t | 45.6 | |
| КГА+ C(SiO$_{3}^{{2 - }}$) 20% | 1 | 0–5 | С(Са2+) = 7.37 × 10–4t + + 1.29 × 10–3 | 7.37 | C(Ca2+) = 8.04 × 10–5t + + 2.16 × 10–4 | 8.04 |
| 2 | 6–15 | С(Са2+) = 3.94 × 10–3 + + 3.66 × 10–3e1.12E–02t | 39.4 | C(Ca2+) = 4.27 × 10–4 + 4.17 × 10–4e3.27E–03t | 42.7 | |
| КГА+ C(SiO$_{3}^{{2 - }}$) 25% | 1 | 0–5 | С(Са2+) = 6.92 × 10–4t + + 2.46 × 10–3 | 6.92 | C(Ca2+) = 8.27 × 10–5t + 1.67 × 10–4 | 8.27 |
| 2 | 6–15 | С(Са2+) = 4.34 × 10–3 + + 4.27 × 10–3e4.74E–03t | 43.4 | C(Ca2+) = 3.74 × 10–4 + + 3.57 × 10–4e6.72E–03t | 37.4 | |
| КГА+ C(SiO$_{3}^{{2 - }}$) 30% | 1 | 0–5 | С(Са2+) = 7.46 × 10–4t + + 1.30 × 10–3 | 7.46 | C(Ca2+) = 1.15 × 10–4t + 8.99 × 10–5 | 11.5 |
| 2 | 6–15 | С(Са2+) = 4.19 × 10–3 + + 3.98 × 10–3e9.69E–03t | 41.9 | C(Ca2+) = 3.61 × 10–5 + + 3.53 × 10–4e3.37E–03t | 36.1 | |
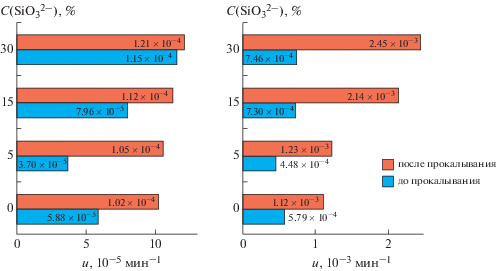
При сравнении скоростей растворения кремнийсодержащих образцов до и после прокаливания установлено, что более растворимыми являются порошки, подвергшиеся термической обработке (рис. 6). Это обусловлено тем, что после прокаливания образцы представляют смесь из двух фаз (Si-ГА и β-ТКФ), β-ТКФ является более растворимой по сравнению с ГА. Двухфазные композиционные материалы, состоящие из Si-ГА и β-ТКФ, характеризуются высокой степенью биорезорбируемости, что позволяет использовать их в медицине для создания на их основе материалов, скорость резорбции которых можно контролировать за счет различной растворимости составляющих компонентов.
ЗАКЛЮЧЕНИЕ
В ходе эксперимента установлено, что при варьировании концентрации силикат-ионов образуется кремнийсодержащий карбонатгидроксилапатит А-типа. Показано, что с возрастанием концентрации силикат-ионов в исходном реакционном растворе уменьшается содержание фосфат-ионов в составе синтезированных образцов, что говорит о возможном изоморфном замещении. Изучены скорости растворения порошков в 0.9%-ном NaCl-растворе и ацетатном буфере до и после термообработки. Установлено, что наибольшей скоростью растворения обладают образцы с максимальной концентрацией силикат-ионов, подвергшиеся термической обработке.
Полученные двухфазные композиционные материалы, состоящие из Si-ГА и β-ТКФ, характеризуются высокой степенью биорезорбируемости, что позволяет использовать их как биосовместимые материалы в медицине.
Список литературы
Баринов С.М., Комлев С.В. // Успехи химии. 2010. № 1. С. 15.
Dorozhkin S.V. // Funct. Biomater. 2015. P. 708.
Голованова О.А. Патогенные минералы в организме человека. Омск: Изд-во ОмГУ, 2006. 400 с.
Surmenev R.A., Surmeneva M.A., Ivanova A.A. // Acta Biomat. 2014. V. 10. P. 557.
Dorozhkin S.V. // Ceram. Int. 2016. V. 42. P. 6529.
Pietak A.M., Reid J.W., Stott M.J., Sayer M. // Biomater. 2007. V. 28. P. 4023.
Gayathri N., Nagarajan L. // Int. J. Sci. Research (IJSR). 2016. V. 5. № 3. P. 850.
Barinov S.M., Komlev V.S. Calcium phosphate based bioceramics for bone tissue engineering. Zuerich: Trans. Tech. Publ., 2008. 166 p.
Комлев В.С., Фадеева И.В., Гурин А.Н. и др. // Журн. неорган. материалы. 2009. Т. 45. № 3. С. 1.
Цыгановкина Е.В., Орлов М.А. // Успехи в химии и химической технологии. 2016. Т. 30. № 1. С. 85.
Gurin A.N., Komlev V.S., Fadeeva I.V., Barinov S.M. // Stomatologiia (Mosk). 2011. № 5. V. 90. P. 64.
Гурин А.Н., Григорьянц Л.А., Григорьян А.С. и др. // Медицинский алфавит. Стоматология. 2010. № 1. С. 32.
Porter A.E. // J. Biomater. 2003. V. 24. P. 4609.
Мансурова Л.А., Федчишин О.В., Трофимов В.В. и др. // Сибирский медицинский журн. 2009. № 7. С. 16.
Saki M. // J. Yakhteh. 2009. V. 11. № 1. P. 55.
Гурин А.Н., Комлев В.С. // Медицинский алфавит. Стоматология. 2011. Т. 3. С. 26.
Белецкий Б.И., Свентская Н.В. // Стекло и керамика. 2009. № 3. С. 26.
Pietak A.M., Reid J.W., Stott M.J., Sayer M. // Biomater. 2007. V. 28. P. 4023.
Сурменева М.А., Сурменев Р.А., Чайкина М.В. // Физика и химия обработки материалов. 2012. № 3. С. 51.
Zheng Y. // Ceram. Int. 2014. V. 40. P. 14661.
Солоненко А.П., Голованова О.А. // Журн. неорган. хим. 2014. Т. 59. № 11. С. 1472.
Солоненко А. П. // Стекло и керамика. 2016. № 10. С. 37.
Солоненко А.П., Боксгорн В.В. // Изв. РАН. Сер. хим. 2017. № 3. С. 439.
Maria V.R. // J. Mater. Chem. 2005. V. 15. P. 1509.
Ле Ван Тхуан. Дис. “Коллоидно-химические аспекты синтеза гидроксилапатита, модифицированного силикат-ионами” канд. техн. наук. Белгород, 2015. 205 с.
Методика измерений титриметрическим методом с трилоном Б. РД 52.24.403-2018.
Метод определения содержания полифосфатов ГОСТ 18309-72.
Вересов А.Г., Путляев В.И., Третьяков Ю.Д. // Рос. хим. журн. 2000. Т. 94. № 6. Ч. 2. С. 32.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография