

Кристаллография, 2020, T. 65, № 6, стр. 907-913
Кристаллографические исследования мутантных форм и комплексов олигопептидазы В из Serratia proteamaculans
Д. Е. Петренко 1, *, А. Ю. Николаева 1, В. А. Лазаренко 1, П. В. Дороватовский 1, В. И. Тимофеев 1, 2, А. В. Власкина 1, Д. А. Корженевский 1, А. Г. Михайлова 3, К. М. Бойко 4, Т. В. Ракитина 1, 3, **
1 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
2 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
3 Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
Москва, Россия
4 Институт биохимии им. А.Н. Баха,
Федеральный исследовательский центр “Фундаментальные основы биотехнологии”
Москва, Россия
* E-mail: dmitry.e.petrenko@gmail.com
** E-mail: taniarakitina@yahoo.com
Поступила в редакцию 04.06.2020
После доработки 04.06.2020
Принята к публикации 11.06.2020
Аннотация
Выполнены структурные исследования бактериальной олигопептидазы В (ОрВ), принадлежащей к наименее исследованной группе ферментов семейства пролилолигопептидаз. С целью получения пространственных структур OpB из Serratia proteamaculans (PSP), находящейся на разных стадиях каталитического цикла, осуществлен подбор условий кристаллизации мутантных форм фермента, его комплексов с пептидами, имитирующими субстраты и продукты катализа, а также комплекса с ингибитором – аналогом переходного состояния. Проведены наработка кристаллов, рентгеноструктурные эксперименты и предварительный анализ дифракционных наборов. Собранные дифракционные наборы позволяют получить структурные данные, необходимые для описания каталитического цикла бактериальных ОpB.
ВВЕДЕНИЕ
Олигопептидазы В (OpB) – это трипсиноподобные сериновые пептидазы, относящиеся к семейству пролилолигопептидаз (POP). Характерной особенностью POP является двухдоменная 3D-архитектура, включающая в себя С-концевой α/β-гидролазный каталитический домен и N-концевой 7–8-лопастный β-пропеллерный домен, препятствующий проникновению субстратов размером более 3 кДа к каталитической триаде, локализованной во внутренней полости границы между доменами [1]. Олигопептидазы В найдены в бактериях, протозойных паразитах (трипаносомах и лейшманиях) и растениях [2]. Известно, что они являются факторами патогенеза при паразитарных и некоторых бактериальных инфекциях и являются потенциальными мишенями для разработки терапевтических средств [3]. В острых фазах заболевания эти трипсиноподобные ферменты попадают в кровь больных, где гидролизуют ряд физиологически важных пептидов, включая предсердный натрийуретический фактор, что вызывает серьезные нарушения в системе кроветворения [4]. Кроме того, было показано, что именно OpB обеспечивают резистентность бактерий к антимикробным пептидам, обогащенным основными аминокислотными остатками [5]. Поиск специфических ингибиторов OpB для последующего дизайна лекарственных средств, в том числе антипаразитарных препаратов, требовал знания пространственной структуры данного фермента. Такие данные, полученные для ферментов из лейшмании Leishmania major и трипаносомы Trypanosoma brucei, позволили предложить механизм каталитической активации ОpB простейших [6, 7]. Однако из-за аминокислотных замен в ключевых позициях реализация данного механизма в бактериальных ОpB была маловероятна [8–11]. В 2019 г. получена первая пространственная структура бактериальной ОpB из Serratia Proteamaculans (PSP, PDB ID 6TF5) [12]), которая должна была прояснить, каким образом происходит активация бактериальных ферментов данного класса. Однако анализ структуры показал, что фермент в кристалле имеет каталитически неактивную конформацию, в которой остатки активного центра разобщены между собой.
Цель данной работы – получение и рентгеноструктурный анализ кристаллов фермента, находящегося на разных стадиях каталитической активации. Для этого осуществлен подбор условий кристаллизации мутантных форм PSP, а также комплексов PSP с рядом лигандов, проведена наработка кристаллов и их рентгеноструктурный анализ (РСА). Выбор точек мутагенеза, а также лигандов для получения комплексов проводили на основе результатов предшествующих исследований нативной и рекомбинантной PSP [13–18]. Дифракционные наборы были собраны для каталитически неактивного мутанта PSP и мутанта, обладающего повышенной эффективностью катализа, для комплексов PSP с аналогами субстратов и продуктов катализа, а также с необратимым ингибитором – аналогом переходного состояния (табл. 1). Предварительный анализ рентгеноструктурных данных показал, что во всех случаях кристаллы принадлежат одной и той же пространственной группе, что указывает на то, что в отличие от протозойных аналогов каталитическая активация PSP происходит без значительных изменений взаимного расположения доменов.
Таблица 1.
Список исследуемых мутантных форм и комплексов PSP
| Образец | Описание | Цель исследований |
|---|---|---|
| PSP_S532A | Каталитически не активный мутант | Изучение влияния функционально значимых мутаций на конфигурацию активного центра и общий фолдинг белка |
| PSP_E125A | Мутант с увеличенной каталитической активностью | |
| PSP_S532A + ZAKIR | Комплекс с аналогом субстрата | Получение и исследование структуры PSP в каталитически активном состоянии |
| PSP + ZAAK | Комплексы с аналогами продуктов каталитической реакции, различающимися аминокислотными остатками в P1- и P2-позициях1 | Изучение выхода продукта реакции из активного центра и влияния аминокислотных остатков в P1- и Р2-позициях |
| PSP + ZALR | ||
| PSP + ZADR | ||
| PSP + ZAAR | ||
| PSP-TLCK | Комплекс с необратимым ингибитором – аналогом переходного состояния | Получение и исследование структуры PSP в каталитически активном состоянии |
МАТЕРИАЛЫ И МЕТОДЫ
Сайт-направленный мутагенез S532A проводили согласно [9, 10]. Синтетические олигонуклеотидные праймеры для замены кодона (5'-CACGGCGGCAAGTAAGGGCGTTTCAAAGC-3') и отбора мутантных клонов (5'-GGCTTTGAAACGC CCTT-3') были синтезированы компанией Евроген (Москва, Россия). Восемнадцать циклов полимеразной цепной реакции (ПЦР) проведены на шаблоне PSP-экспрессирующей плазмиды [9, 10] с использованием набора для ПЦР Tersus Plus (Евроген, Москва, Россия) в соответствии с рекомендациями производителя. ПЦР-продукт обрабатывали эндонуклеазой рестрикции DpnI (Thermo Fisher Scientific, Массачусетс, США) и трансформировали в E. coli. Мутантные клоны отбирали методом ПЦР на колониях с использованием Taq ДНК-полимеразы (Евроген, Москва, Россия). Плазмидную ДНК, выделенную из мутантных клонов, секвенировали для контроля отсутствия случайных мутаций, связанных с ПЦР.
Получение рекомбинантных белков. Получение рекомбинантной PSP и ее мутантов (табл. 1) проводили в системе экспрессии E. coli, как описано в [8–10]. При выделении белка использовали металло-хелатную афинную хроматографию, проводимую на Ni-NTA. Концентрирование белка проводили на ультрафильтрационной колонке Centricon (Merck-Millipore, США), оценку чистоты препарата осуществляли с помощью электрофореза в ДСН-ПААГ на ячейке для электрофореза Mini-Protean 3 (Bio-Rad, США) с последующим окрашиванием гелей с помощью Coomassie G-250. Определение концентрации белка проводили методом Брэдфорда с помощью набора Bio-Rad Protein Assay и стандарта БСА.
Кристаллизация. Подбор условий кристаллизации осуществляли методом диффузии в парах в варианте “сидячей капли”. Использовали роботизированную систему кристаллизации фирмы “Rigaku” (Япония) Ресурсного центра молекулярной и клеточной биологии (РЦ МКБ) НИЦ “Курчатовский институт”, белковые препараты с концентрацией 15–21 мг/мл в буфере (20 мМ Трис pH 8.0, 100 мМ хлорид натрия, 5 мМ спермин), стандартные наборы для кристаллизации глобулярных белков компании Hampton Research (США): Crystal Screen HT, Index HT, PEG/Ion HT, PEGRx HT, набор TOP96 компании Anatrace (США) и кристаллизационные планшеты на 96 лунок (ArtRobbins), каждая из которых содержала три подлунки для варьирования концентрации белка в рамках одного условия кристаллизации. Соотношения белка и противораствора в подлунках были следующими: 1 : 1, 2 : 1 и 1 : 2, суммарный объем белка в лунке – 0.1 мкл для соотношений 1 : 1 и 2 : 1 и 0.2 мкл для соотношения 1 : 2. Объем противораствора составлял 50 мкл. Планшеты инкубировали при температуре +4°С с инспекцией результатов раз в семь дней. Время роста большинства кристаллов составляло от 8 до 14 дней.
Для получения кристаллов комплексов использовали метод сокристаллизации: к раствору белка перед кристаллизацией добавляли соответствующий лиганд (табл. 2) в концентрации 10 мМ. Если рост кристаллов не наблюдался, проводили настаивание кристаллов свободной формы фермента в присутствии 25 мМ лиганда в течение 3 ч.
Таблица 2.
Финальные условия кристаллизации исследуемых мутантных форм и комплексов PSP
| Образец | Условия кристаллизации1 | Криопротектант |
|---|---|---|
| PSP_S532A | 0.2 M Li2SO4, 0.1 M Bis-Tris, pH 5.5, 23% ПЭГ 3350 | Глицерин 25% |
| PSP_E125A | 0.2 M (NH4)2SO4, 0.1 M MES, pH 6.5, 30% ПЭГ 5000, MME | |
| PSP_S532A + ZAKIR, настаивание 3 ч в 25 мM лиганде | 0.2 M Li2SO4, 0.1 M Bis-Tris, pH 5.5, 27% ПЭГ 3350 | |
| PSP + ZAAK, сокристаллизация с 10 мM лигандом | 0.2 M C3H2O4Na2, pH 7.0, 20% ПЭГ 3350 | Паратон |
| PSP + ZALR, сокристаллизация с 10 мM лигандом | 0.2 M C3H2O4Na2, pH 7.0, 24% ПЭГ 3350 | |
| PSP + ZADR, сокристаллизация с 10 мM лигандом | 0.2 M C3H2O4Na2, pH 7.0, 20% ПЭГ 3350 | |
| PSP + ZAAR, сокристаллизация с 10 мM лигандом | 0.2 M C3H2O4Na2, pH 7.0, 20% ПЭГ 3350 | |
| PSP-TLCK | 0.2 M Li2SO4, 0.1 M Tris, pH 8.5, 30% ПЭГ 4000 |
Оптимизацию условий проводили при температуре +4°С методом диффузии в парах в варианте “висячей капли” в 24-луночных планшетах фирмы “VDX” (США). Кристаллизационная капля состояла из 3 мкл белкового раствора, смешанного с осадителем в равных пропорциях и уравновешенного против 400 мкл осадителя в герметичном резервуаре. Финальные условия кристаллизации собраны в табл. 2.
Сбор и обработка дифракционных данных на синхротронном источнике НИЦ “Курчатовский институт”. Перед сбором дифракционных данных кристаллы вылавливали петлей и переносили в криораствор, содержащий паратон, либо кристаллизационный раствор с добавлением глицерина до финальной концентрации в 25% (табл. 2). В случае работы с кристаллами комплексов в криораствор также добавляли соответствующий лиганд в требуемой концентрации.
Сбор дифракционных данных проводили на станции Белок (λ = 0.787 Å) Курчатовского источника синхротронного излучения в прямой геометрии (θ = 0°) при температуре 100 K. Было собрано 350 дифракционных картин с шагом в 1°, расстояние образец–детектор 140 мм. Картины дифракции детектировали с помощью двумерного детектора Rayonix MARCCD.
Данные проиндексированы и проинтегрированы с помощью программы Mosflm из пакета CCP4 [19]. Статистики наборов данных приведены в табл. 3.
Таблица 3.
Кристаллографические данные и параметры съемки кристаллов, полученных для комплекса PSP-TLCK и мутантных белков на синхротронном источнике НИЦ “Курчатовский институт”
| PSP_S532A | PSP_E125A | PSP-TLCK | |
|---|---|---|---|
| Пр. гр. | P212121 | P212121 | P212121 |
| a, b, c, Å; α = β = γ, град | 70.71, 100.40, 108.67; 90 | 68.84, 98.56, 108.26; 90 | 73.32, 101.10, 108.76; 90 |
| Т, K | 100 | 100 | 100 |
| λ, Ǻ | 0.787 | 0.787 | 0.787 |
| Разрешение, Ǻ | 47.8–1.88 (1.93–1.88) | 44.9–2.72 (2.79–2.72) | 30.0–2.30 (2.36–2.30) |
| Число независимых рефлексов | 63 282 (4622) | 20 453 (1476) | 35 505 (2598) |
| Повторяемость | 7.25 (4.31) | 6.18 (5.96) | 7.92 (6.14) |
| Полнота набора, % | 99.80 (99.78) | 99.92 (99.86) | 99.71 (99.01) |
| I/σ (I) | 10.15 (2.09) | 8.45 (2.11) | 7.98 (2.14) |
| Rmrgd-F, % | 4.9 (31) | 6.1 (29) | 5.7 (24) |
Сбор и обработка дифракционных данных на синхротронном источнике SPring-8, Япония. Криопротектирование кристаллов проводили аналогично тому, как описано в предыдущем разделе.
Сбор дифракционных данных проводили на синхротронном источнике SPring-8, Япония (станция BL41XU). Эксперимент выполнен при температуре 100 K. Картины дифракции детектировали с помощью детектора Pilatus 6M. Для расчета стратегии сбора данных использовали программы HKL2000 и Mosflm. Дифракционные данные были обработаны, как описано выше. Статистики наборов данных и обработки приведены в табл. 4.
Таблица 4.
Кристаллографические данные и параметры съемки кристаллов, полученных для комплексов PSP и PSP_S532A на синхротронном источнике SPring-8, Япония
| Белок | PSP | PSP | PSP | PSP | PSP_S532A |
|---|---|---|---|---|---|
| Пептид | ZAAK | ZALR | ZADR | ZAAR | ZAKIR |
| Пр. гр. | P212121 | P212121 | P212121 | P212121 | P212121 |
| a, b, c, Å; α = β = γ, град | 72.45, 100.50, 108.72; 90 | 72.89, 100.44, 108.55; 90 | 72.95, 100.53, 108.88; 90 | 73.09, 100.62, 108.63; 90 | 70.71, 100.45, 108.96; 90 |
| Т, K | 100 | 100 | 100 | 100 | 100 |
| λ, Ǻ | 0.8 | 0.8 | 0.8 | 0.8 | 0.8 |
| Разрешение, Ǻ | 30.0–1.8 (1.9–1.8) | 30.0–1.75 (1.84–1.75) | 30.84–1.72 (1.81–1.72) | 30.0–1.65 (1.74–1.65) | 73.85–2.00 (2.11–2.00) |
| Число независимых рефлексов | 73 343 (10 481) | 80 464 (11 668) | 78 927 (12 166) | 95 311 (13 658) | 54 664 (7867) |
| Повторяемость | 6.43 (6.41) | 4.96 (4.87) | 4.29 (4.67) | 5.84 (6.06) | 7.15 (7.43) |
| Полнота набора, % | 98.99 (98.21) | 99.5 (99.93) | 92.53 (98.83) | 98.57 (97.79) | 99.97 (100.00) |
| I/σ (I) | 10.62 (3.22) | 7.47 (2.53) | 3.34 (2.1) | 9.26 (2.29) | 4.01 (1.46) |
| Rmrgd-F, % | 4.8 (24) | 6.6 (29) | 17.3 (32) | 5.0 (33) | 10.3 (51) |
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
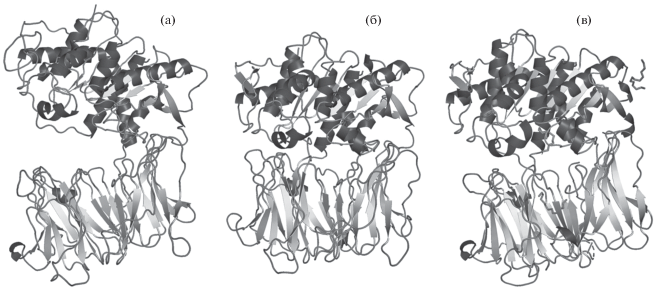
В отличие от своих протозойных аналогов бактериальные OpB плохо поддаются кристаллизации, поэтому ранее для изучения механизма катализа OpB из S. Proteamaculans (PSP) использовались методы моделирования по гомологии и молекулярной динамике [9, 10], а также доступные пространственные структуры близкородственных ферментов из лейшмании L. major и трипаносомы T. brucei [6, 7]. Исходя из структурных данных, полученных для протозойных ферментов, OpB существуют в двух конформациях: открытой (неактивной), в которой каталитический и β-пропеллерный домен, а также аминокислотные остатки активного центра разобщены между собой, и закрытой (активной), в которой два домена сближены, а активный центр собран (рис. 1а, 1б). В то же время анализ пространственных структур, полученных для других представителей семейства POP (дипептидилпептидаз, DPP), показывает, что конформационные перестройки активного центра и активация ферментов могут происходить без изменения положения доменов друг относительно друга [20]. В 2019 г. получена пространственная структура PSP (PDB ID 6TF5), которая стала первой структурой бактериальной ОpB [12]. Анализ структуры показал, что активный центр фермента находится в каталитически неактивной конформации. При этом взаимное расположение доменов друг относительно друга не позволило однозначно определить, находится ли фермент в открытой или закрытой конформации (рис. 1в).
Рис. 1.
Модели открытой (а) и закрытой (б) конформаций PSP, полученные с помощью моделирования по гомологии на основе пространственных структур PDB ID 4BP8 и 4BP9, полученных для OpB Trypanosoma brucei в открытой и закрытой конформациях соответственно. Модель пространственной структуры PSP (PDB ID 6TF5), полученной методом РСА в 2019 г. (в).
Чтобы определить, как происходит активация PSP, требовалось получить пространственные структуры фермента, находящегося на других стадиях каталитической активации; для этого решили провести кристаллографические исследования мутантных форм PSP, а также комплексов фермента с лигандами различной природы (табл. 1).
Для исследований был отобран мутант PSP_E125A, обладающий повышенной эффективностью катализа [9], а также каталитически неактивный мутант PSP_S532A, содержащий аланиновую замену каталитического серина S532. Данная мутантная форма использована для получения комплексов с пептидным субстратом ZAKIR (табл. 1). Ожидается, что связывание субстрата в субстратсвязывающем центре фермента вызовет переход активного центра в каталитически активную конформацию, при этом нарушение каталитической триады не позволит начаться гидролизу и зафиксирует данное состояние.
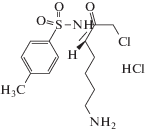
Следующим типом комплекса, выбранным для кристаллизации, был комплекс PSP с низкомолекулярным ингибитором N-альфа-тоцил-L-лизин-хлорометил кетоном (TLCK) (рис. 2), являющимся необратимым ингибитором сериновых протеаз и пептидаз, который способен образовать ковалентную связь одновременно с серином и гистидином каталитического центра, имитируя переходное состояние субстрата в процессе катализа [21].
Финальные комплексы, полученные в настоящей работе, представляли собой комплексы PSP c аналогами продуктов катализа – пептидами вида z-ХХXR/K, где X – любая аминокислота, а z – бензилоксикарбонил (табл. 1). Олигопептидазы B имеют трипсиноподобную субстратную специфичность и гидролизуют пептидную связь после положительно заряженного остатка. С-концевые R/K аминокислотные остатки пептидов, находящихся в кристаллизационной смеси в 100-кратном молярном избытке по сравнению с ферментом, с высокой вероятностью будут взаимодействовать с аминокислотными остатками, ответственными за удаление продуктов реакции из активного центра, а пространственные структуры таких комплексов могут стать иллюстрациями завершающей стадии катализа.
Для всех описанных комплексов подобраны условия кристаллизации (табл. 2) и получены кристаллы (рис. 3). Сравнительный анализ условий кристаллизации показал, что все комплексы PSP c аналогами продуктов катализа кристаллизовались в сходных условиях: 0.2 М малоната натрия, pH 7.0, а в качестве осадителя – 20–30% полиэтиленгликоля (ПЭГ) 3350. Кристаллы PSP_S532A росли в том же ПЭГ при рН 5.5 в присутствии 0.2 М сульфата лития, PSP_E125A – при рН 6.5 и с ПЭГ 5000, а комплекс PSP-TLCK – при рН 8.5 и с ПЭГ 4000.

Рентгеноструктурный анализ полученных кристаллов проводили на синхротронных источниках НИЦ “Курчатовский институт” и SPring-8 (Япония), кристаллографические данные и параметры съемки представлены в табл. 3 и 4 соответственно. Наборы, пригодные для решения пространственных структур, собраны с разрешением 2.72–1.88 Å (синхротрон НИЦ “Курчатовский институт”) и 2.0–1.8 Å (SPring-8).
Таким образом, найдены условия кристаллизации, наработаны кристаллы и проведен сбор и последующий анализ рентгеноструктурных данных для двух мутантных белков и шести комплексов с лигандами. Собранные дифракционные наборы позволят получить недостающую информацию для описания каталитического цикла бактериальных ОpB. Уже на стадии предварительного анализа, исходя из того, что все кристаллы принадлежат пр. гр. P212121, совпадающей с пространственной группой ранее полученной структуры PSP [12], можно предположить, что переход PSP между каталитически активным и неактивным состояниями, скорее всего, не сопровождается сильным расхождением доменов, которое наблюдалось в случае OpB из T. Brucei. Кристаллы, полученные для протозойного фермента в открытой и закрытой конформациях, принадлежали к разным пространственным группам – P3221 и P1211 соответственно [7].
Работы в части кристаллизации белков проведены при поддержке РЦ МКБ НИЦ “Курчатовский институт”, в части определения каталитической активности белков и эффективности ингибиторов – при поддержке Министерства науки и высшего образования РФ в рамках выполнения работ по Государственному заданию ИБХ РАН (АААА-А19-119042690072-1); в части РСА на источнике SPring-8 – при поддержке Федерального космического агентства (проект КЭ (ЦР) “Кристаллизатор”); решение и уточнение структур проводили при частичной поддержке Министерства науки и высшего образования РФ в рамках выполнения работ по Государственному заданию ФНИЦ “Кристаллография и фотоника” РАН.
Список литературы
Fulop V., Bocskei Z., Polgar L. // Cell. 1998. V. 94. P. 161.
Kaushik S., Sowdhamini R. // BMC Genomics. 2014. V. 15. P. 985.
Coetzer T.H., Goldring, J.P., Huson L.E. // Biochimie. 2008. V. 90. P. 336.
Motta F., Azevedo C., Neves B. et al. // Biochimie. 2019. V. 167. P. 207.
Mattiuzzo M., De Gobba C., Runti G. et al. // J. Microbiol. Biotechnol. 2014. V. 24. № 2. P. 160.
McLuskey K., Paterson N.G., Bland N.D. et al. // J. Biol. Chem. 2010. V. 285. № 50. P. 39249.
Canning P., Rea D., Morty R.E., Fülöp V. // PLOS ONE. 2013. V. 8. № 11. e79349.
Михайлова А.Г., Некрасов А.Н., Зинченко А.А. и др. // Биохимия. 2015. Т. 80. № 11. С. 1606.
Mikhailova A.G., Rakitina T.V., Timofeev V.I. et al. // Biochimie. 2017. V. 139. P. 125.
Petrenko D.E., Mikhailova A.G., Timofeev V.I. et al. // J. Biomol. Struct. Dyn. 2019. https://doi.org/10.1080/07391102.2019.1692694
Агапова Ю.К., Талызина А.А., Зейфман Ю.С. и др. // Кристаллография. 2019. Т. 64. № 5. С. 747.
Петренко Д.Е., Николаева А.Ю., Лазаренко В.А. и др. // Кристаллография. 2020. Т. 65. № 2. С. 266.
Михайлова А.Г., Лихарева В.В., Хайруллин Р.Ф. и др. // Биохимия. 2006. Т. 71. № 5. С. 697.
Хайруллин Р.Ф., Михайлова А.Г., Себякина Т.Ю. и др. // Биохимия. 2009. Т. 74. № 10. С. 1427.
Михайлова А.Г., Хайруллин Р.Ф., Демидюк И.В. и др. // Биохимия. 2011. Т. 76. № 4. С. 591.
Михайлова А.Г., Хайруллин Р.Ф., Коломийцева Г.Я. и др. // Биохимия. 2012. Т. 77. № 3. С. 384.
Mikhailova A.G., Khairullin R.F., Demidyuk I.V. et al. // Protein Exp. Purif. 2014. V. 93. P. 63.
Ovchinnikova M.V., Mikhailova A.G., Karlinsky D.M. et al. // Acta Naturae. 2018. V. 10. № 2. P. 65.
Winn M.D., Ballard C.C., Cowtan K.D. et al. // Acta Cryst. D. 2011. V. 67. № 4. P. 235.
Aertgeerts K., Ye S., Tennant M.G. et al. // Protein Sci. 2004. V. 13. P. 412.
Lu D., Fütterer K., Korolev S. et al. // J. Mol. Biol. 1999. V. 292. № 2. P. 361.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография