

Кристаллография, 2021, T. 66, № 5, стр. 807-814
Формирование в растворе структур высокого порядка CBS-пирофосфатазой из D. hafniense
Л. А. Дадинова 1, *, Е. Ю. Сошинская 1, Ю. М. Чесноков 1, 2, Р. А. Камышинский 1, 2, А. Л. Васильев 1, 2, Э. В. Штыкова 1
1 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
2 Национальный исследовательский центр“Курчатовский институт”
Москва, Россия
* E-mail: lubovmsu@mail.ru
Поступила в редакцию 03.09.2020
После доработки 03.09.2020
Принята к публикации 19.10.2020
Аннотация
Для решения вопроса об олигомерном состоянии CBS-пирофосфатазы (CBS-PPase) дикого типа из D. hafniense проведено исследование этого фермента двумя независимыми структурными методами: с помощью малоуглового рентгеновского рассеяния и криогенной просвечивающей электронной микроскопии. Впервые обнаружено формирование в концентрированном растворе белка стабильных структур высокого порядка – крупных спиралевидных ассоциатов. Также впервые показано, что образование таких структур является обратимым процессом и при присоединении лиганда белок переходит в форму тетрамера, в которой обычно существует в разбавленных растворах. Результаты работы важны для понимания функциональных особенностей CBS-PPase, в том числе для понимания патогенеза некоторых заболеваний.
ВВЕДЕНИЕ
Олигомеризация является хорошо известным свойством белков и наблюдается во всех биологических системах [1]. Подсчитано, что по крайней мере 35% всех клеточных белков существует в виде олигомеров [2, 3]. С биологической точки зрения олигомеризация важна, поскольку она сохраняет жизненно важные молекулярные ресурсы, создавая из повторяющихся функционально значимых единиц более крупные полипептидные формирования, обладающие новыми функциональными свойствами, например кооперативностью.
Белковые олигомеры могут быть гомо- или гетеро-олигомерами и варьироваться от димеров до структур высокого порядка: капсидов – внешней белковой оболочки вирусов, и фибрилл – нитевидных белковых структур в клетках и тканях млекопитающих. Некоторые белки образуют только одно конкретное активное олигомерное состояние. Хорошо известным примером такого белка является гемоглобин, существующий в красных кровяных тельцах как гетеро-тетрамер [4]. Другие белки в процессе выполнения своих функций могут находиться в динамическом равновесии между несколькими состояниями с разной активностью, переключаясь между этими состояниями при определенных условиях. Отметим, что во многих случаях такая динамическая сборка и разборка олигомеров играют центральную роль в регуляции активности белка, например в случае актина, который индуцирует подвижность клеток с помощью этого механизма [5].
Образование таких белковых структур высокого порядка, как фибриллы, часто лежит в основе патогенеза нейродегенеративных заболеваний, встречающихся как у человека, так и у животных. Амилоидные фибриллы и олигомеры связаны с большим количеством заболеваний человека, включая болезнь Альцгеймера и прионные болезни. Формирование белковых агрегатов, отражающее конформационную и термодинамическую нестабильность белка, можно рассматривать как общий компонент патогенеза [6]. Недавно стало известно, что даже такие хорошо изученные и полезные белки, как, например, лизоцим, являющийся антибактериальным агентом и разрушающий клеточные стенки бактерий, при определенных условиях могут образовывать патогенные формирования и превращаться в амилоидные структуры [7]. Поэтому важной областью исследования структуры и функции белковых макромолекул является изучение процессов их олигомеризации и формирования структур высокого порядка.
В недавних исследованиях структурных особенностей неорганических CBS-пирофосфатаз (CBS-PPase) из разных бактериальных клеток (D. hafniense, E. lenta и E. harbinense) было показано, что в растворе этот фермент образует устойчивый тетрамер [8, 9], хотя имеется устоявшееся мнение, что CBS-PPase является гомодимером [10]. Отметим, что структурные исследования CBS-PPase и определение их олигомерного состояния очень важны для установления механизмов передачи сигнала между активным и регуляторным центрами, понимания молекулярных основ регуляции ферментов этого семейства и определения механизма подавления их каталитической активности.
CBS-PPase относятся к семейству II растворимых неорганических PPase, которые являются наиболее каталитически активными среди всех типов PPase. Их активные сайты располагаются между DHH- и DHHA2-доменами [11], а автоингибирующая регуляторная вставка в DHH-домене [12]. Эта вставка образована двумя доменами цистатион-β-синтазы (CBS) и одним доменом DRTGG (названного в честь консервативных мотивов Asp-Arg и Thr-Gly-Gly).
Известно, что CBS-домены участвуют в образовании структур более высокого порядка, формируя из двух модулей Бейтмана гомодимерные формы, состоящие уже из четырех CBS-доменов [13]. При этом CBS-домены способны связывать лиганды, в качестве которых в CBS-РРase выступают различные фосфатные производные аденозина [13–15].
Несмотря на достигнутые успехи в изучении структуры и базовых молекулярных механизмов функционирования этих ферментов [16, 17], имеющихся данных недостаточно для установления механизма регуляции белка и определения пути передачи сигнала от регуляторного к активному центру. Вопрос об олигомерном состоянии белка по-прежнему остается открытым из-за противоречивости данных [8–10] и отсутствия структуры высокого разрешения полноразмерного фермента. На данный момент известны лишь несколько кристаллических структур канонических PPase семейства II [18–20] и регуляторной вставки из Clostridium perfringens, в которой пара CBS- и DRTGG-домен образует гомодимер. Но выяснить механизм передачи сигнала между активным и регуляторным центрами невозможно без определения строения полноразмерного фермента, в связи с чем структурный анализ полноразмерной CBS-PPase в условиях, наиболее близких к физиологическим, остается актуальной задачей. Поэтому цель настоящей работы – структурный анализ четвертичной структуры и олигомерного состояния белка CBS-PPasе из D. hafniense (dh-PPase) с помощью взаимодополняющих методов структурной диагностики – малоуглового рентгеновского рассеяния (МУРР) и криогенной просвечивающей электронной микроскопии (крио-ПЭМ).
Результаты работы могут стать шагом к пониманию принципиально важных функциональных особенностей CBS-PPase и способствовать устранению пробелов в понимании патогенеза некоторых заболеваний.
МАТЕРИАЛЫ И МЕТОДЫ
Выделение и очистка CBS-PPasе. Образцы бактериальной неорганической пирофосфатазы dh-PPase предоставлены научной группой из Института физико-химической биологии им. А.Н. Белозерского МГУ им. М.В. Ломоносова. Для приготовления препаратов dh-PPase использовали методики, описанные в [15]. Чистота препаратов была подтверждена SDS-PAGE, как описано в [10]. Концентрацию белка определяли спектрофотометрически, используя значение ${\text{A}}_{{280}}^{{0.1\% }}$, равное 0.478, рассчитанное из аминокислотного состава dh-PPasе в программе ProtParam [21]. Концентрация белка варьировалась от 0.5 до 9 мг/мл. Все эксперименты проводили в 0.1 M Mops-KOH-буфере, pH 7.2, 2 мМ MgCl2, 0.1 мМ CoCl2, 150 мМ KCl.
Эксперимент и анализ данных МУРР. Измерения кривых МУРР проводили в Гамбурге на синхротроне DESY, накопительном кольце PetraIII линии P12. Линия оснащена роботизированной системой для автоматической смены образцов, а также 2D-детектором Pilatus2M (фирма DECTRIS, Швейцария). Интенсивность рассеяния с длиной волны рентгеновского излучения λ = 0.124 нм была измерена в области значений волновых векторов 0.08 < s < 7 нм–1, где s = $(4\pi \sin \theta ){\text{/}}\lambda $, 2θ – угол рассеяния. Для предотвращения искажения результатов из-за возможных радиационных повреждений было снято по 50 кривых рассеяния для буфера (перед серией экспериментов для образца и после) и образца. В ходе обработки полученных данных радиационных повреждений выявлено не было. Первичная обработка полученных данных была проведена с использованием стандартных процедур [22].
Для исключения влияния возможного межчастичного взаимодействия и образования агрегатов также использовали процедуру онлайн эксклюзионной хроматографии в сочетании с малоугловым рентгеновским рассеянием (SEC-SAXS). Данный метод позволяет получить рассеяние от монодисперсного раствора. Эксперимент проводили с использованием колонки Superdex 200 10/30 (GE Healthcare) в режиме непрерывного потока из колонок SEC, элюируемых с использованием периодов воздействия 1 св общей сложности 3600 с (объем одной колонки).
Для получения максимальной эффективности восстановления исходных данных экспериментальные кривые были обрезаны так, чтобы на выходе получилось N = 10–20 шенноновских каналов:
где $\Delta s = \frac{\pi }{{{{D}_{{\max }}}}}$.Исходя из данного условия, для анализа и модельных расчетов использовали наиболее информативный интервал волновых векторов 0.08 < s < < 2 нм–1.
Радиус инерции Rg был получен с использованием приближения Гинье в области малых значений вектора рассеяния s. Из наклона прямолинейного участка $\ln I(s)$ от s2 графика Гинье экспериментальных данных определялась интенсивность рассеяния в нулевой угол I(0), пропорциональная молекулярной массе:
Данное соотношение справедливо в области (sRg) < 1.3.Молекулярную массу (ММ) рассчитывали двумя способами: с помощью Байесовского подхода (MMB) [23] и на основе расчета объема Порода VP (MMP). MMP находили из установленного эмпирического соотношения между VP и ММP, которое для белков равно 1.65 [24].
Функцию распределения по расстояниям p(r) рассчитывали с помощью программы GNOM [25] по уравнению
По функции p(r) определяется максимальный размер белка Dmax из условия p(r) = 0 при r > Dmax.Параметр Dmax необходим для ab initio-моделирования, которое реализовано в программе DAMMIN [26]. Данный алгоритм поиска формы низкого разрешения основан на моделировании отжига в ограниченном объеме (сфере диаметром Dmax), а конечный результат достигается за счет минимизации невязки χ2 с экспериментальными данными:
(4)
${{{{\chi }}}^{2}} = \frac{1}{{N - 1}}\mathop \sum \limits_j {{\left[ {\frac{{{{I}_{{{\text{exp}}}}}({{s}_{j}}) - c{{I}_{{{\text{calc}}}}}({{s}_{j}})}}{{{{\sigma }}({{s}_{j}})}}} \right]}^{2}},$В качестве дополнительного метода восстановления структуры белка был использован гибридный подход (программа CORAL [27]), сочетающий в себе моделирование жесткими телами на основе структур высокого разрешения (PDB ID: 2haw, 3l31) и ab initio-алгоритм для достраивания недостающих фрагментов белка. При этом недостающие аминокислоты моделируются с помощью виртуальных остатков, свернутых в белковоподобную цепь так, чтобы рассеяние от цельной структуры, рассчитанное в программе CRYSOL [28], соответствовало экспериментальному рассеянию, минимизируя невязку χ2.
Полученные двумя способами модели белка проанализированы с помощью программы SUPCOMB [29] для определения степени различия в их пространственной организации. Значение нормализованного пространственного отклонения (NSD), близкое к 1.0, показывает, что модели схожи.
Вспомогательную программуGEN_DAM_body [В.В. Волков, не опубликовано, используется с разрешения автора] применяли для построения спирали с заданными параметрами. Расчет интенсивности рассеяния от полученной модели проводили с помощью программы DAM2IS из программного комплекса ATSAS [27].
Анализ неоднозначности полученных ab initio-моделей выполняли с помощью программы AMBIMETER [30]. Прогнозирование форм белков и их структурную классификацию проводили с помощью DATCLASS [24].
Для анализа количественного состава олигомерной смеси dh-PPase в растворе использовали программу OLIGOMER [22]. С учетом интенсивности рассеяния каждой компоненты в смеси, Ii(s), программа приближает экспериментальную кривую рассеяния к их линейной комбинации, чтобы определить объемную долю wi каждой компоненты. Уравнение
решается относительно wi методом неотрицательных наименьших квадратов, чтобы минимизировать расхождение между экспериментальной и расчетной кривыми рассеяния.Эксперимент по криогенной просвечивающей электронной микроскопии. Исследование проводили с помощью крио-ПЭМ TitanKrios (Thermo Fisher Scientific, США), оснащенного высокочувствительной системой прямого детектирования электронов Falcon II (Thermo Fisher Scientific, США), а также корректором сферических аберраций (Image corrector, CEOS, Германия) при ускоряющем напряжении 300 кВ. Перед крио-ПЭМ-исследованием проводили процедуру витрификации образцов. Электронно-микроскопические медные сетки, покрытые тонким слоем аморфного углерода, были гидрофилизованы в тлеющем разряде (30 с, 25 мА) с помощью установки Pelcoeasi Glow (Ted Pella, США). Далее на сетки наносили 3 мкл препарата с образцом и с помощью установки Vitrobot Mark IV (Thermo Fisher Scientific, США) проводили процедуру витрификации образцов в жидком этане, охлажденном до температуры жидкого азота. В результате частицы образца были зафиксированы в тонком слое аморфного льда, что позволило исследовать их в нативном состоянии и минимизировать радиационные повреждения объекта в ходе дальнейших экспериментов. В течение всего эксперимента влажность в камере системы Vitrobot Mark IV составляла 95–100%, температура 4°С.
Набор данных крио-ПЭМ проводили в автоматизированном режиме с помощью программного обеспечения EPU (Thermo Fisher Scientific, США). Изображения получены при увеличении 59 000 (размер пикселя 1.107 Å), суммарная доза электронов, прошедших через единицу площади образца составила ∼60 э/Å2. Предобработка стеков изображений – коррекция дрейфа, оценка параметров функции передачи контраста (CTF) и выбор одиночных проекций объекта интереса – выполнена с помощью программного пакета Warp [31]. ∼318 000 потенциальных проекций одиночных частиц были выделены из 606 стеков исходных изображений после предобработки и экспортированы в программный пакет СryoSPARC [32] для проведения двумерной классификации. На следующем этапе были отобраны классы, содержащие изображения ∼65 000 частиц dh-PPase, и проведена финальная двумерная классификация.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Поскольку белок CBS-PPase из бактерии D. hafniense был получен при концентрации 1 мг/мл, для определения его олигомерного состояния в растворе образец концентрировали до 9 мг/мл. Кривая малоуглового рассеяния концентрированного раствора dh-PPase (рис. 1, кривая 1) в области малых углов демонстрирует резкое увеличение интенсивности рассеяния и выраженные максимумы в диапазоне значений вектора рассеяния 0.027 < s < 0.25 нм–1. Такой профиль рассеяния отражает присутствие в растворе крупных рассеивающих объектов (кластеров), имеющих строго определенную форму. Наблюдаемые максимумы не исчезают при последовательном разбавлении раствора до концентрации 0.5 мг/мл (рис. 1, кривые 2–4), что говорит о высокой стабильности образованных структур. Для определения их формы по выделенному диапазону данных (0.027 < s < 0.25 нм–1), соответствующему 16 шенноновским каналам, было проведено ab initio-восстановление с помощью программы DAMMIN [26]. Соответствующая функция распределения по расстояниям p(r) представлена на рис. 1в. Максимальный размер Dmax, определенный из функции p(r), равен 210 нм, а поперечное сечение рассеивающего объекта ∼90 нм. Наличие периодически повторяющихся слабо выраженных максимумов на профиле функции p(r) свидетельствовало о присутствии упорядоченных повторяющихся элементов в структуре кластеров dh-PPase. При восстановлении программой DAMMIN формы белка этими периодически повторяющимися элементами оказались витки слегка искаженной спирали, размеры которой соответствовали размерам, определенным из анализа функции p(r), т.е. спиральному телу длиной 210 нм и диаметром 90 нм (рис. 1г). Толщина витка спирали оказалась равной 35 нм.
Рис. 1.
Структурный анализ ассоциатов полноразмерного белка dh-PPase (а); кривые рассеяния от растворов белка dh-PPase с концентрацией 9 (1), 4.5 (2), 2.5 (3), 0.5 мг/мл (4). Анализ структуры ассоциатов: начальная часть (0.027 < s < < 0.25 нм–1) экспериментальной кривой 1 (точки), модельная кривая от ab initio-модели (сплошная линия) и рассеяние от идеальной спирали (пунктирная линия) (б). Функция распределения по расстояниям p(r) для ассоциатов (в). Восстановленное по функции p(r) с помощью программы DAMMIN спиральное тело, идеальная спираль с теми же параметрами (г).
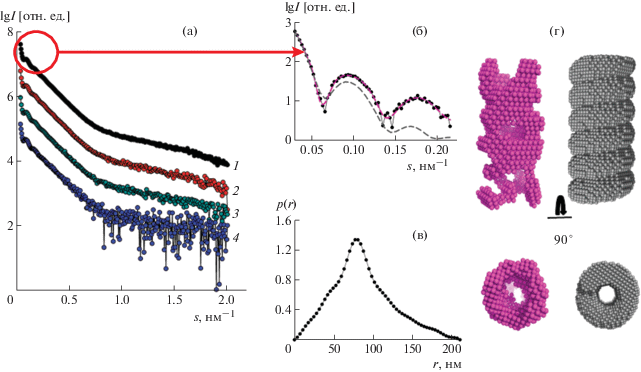
Наличие спиральной структуры подтверждено независимым моделированием с помощью геометрических тел. Для этого была построена шариковая модель спирального тела, внешние размеры которого определялись из анализа функции распределений по расстояниям p(r). От полученной спирали была рассчитана интенсивность малоуглового рассеяния и проведено сравнение с экспериментальными данными МУРР (рис. 1б). Профиль рассеяния от шариковой модели идеальной спирали соответствует профилю экспериментальной кривой (рис. 1б), а имеющееся расхождение обусловлено неидеальностью структуры dh-PPase в растворе, поскольку моделирование геометрическими телами представляет собой метод грубого приближения и не может учитывать все особенности реальных структур, а также подвижность и гибкость их отдельных фрагментов. Однако проведенное моделирование дает основание утверждать, что при концентрировании dh-PPase действительно формирует в растворе большие спиралевидные ассоциаты (рис. 1а, кривые 1–4).
Поскольку в растворе dh-PPase присутствуют крупные белковые ассоциаты-кластеры, для получения данных рассеяния только от минимальных по размеру стабильных форм белка использовали метод SEC-SAXS. После проведения процедуры SEC-SAXS dh-PPase была получена в монодисперсном состоянии, о чем свидетельствуют характерная форма кривой рассеяния (рис. 2а, кривая 1) и график в координатах Гинье с достаточно длинным прямолинейным участком (рис. 2б). Радиус инерции Rg, рассчитанный с использованием приближения Гинье в области малых значений вектора рассеяния s, оказался равен 4.95 нм.
Рис. 2.
Сравнение кривой рассеяния dh-PPase после онлайн эксклюзионной хроматографии (точки) и модельных кривых, полученных с помощью программ DAMMIN (удлиненная пунктирная линия) и CORAL (короткая пунктирная линия) (1); сравнение кривой рассеяния от концентрированного раствора dh-PPase (точки) с модельной кривой, полученной программой OLIGOMER (пунктирная линия) (2) (а). График Гинье (б); график в координатах Кратки (в); функция распределения по расстояниям для dh-PPase (г). Модели dh-PPase, полученные с помощью программ DAMMIN (шарики) и CORAL (ленты – каталитические домены DHH и DHHA2, регуляторные домены CBS и DRTGG, шарики – восстановленные программой CORAL линкеры между доменами) (д).
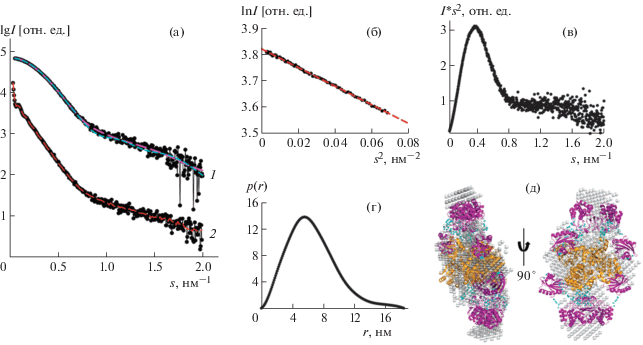
График Кратки (рис. 2в) для этой фракции белка имеет характерный колоколообразный вид, что указывает на то, что образец в целом компактный и имеет ограниченное число разупорядоченных гибких областей [33]. Компактность полученной структуры также подтверждена с помощью программы прогнозирования форм белков по кривой рассеяния DATCLASS [24].
Значения ММ, определенных по экспериментальной кривой, исходя из эмпирического соотношения Породовского объема к ММ белка [24] и обобщенного Байесовского подхода [23], составляют 250 и 243 кДа соответственно. В целом полученные значения ММ и радиуса инерции Rg соответствуют тетрамеру белка (табл. 1).
Таблица 1.
Макромолекулярные характеристики dh-PPase после процедуры онлайн эксклюзионной хроматографии
| Образец | Rg, нм | VP, нм3 | Dmax, нм | MMP, кДа | MMB, кДа |
|---|---|---|---|---|---|
| dh-PPase | 4.95 ± 0.10 | 412 ± 20 | 18.3 ± 1 | 250 ± 10 | 243 (доверительный интервал: 195–264) |
Используя функцию распределения по расстояниям p(r), методом ab initio, реализованном в программе DAMMIN [26], получена форма белка низкого разрешения (рис. 2д, серые шарики), рассеяние от которой хорошо соответствует экспериментальным данным ${{{{\chi }}}^{{2~}}}$ = 1.1 (рис. 2а, кривая 2, пунктирная кривая). Так как полученные структурные характеристики (табл. 1) указывают на то, что белок образует тетрамер в растворе, т.е. димер димеров, при восстановлении формы использовали симметрию P2.
Поскольку решение обратной задачи восстановления трехмерной формы по одномерной кривой рассеяния является неоднозначным, с помощью программы AMBIMETER [30] проведена оценка неоднозначности полученной модели. Как правило, индекс неоднозначности ∼1.5 предполагает, что найдено уникальное, т.е. практически единственное решение, в то время как оценка выше 2.5 говорит о неоднозначном восстановлении формы. В данном случае индекс неоднозначности оказался меньше 1.5, что указывает на уникальность полученной модели.
В качестве дополнительного метода определения структуры фермента использовали гибридное моделирование и программу CORAL [27]. Данный метод позволяет получить более детальную структуру dh-PPase и определить относительное положение каталитической и регуляторной частей белка в растворе. Для моделирования использовали известные структуры высокого разрешения отдельных частей CBS-PPase: регуляторная часть, состоящая из CBS- и DRTGG-домена (PDB ID: 3l31), и каталитическая часть, т.е. DHH- и DHHA2-домены, структура которой была получена в [8]. Поскольку каждая из частей является димерной, они были разделены на мономерные составляющие с условием контакта между соответствующими мономерами, а каталитическая и регуляторная части были соединены между собой достроенными программой CORAL гибкими линкерами. Для получения тетрамера была применена симметрия P2. Полученная модель представлена на рис. 2д и хорошо соответствует экспериментальным данным с χ2 = 1.62.
Как можно видеть, модели, полученные двумя независимыми методами, хорошо согласуются друг с другом (рис. 2д), что подтверждается также количественной оценкой с использованием нормализованного пространственного расхождения NSD [30]. При сравнении моделей, полученных с помощью программ DAMMIN и CORAL, значение NSD равно 1.5. Это свидетельствует о хорошем совпадении структур, полученных разными методами.
Учитывая неоднозначность решения структурных задач в малоугловом рассеянии и противоречивость данных [8–10], необходимо независимое подтверждение существования dh-PPase в растворе в виде тетрамера в качестве минимальной стабильной формы этого белка. С этой целью был использован метод крио-ПЭМ.
На рис. 3 представлены результаты 2D-классификации частиц.
Рис. 3.
Результаты двумерной классификации проекций одиночных частиц dh-PPase. Ширина одиночного изображения (квадрата) – 28 нм.

Анализ данных крио-ПЭМ также приводит к выводу, что dh-PPase представляет собой тетрамер. Однако из-за большого количества доменов, соединенных гибкими фрагментами белковой цепи, наблюдается высокая подвижность доменов, что затрудняет получение трехмерной структуры высокого разрешения. Тем не менее форма проекций частиц, представленных на рис. 3, и их размер соответствуют моделям, полученным по данным МУРР, следовательно, тетрамер является наименьшей устойчивой структурой dh-PPase.
Однако, как показали метод онлайн эксклюзионной хроматографии и последующий анализ, в концентрированном (9 мг/мл) растворе белок существует в виде крупных спиралевидных кластеров. Но можно предположить и одновременное существование какого-то количества тетрамеров dh-PPase. Применение программы OLIGOMER [22] показало, что раствор dh-PPase действительно представляет собой смесь, состоящую из объемной доли wi = 0.93 спиралевидных кластеров и объемной доли wi = 0.07 тетрамеров белка (рис. 2а, кривая 2). Профиль кривой МУРР такой смеси в основном соответствует рассеянию от крупных удлиненных тел. То есть при достаточно высокой концентрации в растворе происходит почти полная ассоциация белка в большие упорядоченные кластеры. Эти ассоциаты являются термодинамически и структурно устойчивыми и не распадаются на отдельные макромолекулы даже при разведении раствора dh-PPase почти в 20 раз (рис. 1а).
Для ответа на вопрос об обратимости такой ассоциации при выполнении белком его функций, например, при присоединении лиганда, к концентрированному раствору dh-PPase был добавлен диаденозинтетрафосфат (Ap4A) в нужной концентрации (1000 мкМ). Оказалось, что в присутствии лиганда спиралевидные кластеры распадаются, а раствор белка представляет собой смесь небольшого количества агрегатов с тетрамерами dh-PPase, т.е. при присоединении субстрата минимальной рабочей единицей фермента действительно является тетрамер [9].
Таким образом, наблюдаемая ассоциация dh-PPase не случайна, обратима и отражает определенные функциональные свойства фермента.
ЗАКЛЮЧЕНИЕ
Полученные результаты структурных исследований двумя независимыми методами, МУРР и крио-ПЭМ, свидетельствуют о тетрамерной структуре dh-PPase в растворе. Этот вывод подтверждается и в процессе взаимодействия фермента с лигандом. Добавление последнего к концентрированному раствору dh-PPase, находящемуся в основном в виде крупных спиралевидных ассоциатов, приводит к исчезновению ассоциатов и получению монодисперсного раствора тетрамера белка. Образование упорядоченных структур высокого порядка этого фермента, как и обратимость этого явления, наблюдались в настоящей работе впервые. Однако природу и значимость этого явления еще только предстоит определить соответствующими методами биохимии и биологии.
Авторы выражают благодарность научной группе из Института физико-химической биологии им. А.Н. Белозерского МГУ им. М.В. Ломоносова и лично В.А. Анашкину за предоставление образцов.
Работа выполнена при поддержке Министерства науки и высшего образования РФ в рамках выполнения работ по Государственному заданию ФНИЦ “Кристаллография и фотоника” РАН.
Список литературы
Mayssam H.A., Barbara I. // Bioorg. Med. Chem. 2005. V. 13. P. 5013. https://doi.org/10.1016/j.bmc.2005.05.037
Jones S., Thornton J.M. // Proc. Natl. Acad. Sci. 1996. V. 93. P. 13. https://doi.org/10.1073/pnas.93.1.13
Goodsell D.S, Olson A.J. // Annu. Rev. Biophys. Biomol. Struct. 2000. V. 29. P. 105. https://doi.org/10.1146/annurev.biophys.29.1.105
Fermi G., Perutz M.F., Shaanan B. et al. // J. Mol. Biol. 1984. V. 175. P. 159. https://doi.org/10.1016/0022-2836(84)90472-8
Ono S. // Int. Rev. Cytol. 2007. V. 258. P. 1. https://doi.org/10.1016/S0074-7696(07)58001-0
Сахаров В.Н., Литвицкий П.Ф. // Вестн. РАМН. 2016. Т. 71. С. 46. https://doi.org/10.15690/vramn635
Granel B., Valleix S., Serratrice J. et al. // Medicine. 2006. V. 85. P. 66. https://doi.org/10.1097/01.md.0000200467.51816.6d
Dadinova L.A., Soshinskaia E.Yu., Jeffries C.M. et al. // Biomolecules. 2020. V. 10. P. 564. https://doi.org/10.3390/biom10040564
Dadinova L.A., Anashkin V.A., Shtykova E.V. // Crystallography Reports. 2020. V. 65. P. 749. https://doi.org/10.1134/S1063774520050053
Parfenyev A., Salminen A., Halonen P. et al. // J. Biol. Chem. 2001. V. 276. P. 24511. https://doi.org/10.1074/jbc.M101829200
Aravind L., Koonin E. // Trends Cell. Biol. 1998. V. 23. P. 17.
Baykov A.A., Anashkin V.A., Salminen A. et al. // FEBS Lett. 2017. V. 591. P. 3225. https://doi.org/10.1002/1873-3468.12877
Anashkin V.A., Salminen A., Tuominen H.K.et al. // J. Biol. Chem. 2015. V. 290. P. 27594. https://doi.org/10.1074/jbc.M115.680272
Jamsen J., Tuominen H., Salminen A. et al. // Biochem. J. 2007. V. 408. P. 327. https://doi.org/10.1042/BJ20071017
Salminen A., Anashkin V.A., Lahti M.et al. // J. Biol. Chem. 2014. V. 289. P. 22865. https://doi.org/10.1074/jbc.M114.589473
Anashkin V.A., Salminen A., Osipova E. et al. // ACS Omega. 2019. V. 4. P. 15549. https://doi.org/10.1021/acsomega.9b01879
Anashkin V.A., Salminen A., Vorobjeva N.N. et al.// Biochem. J. 2016. V. 473. P. 2097. https://doi.org/10.1042/BCJ20160293
Merckel M., Fabrichniy I., Salminen A. et al. // Structure. 2001. V. 94. P. 289. https://doi.org/10.1016/s0969-2126(01)00587-1
Ahn S., Milner A., Fütterer K. et al. // J. Mol. Biol. 2001. V. 313. P. 797. https://doi.org/10.1006/jmbi.2001.5070
Rantanen M., Lehtiö L., Rajagopal L. et al. // Acta Cryst. D. 2007. V. 63. P. 738. https://doi.org/10.1107/S0907444907019695
Gasteiger E., Hoogland C., Gattiker A. et al. The Proteomics Protocols Handbook. N.J.: Humana Press, 2005. 607 p.
Konarev P.V., Volkov V.V., Sokolova A.V. et al. // J. Appl. Cryst. 2003. V. 36. P. 1277. https://doi.org/10.1107/S0021889803012779
Hajizadeh N.R., Franke D., Jeffries C.M.et al. // Sci. Rep. 2018. V. 8. P. 7204. https://doi.org/10.1038/s41598-018-25355-2
Franke D., Petoukhov M.V., Konarev P.V. et al. // J. Appl. Cryst. 2017. V. 50. P. 1212. https://doi.org/10.1107/S1600576717007786
Svergun D.I. // J. Appl. Cryst. 1992. V. 25. P. 495. https://doi.org/10.1107/S0021889892001663
Svergun D.I. // Biophys. J. 1999. V. 76. P. 2879. https://doi.org/10.1016/S0006-3495(99)77443-6
Petoukhov M.V., Franke D., Shkumatov A.V. et al. // J. Appl. Cryst. 2012. V. 45. P. 342. https://doi.org/10.1107/S0021889812007662
Svergun D.I., Barberato C., Koch M.H.J. // J. Appl. Cryst. 1995. V. 28. P. 768. https://doi.org/10.1107/S0021889895007047
Kozin M., Svergun D. // J. Appl. Cryst. 2001. V. 34. P. 33. https://doi.org/10.1107/S0021889800014126
Petoukhov M.V., Svergun D.I. // Acta Cryst. D. 2015. V. 71. P. 1051. https://doi.org/10.1107/S1399004715002576
Tegunov D., Cramer P. // Nat. Methods. 2019. V. 16. P. 1146. https://doi.org/10.1038/s41592-019-0580-y
Punjani A., Rubinstein J., Fleet D. et al. // Nat. Methods. 2017. V. 14. P. 290. https://doi.org/10.1038/nmeth.4169
Jacques D.A., Guss J.M., Svergun D.I. et al. // ActaCryst. D. 2012. V. 68. P. 620. https://doi.org/10.1107/S0907444912012073
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография