

Кристаллография, 2022, T. 67, № 3, стр. 411-418
Изучение свободной энергии связывания пептидных субстратов в активном центре олигопептидазы В из Serratia proteamaculans методом MM-GBSA
Д. Е. Петренко 1, *, В. И. Тимофеев 1, 2, Д. М. Карлинский 3, Д. Д. Плащинская 3, 4, А. Г. Михайлова 3, Т. В. Ракитина 1, 3, **
1 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
2 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
3 Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
Москва, Россия
4 Московский физико-технический институт
Долгопрудный, Россия
* E-mail: dmitry.e.petrenko@gmail.com
** E-mail: taniarakitina@yahoo.com
Поступила в редакцию 12.05.2021
После доработки 26.05.2021
Принята к публикации 26.05.2021
- EDN: KKVFTR
- DOI: 10.31857/S002347612203016X
Аннотация
Проведены моделирование и термодинамические исследования фермент-субстратных комплексов, состоящих из олигопептидазы В (ОрВ) бактерии Serratia proteamaculans дикого типа (PSP) и ее мутантных форм, содержащих аминокислотные замены E125A и D649A, которые увеличивают эффективность катализа и изменяют вторичную субстратную специфичность соответственно, и пептидных субстратов GRRG и GKRG, содержащих аргинин в P1-положении и аргинин или лизин в Р2-положениях соответственно. Свободные энергии связывания пептидных субстратов в активных центрах ферментов, вычисленные методом, представляющим собой комбинацию молекулярной механики и решения обобщенного уравнения Борна (MM-GBSA), сопоставлены с эффективностями гидролиза (kcat/Km), экспериментально измеренными для соответствующих фермент-субстратных пар. Показано, что свободная энергия связывания в комплексе PSP/GRRG почти в 2 раза больше, чем в комплексе PSP/GKRG, что хорошо коррелирует с повышенной в 2.7 раза эффективностью гидролиза. Аминокислотные замены E125A и D649A приводили к увеличению свободной энергии связывания в обоих типах комплексов, что соответствовало повышенной по сравнению с PSP дикого типа каталитической эффективности PSP-E125A в отношении обоих субстратов и PSP-D649A в отношении лизинсодержащего субстрата. Полученные результаты показывают, что метод MM-GBSA позволяет прогнозировать специфичность бактериальных ОрВ в отношении пептидных субстратов, а также влияние аминокислотных замен на эффективность гидролиза, поэтому он может быть использован в исследованиях механизмов каталитической активации и при моделировании низкомолекулярных ингибиторов бактериальных ОрВ.
ВВЕДЕНИЕ
В термодинамике свободной энергией называют количество внутренней энергии системы, которая может быть использована для выполнения работы. Изменение свободной энергии (ΔG) определяет направленность термодинамических процессов и управляет такими молекулярными процессами, как свертывание белков, молекулярные взаимодействия и химические превращения [1]. Таким образом, определение ΔG является одной из важных задач в биомолекулярных исследованиях белков и их комплексов с другими макромолекулами или низкомолекулярными лигандами (кофакторами, субстратами, ингибиторами). Поскольку экспериментальные измерения термодинамических свойств биомолекулярных систем часто являются дорогостоящими и трудоемкими, точные теоретические расчеты ΔG с помощью методов вычислительной биологии становятся все более востребованными при изучении макромолекулярных комплексов и рациональном дизайне лекарств. В настоящее время наиболее широко используемым инструментом для определения ΔG молекулярных систем являются подходы, основанные на моделировании методами молекулярной динамики (МД), которая позволяет не только моделировать физические процессы, происходящие в системе на атомарном уровне, но и выявлять термодинамические характеристики системы, определяющие ее устойчивость в различных состояниях. Наиболее популярными при вычислении ΔG являются метод, сочетающий использование молекулярной механики и решение уравнения Пуассона–Больцмана (ММ-PBSA), и метод, сочетающий использование молекулярной механики и решение обобщенного уравнения Борна (ММ-GBSA), поскольку они более точны, чем большинство скоринговых функций молекулярного докинга, но при этом не требуют значительных вычислительных мощностей [2].
Олигопептидазы В (OpB), представляющие собой сериновые пептидазы и обладающие трипсиноподобной субстратной специфичностью (КФ 3.4.21.83), найдены в бактериях, паразитических простейших и некоторых растениях [3]. OpB относятся к подсемейству S9А семейства пролил-олигопептидаз (РОР) [4]. Все представители семейства РОР состоят из С-концевого каталитического α/β-гидролазного домена и N-концевого β-пропеллерного регуляторного домена, ограничивающего доступ высокомолекулярных субстратов к каталитической триаде. Два домена соединены шарнирным участком, что позволяет ферментам переходить из каталитически неактивной (открытой) конформации, в которой домены и аминокислотные остатки каталитической триады разобщены, в каталитически активную (закрытую) конформацию, в которой сами домены и остатки каталитической триады сближены [5].
OpВ являются известными факторами патогенеза тяжелых паразитарных заболеваний и некоторых бактериальных инфекций [3, 6]. Данные пептидазы также участвуют в защите бактерий от антимикробных пептидов [7]. Эти факты, а также отсутствие ОрВ у млекопитающих делает эти ферменты перспективными мишенями для разработки противопаразитарных и антибактериальных препаратов. Моделирование низкомолекулярных ингибиторов требует знания пространственных структур ферментов-мишеней, имеющих каталитически активные конформации, и знания механизмов каталитической активации.
В пространственных структурах бактериальных ОpВ, полученных на сегодня, фермент находился в переходной конформации, в которой, несмотря на сближение доменов, каталитическая триада была разобщена [8, 9]. Однако структуры закрытых конформаций были получены для ферментов простейших, а также для бактериальных пролилэндопептидаз (РЕР), относящихся к тому же подсемейству S9А, что и ОрВ; оба типа ферментов были закристаллизованы в комплексах с ковалентно связанными ингибиторами – аналогами переходного состояния [5, 10–12]. Знание пространственных структур OpB из лейшмании Leishmania major (PDBID 2XE4, [10]) и трипаносомы Trypanosoma brucei (PDBID 4ВР9, [5]) в комплексах с антипаином, а также бактериальных РЕР из Myxococcus xanthus (PDBID 2BKL, [11]) и Aeromonas punctata (PDBID 3IVM, [12]) в комплексах с бензилоксикарбонил-Р/A-пролиналом позволяет моделировать каталитически активные конформации ОрВ бактерий. Однако механизм каталитической активации, предложенный в [5] для ОрВ простейших и бактериальных РЕР, не может быть автоматически перенесен на ОрВ бактерий, так как в последних необходимые для реализации данного механизма аминокислотные остатки границы между доменами не являются консервативными [13]. Ранее, комбинируя методы молекулярного моделирования и динамики с направленным мутагенезом, проведенным для подтверждения результатов вычислительных методов, были идентифицированы альтернативные аминокислотные детерминанты каталитической активации, находящиеся на границе между доменами ОрВ из бактерии Serratia proteamaculans (PSP), взятой в качестве модельного бактериального фермента [13, 14].
В настоящей работе с целью изучения прогностической значимости термодинамических расчетов методом MM-GBSA и их применимости для исследования механизма каталитической активации бактериальных ОрВ провели сравнительные исследования модельных фермент-субстратных комплексов, состоящих из PSP дикого типа или мутантных форм данного фермента с измененной активностью и вторичной субстратной специфичностью и тетрапептидов GRRG и GKRG. Рассчитанная методом MM-GBSA свободная энергия связывания пептидных субстратов была сопоставлена с эффективностью катализа, экспериментально измеренной для каждой фермент-субстратной пары. Выявлено, что увеличение ΔG связывания пептида в активном центре олигопептидаз, как правило, коррелирует с повышенной эффективностью катализа, что указывает на применимость метода MM-GBSA для прогнозирования каталитической эффективности бактериальных ОрВ в отношении различных субстратов, а также изучения влияния аминокислотных замен на каталитическую активность.
МАТЕРИАЛЫ И МЕТОДЫ
Построение начальных моделей комплекса. Модель PSP с закрытой конформацией построена как описано в [13], модели комплексов PSP с тетрапептидами GRRG и GKRG – как описано в [15]. Модели комплексов мутантных форм PSP получены с помощью программы PyMol, как описано в [12].
МД-моделирование. Полученные модели олигопептидаз в комплексах с тетрапептидами GRRG и GKRG использованы как стартовые для проведения МД-экспериментов. Для обеих систем моделирование проводили с помощью программы GROMACS 2020.3 [16]. В качестве силового выбрано поле ff14SB с улучшенной точностью моделирования боковых цепей аминокислотных остатков [17]. В качестве модели воды выбрана модель TIP3P, как наиболее подходящая для использования с силовым полем ff14SB. Молекула белка была помещена в ячейку в форме параллелепипеда таким образом, чтобы расстояние от молекулы белка до краев ячейки составляло 1.5 нм. Ячейка была заполнена молекулами воды. Кроме того, в каждую из систем было добавлено 0.15 мМ КCl, а также ионы K+ для уравновешивания заряда системы. На первом этапе моделирования для каждой из систем проведена минимизация энергии с шагом 1 фс до максимальной силы 1000.0 кДж/моль–1 нм–1. Далее системы были уравновешены при температуре 298 K и давлении 1 атм путем моделирования в NVT и NTP (100 пс в каждом) соответственно. Температуру и давление в системах контролировали с использованием модифицированного термостата Берендсена [18] и баростата Паринелло–Рахмана [19] с временными константами tau_t = = 0.1 и tau_p = 2 пс соответственно. Продуктивное 50 нс МД-моделирование для каждой системы проводили в изотермоизобарическом ансамбле с шагом в 2 фс. Алгоритм LINCS [20] использовали для ограничения водородных связей. Дальние электростатические взаимодействия рассчитывали с использованием схемы суммирования по Эвальду [21]. Ван-дер-Ваальсовы и кулоновские взаимодействия были усечены до 1.4 нм, что является оптимальным для используемого силового поля [17]. Поскольку при моделировании выполнялись периодические граничные условия, были выполнены перецентрирование белковых молекул и их возврат в ячейку моделирования с использованием команды gmxtrjconv программного пакета GROMACS с флагом-pbcnojump.
Расчет энергии взаимодействия тетрапептидных субстратов с ферментами. Для расчета ΔG взаимодействия тетрапептидов GRRG и GKRG с модельными ферментами использовали метод MM-GBSA [22]. Расчет осуществляли с использованием программы gmx_MMPBSA версия v1.4.0 (http://doi.org/10.5281/zenodo.4569307), которая с помощью программного пакета Amber Tools 2020 и программы MMPBSA.py [23] позволяет применять метод MM-GBSA для траекторий, полученных с использованием программного пакета GROMACS. Для расчета ΔG использовали 5000 структур для каждой из смоделированных систем. При вычислениях использовали модифицированную обобщенную модель Борна [24].
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Ранее были выявлены заряженные аминокислотные остатки PSP, участвующие в формировании границ между доменами. Замены этих остатков на аланин моделировали активность и вторичную субстратную специфичность фермента [13]. Данные о каталитической эффективности PSP дикого типа и ее мутантных вариантов в отношении п-нитроанилидных субстратов, содержащих остатки аргинина в P1-положениях и остатки аргинина или лизина в Р2-положениях, позволяли исследовать прогностический потенциал метода MM-GBSA, основанного на использовании МД-эксперимента, для определения изменения свободной энергии [2] и его применимость для исследования механизмов каталитической активации и поиска ингибиторов бактериальных ОрВ.
Метод MM-GBSA позволяет сравнить свободную энергию сольватированной молекулы в двух разных состояниях (связанном и свободном). Величина ΔG при связывании лиганда белком-акцептором может быть рассчитана по формуле
(1)
$\Delta G = \Delta {{G}_{{{\text{вак}}}}} + \Delta {{G}_{{{\text{ком}}}}}--(\Delta {{G}_{{{\text{лиг}}}}} + \Delta {{G}_{{{\text{рец}}}}}),$(2)
$\Delta {{G}_{{{\text{cолв}}}}} = \Delta {{G}_{{{\text{эл}}}}} + \Delta {{G}_{{{\text{гидр}}}}},$Впервые такой подход был применен в [22], где использовались расчеты на основе уравнения Пуассона–Больцмана (метод ММ-РBSA). Позднее для ускорения расчетов стали использовать обобщенное уравнение Борна [25, 26]. В настоящей работе применен усовершенствованный метод MM-GBSA, разработанный для макромолекул, в котором борновские радиусы масштабируются для учета межузельных пространств между атомными сферами [24, 27].
В качестве белков-акцепторов выбраны PSP дикого типа и мутантные формы фермента PSP-E125A и PSP-D649A, обладающие повышенной активностью и измененной вторичной субстратной специфичностью соответственно. Известно, что ОрВ, включая PSP, гидролизуют пептидную связь, образованную карбоксильной группой положительных аминокислотных остатков (R или К), находящихся в Р1-положении субстрата. Эффективность гидролиза увеличивается, если предшествующий остаток, занимающий P2-положение, тоже положительный. Все охарактеризованные ОрВ, включая PSP и PSP-E125A, предпочитают субстраты с остатком аргинина в P2-положении, тогда как PSP-D649A эффективнее гидролизует субстраты с остатком лизина в P2-положении. В качестве лигандов использованы тетрапептиды GRRG и GKRG, содержащие остаток аргинина в P1-положениях и остатки аргинина или лизина в Р2-положениях соответственно. Для всех шести систем проведено МД-моделирование продолжительностью по 50 нс.
Первоначальный анализ траекторий включал в себя расчет среднеквадратичных отклонений (СКО) Cα-атомов от начального положения (рис. 1), изменения радиусов инерции комплексов (рис. 2), а также анализ подвижности отдельных аминокислотных остатков в процессе МД-моделирования (рис. 3).
Рис. 1.
Среднеквадратичные отклонения (СКО) Cα-атомов ферментов в комплексах с пептидами GRRG (1) и GKRG (2) в процессе МД-моделирования, вычисленные для PSP дикого типа (а) и мутантных форм PSP-E125A (б) и PSP-D649A (в).

Рис. 2.
Радиусы инерции ферментов в комплексах с пептидами GRRG (1) и GKRG (2) в процессе МД-моделирования, вычисленные для PSP дикого типа (а) и мутантных форм PSP-E125A (б) и PSP-D649A (в).
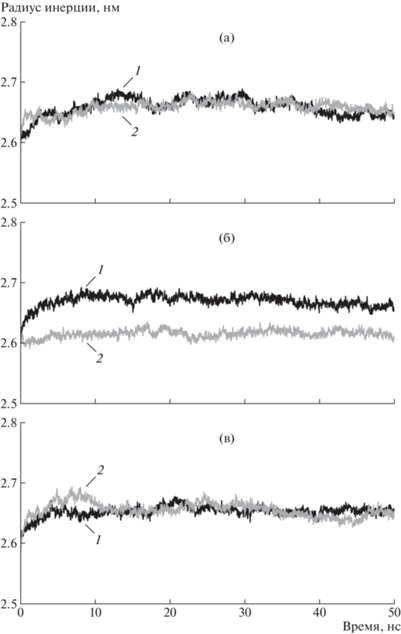
Рис. 3.
Среднеквадратичные флуктуации (СКФ) Cα-атомов аминокислотных остатков ферментов в комплексах с пептидами GRRG (1) и GKRG (2) в процессе МД-моделирования, вычисленные для PSP дикого типа (а) и мутантных форм PSP-E125A (б) и PSP-D649A (в).

Из графика на рис. 1а видно, что в случае PSP дикого типа сначала (через 1.5 нс моделирования) величины СКО для обеих систем (комплексы с GRRG и GKRG) стабилизируются на уровне около 0.275 нм, затем (на 6-й нс) опять наблюдается увеличение СКО до 0.35 нм (комплекс с GKRG) и до 0.45 нм (комплекс с GRRG). В конце моделирования (начиная с 43 нс) значения СКО в обеих системах стабилизируются на уровне 0.33 нм. В случае PSP-E125A обе системы стабилизируются на 15 нс, при этом величины СКО составляют 0.29 и 0.42 нм для комплекса с GKRG и GRRG соответственно (рис. 1б). Комплекс PSP-D649A/ GKRG стабилизируется на 10 нс при значении СКО 0.31 нм, а комплекс PSP-D649A/GRRG – на 20 нс, СКО 0.36 нм (рис. 1в). Учитывая размер моделируемых комплексов (молекулярная масса PSP 76 кДа), можно сказать, что значения СКО свидетельствуют о высокой устойчивости систем, при этом комплексы с пептидом GKRG немного более стабильны при МД-моделировании.
Рисунки 2а и 2в показывают, что в случае PSP дикого типа и PSP-D649A радиусы инерции обеих систем практически не меняются, находясь в диапазоне от 2.6 до 2.65 нм, что указывает на то, что во время МД-моделирования компактность систем существенно не меняется. Это, в свою очередь, свидетельствует о том, что в процессе моделирования не происходит существенного движения доменов друг относительно друга. В то же время радиус инерции комплекса PSP-D649A/GRRG немного увеличивается, оказываясь в диапазоне 2.65–2.7 нм (рис. 2б), что коррелирует с увеличением СКО (рис. 1б) и указывает на незначительное расхождение доменов.
Из графиков на рис. 3 следует, что в случае PSP дикого типа и обоих мутантных белков значения среднеквадратичных флуктуаций (СКФ) Cα-атомов аминокислотных остатков каталитического домена PSP (С-концевая область белка, начиная с 410 аминокислоты) во всех системах не превышают 0.4 нм. Более высокие (>0.4 нм) значения RMSF Cα-атомов наблюдаются только в области петлевых участков пропеллерного домена.
Таким образом, все исследуемые комплексы являются стабильными и позволяют применить метод MM-GBSA для расчета энергии связывания пептидов. Рассчитанные с помощью MM-GBSA изменения свободной энергии при связывании тетрапептидов GRRG и GKRG молекулами PSP дикого типа и мутантными формами фермента приведены в табл. 1.
Таблица 1.
Изменения свободной энергии (ΔG) при взаимодействии PSP дикого типа и ее мутантных форм с тетрапептидами GKRG и GRRG
| Белки и пептиды | Расчетные значения ΔG, ккал/моль | П-нитро-анилидные субстраты | Эффективности гидролиза* kcat/Km, µМ–1 мин–1 |
|---|---|---|---|
| PSP | |||
| GKRG | –16.6 ± 5.1 | Z-KR-pNA | 162 |
| GRRG | –29.8 ± 5.7 | Z-RR-pNA | 431 |
| PSP-E125A | |||
| GKRG | –38.3 ± 7.4 | Z-KR-pNA | 299 |
| GRRG | –34.6 ± 8.0 | Z-RR-pNA | 972 |
| PSP-D649A | |||
| GKRG | –33.0 ± 5.1 | Z-KR-pNA | 357 |
| GRRG | –33.5 ± 6.4 | Z-RR-pNA | 184 |
Из табл. 1 следует, что значение ΔG для комплекса PSP/GRRG примерно в 2 раза больше, чем для комплекса PSP/GKRG, что указывает на более сильное связывание фермента дикого типа с первым пептидом. Это коррелирует с ранее полученными данными о большем количестве межмолекулярных контактов, возникающих между PSP и тетрапептидом с остатком аргинина в Р2-положении [15]. Полученные в [13, 14] кинетические параметры гидролиза п-нитроанилидных субстратов: Z-RR-pNA и Z-KR-pNA (Z – бензилоксикарбонил), которые можно рассматривать как аналоги тетрапептидов GRRG и GKRG, подтверждают, что PSP гидролизует субстрат с аргинином в Р2-положении в 2.7 раза эффективнее, чем с лизином, что указывает на высокую точность расчетов методом MM-GBSA в случае фермента дикого типа.
Наиболее сильное связывание пептидов, исходя из расчетных данных, наблюдается в комплексах PSP-E125A, что коррелирует с более высокими значениями эффективности гидролиза обоих субстратов по сравнению с ферментом дикого типа (табл. 1). В случае субстрата, содержащего лизин в Р2-положении, 2.3-кратное усиление связывания в комплексе PSP-E125A/GKRG по сравнению с комплексом PSP/GKRG хорошо коррелирует с 1.8-кратным увеличением эффективности гидролиза в первой фермент-субстратной паре. В случае субстрата, содержащего аргинин в Р2-положении, увеличение связывания в комплексе PSP-E125A/GRRG по сравнению с комплексом PSP/GRRG составляет только 14%, тогда как каталитическая эффективность мутантного белка в отношении данного субстрата увеличивается в 2.2 раза.
Расчетные энергии связывания пептидов в комплексах PSP-D649A/GKRG и PSP-D649A/ GRRG увеличены в 2 раза и на 11% соответственно по сравнению с PSP-содержащими комплексами (табл. 1). В первом случае это хорошо коррелирует с кинетическими данными; каталитическая эффективность PSP-D649A в отношении Z-KR-pNA в 2.2 раза выше, чем у фермента дикого типа. Во втором случае расчетная величина ΔG оказалась выше, чем ожидалось согласно кинетическим данным. В этой связи нельзя не отметить, что для обоих мутантных белков точность предсказания вторичной субстратной специфичности была заметно более низкой, чем для фермента дикого типа. Можно предположить, что при введении мутаций стабилизация оптимальной конформации белка занимает определенное время, и расчет энергии по полной МД-траектории начинается еще до полной стабилизации системы, что и вносит ошибку. В то же время для большинства фермент-субстратных пар влияние мутации на величину свободной энергии связывания субстрата в активном центре фермента коррелировало с кинетическими данными.
Таким образом, результаты расчетов изменения свободной энергии при образовании фермент-субстратных комплексов в целом коррелируют с экспериментальными кинетическими данными. Это подтверждает целесообразность использования метода MM-GBSA для виртуальной оценки специфичности бактериальных ОрВ в отношении различных субстратов и изучения влияния аминокислотных замен на каталитическую эффективность ферментов, что в совокупности указывает на перспективность применения данного метода для решения таких задач, как изучение механизма каталитической активации бактериальных ОрВ и прогнозирование эффективности низкомолекулярных ингибиторов.
Работа выполнена при финансовой поддержке Российского научного фонда (проект № 21-74-20154) в части моделирования, МД-экспериментов и термодинамических расчетов. Сравнительный анализ методов ММ-РBSA и ММ-GBSA проведен при поддержке Министерства науки и высшего образования РФ в рамках выполнения работ по Государственному заданию ФНИЦ “Кристаллография и фотоника” РАН.
Список литературы
Christ C.D., Mark A.E., van Gunsteren W.F. // J. Comput. Chem. 2009. V. 31. P. 1569. https://doi.org/10.1002/jcc.21450
Wang E., Sun H., Wang J. et al. // Chem. Rev. 2019. V. 119. № 16. P. 9478. https://doi.org/10.1021/acs.chemrev.9b00055
Motta F.N., Azevedo C.S., Neves B.P. et al. // Biochimie. 2019. V. 167. P. 207. https://doi.org/10.1016/j.biochi.2019.10.006
Polgár L. // Cell. Mol. Life Sci. 2002. V. 59. P. 349. https://doi.org/10.1007/s00018-002-8427-5
Canning P., Rea D., Morty R.E. et al. // PloS One. 2013. V. 8. № 11. P. e79349. https://doi.org/10.1371/journal.pone.0079349
Coetzer T.H., Goldring J.P., Huson L.E. // Biochimie. 2008. V. 90 P. 336. https://doi.org/10.1016/j.biochi.2007.10.011
Mattiuzzo M., Gobba C., Runti G. et al. // J. Microbiol. Biotechnol. 2014. V. 24 (2). P. 160. https://doi.org/10.4014/jmb.1310.10015
Petrenko D.E., Timofeev V.I., Britikov V.V. et al. // Biology. 2021. V. 10. № 10. P. 1021. https://doi.org/10.3390/biology10101021
Timofeev V.I., Petrenko D.E., Agapova Y.K. et al. // Crystals. 2021. V. 11. № 11. P. 1438. https://doi.org/10.3390/cryst11111438
McLuskey K., Paterson N.G., Bland N.D. et al. // J. Biol. Chem. 2010. V. 285. № 50. P. 39249. https://doi.org/10.1074/jbc.M110.156679
Shan L., Mathews I.I., Khosla C. // Proc. Natl. Acad. Sci. USA. 2005. V. 102. P. 359. https://doi.org/10.1073/pnas.0408286102
Li M., Chen C., Davies D.R. et al. // J. Biol. Chem. 2010. V. 285. P. 21487. https://doi.org/10.1074/jbc.M109.092692
Mikhailova A.G., Rakitina T.V., Timofeev V.I. et al. // Biochimie. 2017. V. 139. P. 125. https://doi.org/10.1016/j.biochi.2017.05.013
Petrenko D.E., Mikhailova A.G., Timofeev V.I. et al. // J. Biomol. Struct. Dyn. 2020. V. 38. № 16. P. 4868. https://doi.org/10.1080/07391102.2019.1692694
Agapova Y.K., Talyzina A.A., Zeifman Y.S. et al. // Crystallography Reports. 2019. V. 64 № 5. P. 758. https://doi.org/10.1134/S106377451905002X
Abraham M.J., Murtola T., Schulz R. et al. // SoftwareX. 2015. V. 1–2. P. 19. https://doi.org/10.1016/j.softx.2015.06.001
Maier J.A., Martinez C., Kasavajhala K. et al. // J. Chem. Theory. Comput. 2015. V. 11. № 8. P. 3696. https://doi.org/10.1021/acs.jctc.5b00255
Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F. et al. // J. Chem. Phys. 1984. V. 81. № 8. P. 3684. https://doi.org/10.1063/1.448118
Parrinello M., Rahman A. // J. Chem. Phys. 1982. V. 76. № 5. P. 2662. https://doi.org/10.1063/1.443248
Hess B., Bekker H., Berendsen H.J.C. et al. // J. Comput. Chem. 1997. V. 18. P. 1463. https://doi.org/10.1002/(SICI)1096-987X(199709)18:12< 1463::AID-JCC4>3.0.CO;2-H
Darden T., York D., Pedersen L. // J. Chem. Phys. 1993. V. 98. P. 10089. https://doi.org/10.1063/1.464397
Kollman P.A., Massova I., Reyes C. et al. // Acc. Chem. Res. 2000. V. 33. № 12. P. 889. https://doi.org/10.1021/ar000033j
Miller B.R., McGee T.D., Swails J.M. et al. // J. Chem. Theory Comput. 2012. V. 8. № 9. P. 3314. https://doi.org/10.1021/ct300418h
Onufriev A., Bashford D., Case D.A. // Proteins. 2004. V. 55. P. 383. https://doi.org/10.1002/prot.20033
Srinivasan J., Trevathan M.W., Beroza P. et al. // Theor. Chem. Acc. 1999. V. 101. P. 426. https://doi.org/10.1007/s00214990m185
Still W.C., Tempczyk A., Hawley R.C. et al. // J. Am. Chem. Soc. 1990. V. 112. P. 6127. https://doi.org/10.1021/ja00172a038
Onufriev A., Bashford D., Case D.A. // J. Phys. Chem. B. 2000. V. 104. P. 3712. https://doi.org/10.1021/jp994072s
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография