

Кристаллография, 2022, T. 67, № 4, стр. 597-607
Структура, состав и свойства образцов хлопкового пуха в зависимости от методов карбонизации
В. Е. Асадчиков 1, И. Г. Дьячкова 1, *, Д. А. Золотов 1, А. С. Кумсков 1, А. Л. Васильев 1, 2, В. В. Берестов 3
1 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
2 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
3 НИИ Перспективных материалов и технологий
Москва, Россия
* E-mail: dyahkova.i@crys.ras.ru
Поступила в редакцию 21.12.2021
После доработки 21.12.2021
Принята к публикации 29.12.2021
- EDN: TSVKXR
- DOI: 10.31857/S002347612204004X
Аннотация
Представлены результаты исследований структуры, элементного и фазового состава, адсорбционной активности образцов хлопкового пуха, подвергнутых СВЧ- и термической карбонизации. Установлено, что в процессе СВЧ-карбонизации примеси неравномерно осаждаются на волокнах поверхностных слоев, практически не загрязняя фибриллы. При СВЧ-нагреве происходит более активное развитие пористой структуры волокна. В термически карбонизированном образце содержание аморфной фазы больше. Идентифицированы наиболее вероятные кристаллические фазы, образовавшиеся в образцах в процессе карбонизации, – фазы графита с гексагональной и углерода с тетрагональной решетками. Адсорбционная активность СВЧ-карбонизированного образца почти в 3 раза выше, чем термически карбонизированного, при значительно меньшем времени нагрева и большем выходе готового материала. Полученные результаты продемонстрировали перспективность СВЧ-метода карбонизации.
ВВЕДЕНИЕ
Технологии получения и хранения энергии являются самыми перспективными в энергетической отрасли во всем мире. Одно из активно развивающихся направлений – системы накопления энергии на основе суперконденсаторов. Суперконденсаторы используют электростатическую адсорбцию ионов на двухслойных поверхностях электролита/электрода [1, 2]. Благодаря высокой удельной поверхности (более 3000 м2/г) и настраиваемой текстуре поверхности активированные углеродные материалы стали важным классом пористых материалов, применяемых в качестве электродов для суперконденсаторов [3–5]. Суперконденсаторы характеризуются практически неограниченной цикличностью (более 100 000 циклов), слабой температурной зависимостью и, что особенно важно, чрезвычайно высокой электрической мощностью и плотностью энергии [1]. В то же время для повышения конкурентоспособности суперконденсатора по сравнению с батареями нужно оптимизировать технологию получения материала электродов. Это необходимо для повышения плотности энергии, а также увеличения как гравиметрической, так и объемной характеристик суперконденсаторов. Поскольку в качестве сырья для активированных углеродных материалов можно использовать различные естественные материалы – ископаемый уголь, растительную биомассу (скорлупу орехов, древесину и ее компоненты, торф и т.д.), а также недорогие полимеры [6, 7], важно преобразовывать эти материалы для получения оптимальных функциональных физических и химических свойств.
Таким образом, актуальная задача – исследование влияния параметров формирования активированных углеродных материалов на их структуру и функциональные характеристики, а также параллельный поиск новых дешевых углеродсодержащих материалов и изучение их свойств.
Анализ проводимых в данной области исследований [8–10] показал, что наиболее перспективным сырьем для активированных углеродных материалов является хлопковый пух, получаемый из отходов текстильной промышленности. Важными критериями такого выбора послужили низкая стоимость и доступность хлопкового волокна.
Технологический процесс получения активированных углеродных материалов включает в себя несколько этапов. Основными из них являются карбонизация исходного сырья для получения углеродного материала и активация, направленная на развитие его пористой структуры [11]. Для карбонизации углеродсодержащих материалов обычно используют термический нагрев [12, 13]. В последнее время предложен новый метод – сверхвысокочастотный (СВЧ) нагрев [3]. Отметим, что оба метода имеют ряд преимуществ и недостатков [9]. Несмотря на то что карбонизация – это начальный этап получения активированных углеродных материалов, свойства получаемых карбонизированных образцов в значительной степени влияют на конечные свойства активированного углерода. В свою очередь процесс карбонизации зависит от ряда факторов: природы и агрегатного состояния углеродного сырья, методов и условий карбонизации. В настоящее время достаточно хорошо изучено влияние различных видов исходного сырья, получаемых из отходов сельскохозяйственного производства, на физико-химические свойства углеродных материалов. Однако практически отсутствуют данные об исследованиях структуры и свойств материалов при использовании разных методов на этапе карбонизации.
Целью настоящей работы было изучение структуры, морфологии поверхности, элементного и фазового состава, а также адсорбционной активности образцов карбонизированного хлопкового пуха, сравнительный анализ влияния термического и СВЧ-метода карбонизации на структуру и свойства получаемых образцов. Проведенные исследования помогут установить корреляцию между структурными и функциональными характеристиками карбонизированных образцов и методами карбонизации. Это, в свою очередь, позволит оценить перспективность предложенного СВЧ-метода карбонизации для оптимизации технологического процесса получения активированных углеродных материалов.
ОБРАЗЦЫ И МЕТОДЫ ИССЛЕДОВАНИЙ
Материал, методы и режимы карбонизации были выбраны исходя из анализа имеющейся на данный момент литературы и результатов собственных предварительных исследований.
Описание исследуемых образцов. В качестве исходного для процесса карбонизации материала использовали хлопковый пух, представляющий собой отходы текстильного производства с длиной волокон, не превышающей 15 мм. Для более интенсивной карбонизации сырья образцы пропитывали раствором ортофосфорной кислоты H3PO4 концентрацией 5% в течение 1 ч. В этом случае дегидратирующий агент (H3PO4), взаимодействуя с хлопковым пухом, катализирует реакции конденсации и дегидратации, уменьшая тем самым содержание кислорода и увеличивая выход карбонизированного материала [14].
Далее перед проведением термической карбонизации пропитанные образцы высушивали в сушильном шкафу в течение суток, а перед СВЧ-карбонизацией выдерживали во влажном состоянии 24 ч, затем отжимали до массы ~10 г. Карбонизацию образцов стандартным методом [12] осуществляли в термической печи при температуре 670°С в течение 120 мин. СВЧ-карбонизацию образца хлопкового пуха массой 3 г проводили в СВЧ-камере размером 0.6 × 0.6 × 0.6 м при мощности ~500 Вт в среде инертного газа Ar в течение 10 мин. После процесса карбонизации все образцы взвешивали на лабораторных весах II класса точности. Отметим, что в результате карбонизации образцы приобретали темно-графитовый цвет с сохранением исходной формы и потерей массы ~75% относительно исходной при СВЧ-карбонизации и ~80% при термической.
Оптическая микроскопия. Образцы хлопкового пуха исследованы в оптическом микроскопе Биолам М-1 (ЛОМО, Россия) в проходящем и отраженном свете при освещении в режиме светлого и темного поля. Разрешение прибора близко к 0.05 мкм. Увеличение объективов планахроматической коррекции составляло 10, 40 и 100 крат. Для фиксации изображений использовали камеру ТС-1000 (ЛОМО, Россия) с чувствительным элементом 10 Mп, 1/2" CMOS и максимальным разрешением 3664 × 2748.
Рентгеновская томография. Томографические исследования образцов хлопкового пуха проводили на рентгеновском микротомографе (ФНИЦ “КиФ” РАН, Россия) [15]. В конструкции микротомографа в качестве кристалла-монохроматора использован пиролитический графит (поле зрения около 1 × 2 см), что позволяет проводить измерения коэффициента линейного ослабления рентгеновского излучения µ. Измерения проводили при 8.04 кэВ (характеристическая линия CuKα1), так как при этой энергии исследуемые образцы, с одной стороны, прозрачны для сохранения высокого соотношения сигнал/шум на изображениях, а с другой – достаточно поглощают излучение, чтобы наблюдать контраст структуры. Геометрия проведенных экспериментов следующая: расстояние источник–образец 1200 мм, расстояние образец–детектор 10 мм. Условия зондирования: ускоряющее напряжение 40 кВ, ток 40 мА. При проведении томографических исследований образцы вращали относительно фиксированной оси (вертикальной), измеряли 400 проекций с шагом 0.5° и экспозицией 3.5 с на кадр. Для регистрации изображений использовали высокоразрешающий рентгеновский детектор XIMEA-xiRay11 (XIMEA, Словакия), позволяющий получать изображения с разрешением 9 мкм при поле зрения 36 × 24 мм. Обработку томографических данных проводили алгебраическим методом реконструкции.
Рентгенофлуоресцентный анализ. Элементный состав карбонизированных образцов хлопкового пуха определяли методом рентгенофлуоресцентного анализа (РФлА) на рентгеновском дифрактометре с использованием детектора-спектрометра X-123SDD (Amptek, США) [16]. Преимуществом метода РФлА является его неразрушающий характер. Относительная простота метода позволяет оперативно получать качественную информацию об элементном составе образцов.
В качестве источника излучения использована рентгеновская трубка с медным анодом, в качестве монохроматора – высокосовершенный кристалл Si(111). После отражения от кристалла-монохроматора пучок рентгеновских лучей был сколлимирован до размеров пятна на образце ~150 мкм по вертикали и ~5 мм по горизонтали. Энергия излучения CuKα1 составляла 8.04 кэВ. Рентгеновские лучи, попадая на образец, вызывают вторичное флуоресцентное излучение, соответствующее атомам образца, максимальная энергия которого ниже энергии возбуждающего излучения. Однако линия, соответствующая энергии зондирующего излучения (в данном случае CuKα1), также, как правило, проявляется. Нижний предел измерений ~1 кэВ ограничен параметрами чувствительного элемента детектора. Энергетическое разрешение спектров РФлА исследуемых образцов составило ~130 эВ.
Электронная микроскопия. Структура отдельных волокон и одиночных фибрилл карбонизированных образцов хлопкового пуха была исследована методом электронной микроскопии в растровом электронно-ионном микроскопе (РЭИМ) Scios (Thermo Fisher Scientific, США) и в просвечивающем электронном микроскопе (ПЭМ) Tecnai Osiris (Thermo Fisher Scientific, США) при ускоряющем напряжении 200 кВ.
Рентгенофазовый анализ. Для определения фазового состава, а также оценки соотношения кристаллической и аморфной фаз карбонизированные образцы хлопкового пуха исследовали на рентгеновском дифрактометре Rigaku MiniFlex 600 (Rigaku, Япония). Источником излучения служила рентгеновская трубка с медным анодом (CuKα, λ = 1.54 нм) при ускоряющем напряжении 40 кВ и токе 15 мА. Измерения проводили в геометрии Брэгга–Брентано в режиме θ–2θ-сканирования. Интервал углов 2θ составлял 5°–60°, шаг 0.01°, экспозиция 1 с на шаг. Для идентификации фаз применяли базу данных ICDD PDF-4 [17]. Дополнительно были проведены исследования на дифрактометре X’PERT PRO (PANalytical B.V., Нидерланды) при ускоряющем напряжении 40 кВ и токе 40 мА (излучение CuKα, λ = 1.54 нм). Образцы предварительно истерли в порошок. Для идентификации фаз применяли базу данных ICSD [18].
Исследование адсорбционной активности. Адсорбционную активность образцов хлопкового пуха после карбонизации определяли по индикатору – метиленовому синему (МС) [19]. Навеску 0.1 г предварительно высушенного исследуемого материала помещали в колбу с 25 мл раствора индикатора, закрывали и взбалтывали в течение 20 мин со скоростью 1200 об./мин. Далее суспензию центрифугировали в течение 15 мин, отбирали около 1 мл осветленного раствора, отстаивали в течение 15 мин и разбавляли водой до 10 мл. Разбавленный раствор заливали в кювету (толщина поглощающего слоя 1 мм) и снимали спектр пропускания в диапазоне от 400 до 800 нм. Оптическую плотность исследуемого раствора определяли по формуле
где I0 – интенсивность проходящего светового потока в дистиллированной воде, I – интенсивность потока в исследуемом разбавленном растворе; длина волны 665 нм.Адсорбционную активность исследуемых образцов по индикатору вычисляли по формуле
где C1 – массовая концентрация исходного раствора индикатора [мг/л]; C2 – массовая концентрация раствора после контактирования с образцом [мг/л]; K – коэффициент разбавления раствора (в данном случае K = 10); m – масса навески образца [г]; 0.025 – объем раствора индикатора, взятого для осветления [л].Остаточную массовую концентрацию индикатора в осветленном растворе определяли по полученному значению оптической плотности раствора с использованием градуировочного графика зависимости оптической плотности от массовой концентрации растворов сравнения:
где b и k – коэффициенты линейной функции y = = kx + b, полученные линеаризацией градуировочного графика.РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Структурные исследования. На изображениях исследуемых образцов, полученных в оптическом микроскопе (рис. 1), отчетливо проявляются отдельные волокна хлопкового пуха толщиной 5–20 мкм и длиной 10–15 мм. Анализ изображений позволил сделать следующие заключения.
Рис. 1.
Изображения образцов хлопкового пуха в оптическом микроскопе: а – исходное волокно; б – пропитанный 5%-ным раствором ортофосфорной кислоты H3PO4; в – карбонизированный СВЧ-методом; г – карбонизированный термическим методом.
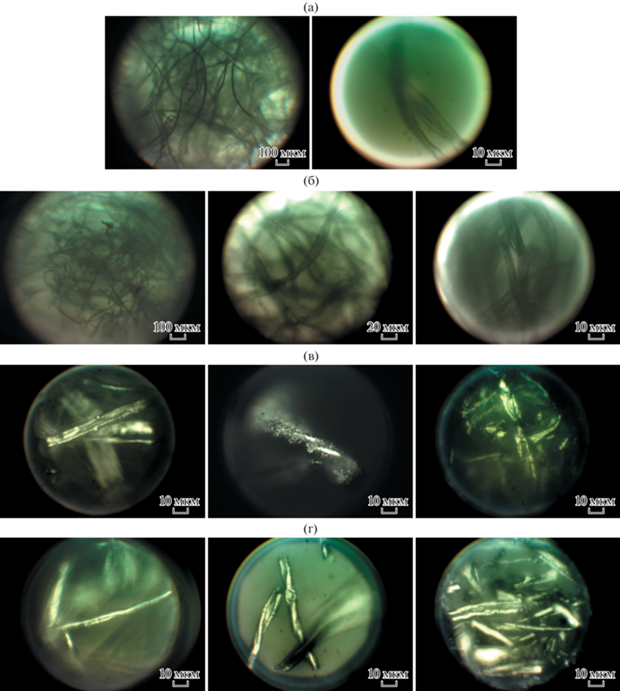
Исходный образец (рис. 1а) и образец, пропитанный раствором ортофосфорной кислоты (рис. 1б), состоят из однородных волокон с загрязнениями в виде микрочастиц на их поверхностях в незначительной концентрации. Отметим, что наличие различных микроэлементов и минеральных веществ характерно для сырого хлопка. Хорошо видна фибриллярная структура некоторых волокон (рис. 1б).
Состояние отдельного волокна в СВЧ-карбонизированном образце (рис. 1в) изменяется, на нем появляются мелкоразмерные наросты или налет относительно большой концентрации. Выявлены также и крупные наросты меньшей концентрации. Обнаружены нарушения структуры части волокон, вздутия, что, вероятно, обусловлено некоторым увеличением диаметра волокна на отдельных участках. В этих местах образуются поперечные изломы и разрушаются стенки волокон, что приводит к появлению пор размером порядка единиц микрон. По-видимому, при резком росте температуры образца в ходе СВЧ-нагрева также резко растет давление в каналах волокон, вызванное образованием летучих соединений, что и приводит к подобной деградации хлопкового волокна.
После термической карбонизации (рис. 1г) структура волокна более однородна. Однако наблюдаются волокна c ярко выраженными продольными и поперечными трещинами разного размера. Видимых наростов и налетов загрязнений на поверхности волокон не наблюдается.
Результаты рентгенотомографических исследований образцов хлопкового пуха представлены на рис. 2. Из анализа томограммы исходного образца (рис. 2а) можно заключить, что его структура относительно однородна. В целом образец достаточно рыхлый. Максимальное значение коэффициента поглощения (µ = 0.33 мм–1) наблюдается в наиболее загрязненной области образца. Из анализа представленных томографических реконструкций можно заключить, что волокно, пропитанное ортофосфорной кислотой (рис. 2б), более плотное и имеет больший коэффициент поглощения µ = 0.56 мм–1, чем исходное (рис. 2а), что, возможно, связано со слипанием материала.
Рис. 2.
Результаты трехмерной томографической реконструкции для образцов хлопкового пуха: а – исходное волокно; б – пропитанный 5%-ным раствором ортофосфорной кислоты H3PO4; в – карбонизированный СВЧ-методом; г – карбонизированный термическим методом.
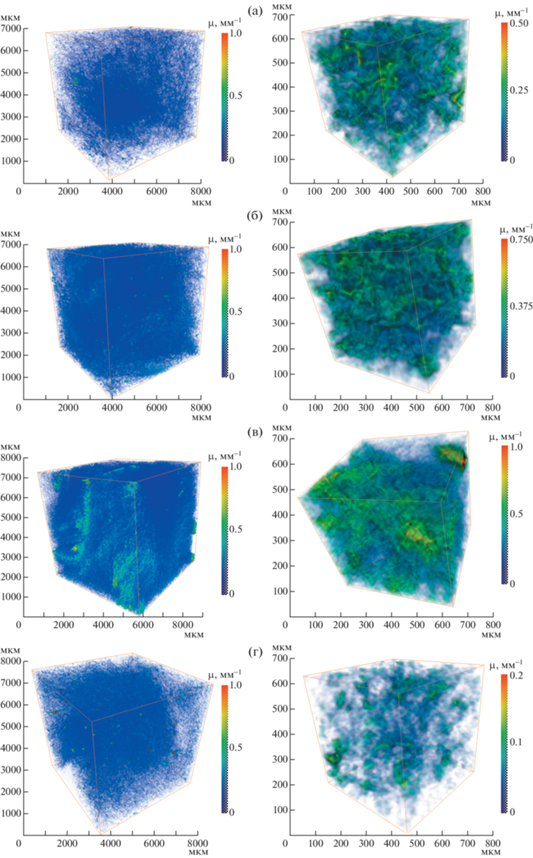
Структура образца, карбонизированного СВЧ-методом (рис. 2в), менее однородна, волокнистость и рыхлость выражены не так явно, как в исходных образцах. В целом преобладает объем области с бóльшим, чем у исходного образца, коэффициентом поглощения. Более плотные по поглощению области размером до нескольких миллиметров неравномерно распределены по объему исследуемого образца. Возможно, это спекшееся волокно и мелкие поглощающие включения примесной фазы, образовавшиеся в результате возрастания температуры образца в процессе относительно быстрого СВЧ-нагрева. Максимальное значение коэффициента поглощения µ = 0.98 мм–1.
Анализ томографического изображения образца, карбонизированного термическим методом (рис. 2г), показал, что его структура однородна и наиболее близка к исходному образцу (рис. 2а). Можно отметить увеличение по сравнению с исходным образцом концентрации мелких включений примесной фазы (точки) во всем объеме. Однако концентрация таких включений ниже, чем в СВЧ-карбонизированном образце (рис. 2в). Образец, карбонизированный термическим методом, характеризуется максимальным значением µ = 0.27 мм–1 – самым низким среди других образцов. Это можно объяснить усыханием самого волокна (потеря массы ~80% относительно исходной по данным взвешивания на лабораторных весах), а также удалением большей части загрязнения в процессе термической обработки.
РЭИМ-исследования СВЧ- и термически карбонизированных образцов показали, что структуры их отдельных волокон и фибрилл схожи (рис. 3). Образцы представляют собой закручивающиеся к центру волокна (рис. 3а, 3д), диаметр волокон 10–20 мкм (рис. 3б). На поверхности некоторых волокон хорошо просматривается фибриллярная структура (рис. 3е). Фибриллы расположены параллельно друг другу и винтообразно закручиваются под углом относительно оси волокна (рис. 3в, 3д). Диаметр фибрилл составляет 300–500 нм (рис. 3г).
Рис. 3.
РЭИМ-изображение волокон образцов, карбонизированных СВЧ- (а–г) и термическим (д, е) методами: а – общий вид; б – единичное волокно шириной 11.31 мкм; в – фибриллярная структура; г – изображение кромки волокна толщиной 349 нм; д – фибриллярная структура; е – изображение края волокна.

ПЭМ отдельных фибрилл показала, что они в основном однородны, имеют вытянутую форму и проявляют контраст, характерный для аморфных объектов. Диаметр фибрилл СВЧ-карбонизированного волокна составил 60–300 нм (рис. 4а), термически карбонизированного – 30–300 нм (рис. 4б). Контраст на ПЭМ-изображениях фибрилл, отчетливее выраженный в случае СВЧ-карбонизированного образца, указывает на возможное присутствие нанопор (рис. 4). Можно полагать, что уже на стадии карбонизации раскрытие закупоренных пор и развитие пористой структуры материала более активно происходит при СВЧ-нагреве. В фибриллах волокон, карбонизированных термическим методом, обнаружены также включения, в целом однородно распределенные по объему (рис. 4б). По-видимому, при термическом нагреве успевает проходить диффузия части примеси, находящейся на поверхности и в межволоконном пространстве, непосредственно в волокно, в то время как при быстром СВЧ-нагреве этого не происходит. В последнем случае часть загрязняющей волокно примеси коагулирует в конгломераты, неоднородно расположенные в межволоконном пространстве. Это предположение подтверждается результатами рентгеновской томографии (рис. 2в, 2г).
Рис. 4.
ПЭМ-изображение одиночных фибрилл в образце, карбонизированном: а – СВЧ-методом; б – термическим методом (стрелками показаны нанопоры).

Исследования элементного и фазового составов. Результаты исследования элементного состава образцов хлопкового пуха методом РФлА представлены на рис. 5 (спектры разнесены по оси интенсивности для наглядности). Отметим, что пики Ar (воздух), Fe (паразитный пик) и Cu (падающее излучение) являются инструментальными и не подлежат учету. Поскольку нижний предел измерений по энергии составляет ~1 кэВ, легкие элементы, такие как C, O, зарегистрировать не удается.
Рис. 5.
Рентгенофлуоресцентные спектры образцов: 1 – пропитанного 5%-ным раствором ортофосфорной кислоты H3PO4; 2–1, 2–2 – карбонизированного СВЧ-методом (1, 2 – разные области образца); 3 – карбонизированного термическим методом.
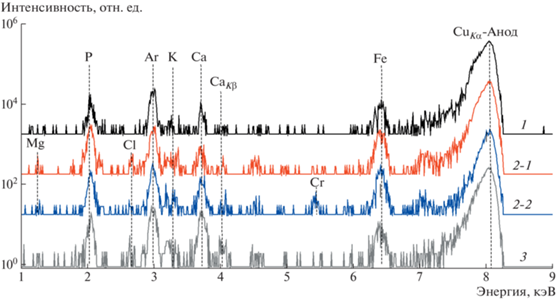
Расшифровка полученных спектров выявила наличие во всех исследуемых образцах следующих элементов: P, K, Ca. В образце, карбонизированном СВЧ-методом, также обнаружены Mg, Cl и Cr, а в образце, карбонизированном термическим методом, – Cl. Анализ полученных спектров позволяет отметить следующее. Распределение найденных элементов по объему исследуемых образцов неравномерное, о чем свидетельствуют разные интенсивности пиков спектров 2–1 и 2–2, полученных от разных областей одного и того же образца, а также пик Cr, выявленный только в спектре 2–2. Кроме того, максимальная концентрация фосфора обнаружена в образце, карбонизированном СВЧ-методом, а кальция – в термически карбонизированном образце.
На следующем этапе работы с целью оценки соотношения аморфной и кристаллической фаз в образцах хлопкового пуха, карбонизированных двумя методами, и их сравнительного анализа были проведены рентгенофазовые исследования. Полученные дифрактограммы образцов хлопкового пуха двух типов представлены на рис. 6. Проявляются широкий (размытый) пик аморфной фазы (2θ = 15°–30°) и слабоинтенсивные пики кристаллических фаз. По базе данных ICDD PDF-4 были определены наиболее вероятные кристаллические фазы, образовавшиеся в образцах в процессе СВЧ– и термической карбонизации. На дифрактограммах (рис. 6а) отчетливо видны пики 0002 и $10\bar {1}0$ графита с гексагональной кристаллической решеткой (PDF 01-077-7164) [20] и пики 110 и 020 углерода с тетрагональной кристаллической решеткой (PDF 00-054-0501) [21], которые были также обнаружены в [22].
Рис. 6.
Дифрактограммы образцов волокон (а) и порошков (б) хлопкового пуха, карбонизированных: 1 – СВЧ-методом; 2 – термическим методом.

На рис. 6б представлены дифрактограммы порошкообразных образцов после СВЧ- и термической карбонизации. Хорошо различимы пики, характеризующие матрицу аморфного вещества. Однако пики углерода не обнаружены. Был выявлен наиболее вероятный состав аморфных фаз: гидрат дигидроортофосфата кальция Ca(H2PO4)2H2O (PDF 01-075-1521) в случае СВЧ-карбонизации и дигидропирофосфат кальция CaH2P2O7 (PDF 00-003-0421) в случае термической карбонизации. Наличие этих фаз хорошо согласуется с результатами РФлА, показавшего присутствие кальция и фосфора в относительно высокой концентрации. Отличие лишь в гидрофильной части, возможно, указывает на различия в обработке исходного волокна СВЧ- и термическим методами.
Сравнивая дифрактограммы (рис. 6а, 6б), можно заключить, что в образце, карбонизированном термическим методом, аморфной фазы больше, чем в образце, карбонизированном СВЧ-методом. Результаты исследований образцов в виде волокон заметно отличаются от результатов порошкообразных образцов. Это можно объяснить тем, что графит, по всей видимости, присутствует внутри самого волокна либо в виде его покрытия. В случае порошкообразных образцов графит равномерно распределен по объему порошка, в результате его пики не заметны на фоне спектра аморфной матрицы. Собственно матрица идентифицируется лучше ввиду отсутствия локализованного графита.
Кроме того, в процессе карбонизации в обоих образцах произошло формирование графитоподобных микрокристаллитов, о чем свидетельствуют обнаруженные кристаллические фазы. Согласно [11], размер и ориентация графитовых кристаллитов определяют текстуру, пористость, удельную поверхность (при последующей активации), емкость и электропроводность электродного материала.
Адсорбционная активность (емкость). На рис. 7 представлены спектры пропускания светового потока в диапазоне длин волн от 400 до 800 нм для чистой дистиллированной воды (кривая 1) и растворов МС, осветленных образцами, карбонизированными СВЧ- (кривая 2) и термическим методами (кривая 3). Интенсивность светового потока, проходящего через дистиллированную воду на длине волны 665 нм, определяли для расчета оптической плотности D исследуемых растворов по формуле (1).
Рис. 7.
Спектры пропускания: 1 – дистиллированной воды; 2 – раствора МС, осветленного образцом, карбонизированным СВЧ-методом; 3 – раствора МС, осветленного образцом, карбонизированным термическим методом.
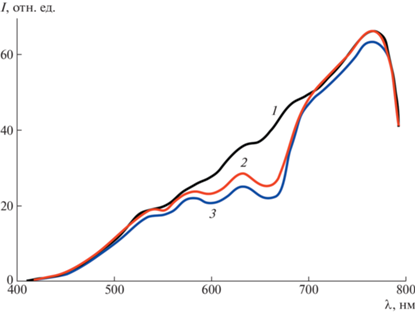
При сравнении спектров пропускания 2 и 3 на длине волны 665 нм, соответствующей максимальному поглощению проходящего светового потока раствором МС, видно, что раствор МС, осветленный СВЧ-карбонизированным образцом, характеризуется бóльшим пропусканием, чем раствор, осветленный образцом, карбонизированным термическим методом (спектр 3).
Значения адсорбционной активности Х СВЧ- и термически карбонизированных образцов, рассчитанные по формуле (2) по полученным значениям оптической плотности D осветленных растворов, составили 126 и 47 мг/г соответственно. Таким образом установлено, что адсорбционная активность СВЧ-карбонизированного образца почти в 3 раза превосходит адсорбционную активность термически карбонизированного образца.
ЗАКЛЮЧЕНИЕ
Исследованы образцы хлопкового пуха, пропитанного раствором ортофосфорной кислоты H3PO4, и проведен сравнительный анализ влияния СВЧ- и термического методов карбонизации на их структуру, морфологию поверхности, элементный и фазовый составы, а также адсорбционную активность.
Установлено, что в результате СВЧ-воздействия, т.е. быстрого нагрева, часть примеси коагулирует в конгломераты, неравномерно расположенные на поверхности волокон в виде наростов фосфорсодержащих (максимальная концентрация фосфора) продуктов пиролиза. В результате повышается среднее по всему волокну значение µ, но не происходит загрязнения самого волокна, в частности фибрилл. После термического нагрева образец имеет самое низкое из максимальных значение µ = 0.27 мм–1, что связано как с усыханием самого волокна (потеря массы ~80% относительно исходной по данным взвешивания на лабораторных весах), так и с отсутствием крупных примесных скоплений продуктов пиролиза в межволоконном пространстве. Однако, по всей видимости, происходит диффузия части примеси, находящейся на поверхности и в межволоконном пространстве, непосредственно в волокно, в результате чего формируются нановключения в фибриллах термически карбонизированного образца.
Результаты ПЭМ-исследований дают основание считать, что более активно раскрытие закупоренных пор и развитие пористой структуры материала уже на стадии карбонизации происходит при СВЧ-нагреве.
Идентифицирован наиболее вероятный состав аморфных фаз: гидрат дигидроортофосфата кальция Ca(H2PO4)2H2O в случае СВЧ-карбонизации и дигидропирофосфат кальция CaH2P2O7 в случае термической карбонизации. Оценки соотношения аморфной и кристаллической фаз показали, что в образце, карбонизированном термическим методом, аморфной фазы больше, чем в образце, карбонизированном СВЧ-методом. Обнаружено, что в процессе карбонизации в обоих образцах произошла локализация графитоподобных микрокристаллитов с гексагональной решеткой, а также образовалась тетрагональная фаза углерода.
Наблюдается меньшая адсорбционная активность термически карбонизированного образца. Это, видимо, обусловлено его менее развитой пористой структурой в сочетании с большей аморфностью и наличием включений в фибриллах. Хотя в этом случае степень загрязнения межволоконного пространства меньше, чем при СВЧ-обработке. Установлено, что адсорбционная активность такого образца почти в 3 раза меньше, чем СВЧ-карбонизированного образца. Время карбонизации образцов СВЧ-методом составило 10 мин с выходом материала ~25 мас. %, а термическим методом – 120 мин с незначительно меньшим выходом материала (~20 мас. %).
Проведенные сравнительные исследования двух методов карбонизации углеродных материалов дают основание считать СВЧ-метод весьма перспективным и требующим дальнейшего изучения.
Авторы выражают благодарность И.С. Волчкову за помощь в проведении рентгеноструктурных исследований и В.В. Артемову за помощь в проведении РЭИМ-исследований.
Работа выполнена при поддержке Министерства науки и высшего образования в рамках госзадания ФНИЦ “Кристаллография и фотоника” РАН в части оптических и рентгеновских исследований, в рамках задания Минобрнауки России FNER-2021-0001 в части получения карбонизированных углеродных образцов и исследования адсорбционной активности. Электронно-микроскопические исследования и рентгенофазовый анализ выполнены на оборудовании ЦКП ФНИЦ “Кристаллография и фотоника РАН”.
Список литературы
Chmiola J., Yushin G., Dash R., Gogotsi Y. // J. Power Sources. 2006. V. 158. P. 765.
Barbieri O., Hahn M., Herzog A., Kötz R. // Carbon. 2005. V. 43. P. 1303.
Kumar S., Grekov D., Pré P., Alappat B.J. // Renew. Sust. En. Rev. 2020. V. 124. P. 109743.
Wang L., Schnepp Z., Titirici M.M. // J. Mater. Chem. 2013. V. 1. P. 5269.
Elzbieta F., Francois B. // Carbon. 2001. V. 39. P. 937.
Simon P., Gogotsi Y. // Nature. Mater. 2008. V. 7. P. 845.
Lipka S.M., Swartz C.R. // Energeia. 2007. V. 18. P. 1.
Villota E.M., Lei H., Qian M. et al. // ACS Sust. Chem. Eng. 2018. V. 6. P. 1318.
Ao W., Fu J., Mao X. et al. // Ren. Sust. En. Rev. 2018. V. 92. P. 958.
Singh G., Lakhi K.S., Sil S. et al. // Carbon. 2019. V. 148. P. 164.
Шорникова О.Н., Максимова Н.В., Авдеев В.В. Связующие для полимерных композиционных материалов. М.: МГУ, 2010. 52 с.
Salitra G., Soffer A., Eliad L. et al. // J. Electrochem. Soc. 2000. V. 147. P. 2486.
Li Z., Guo D., Liu Y. et al. // Chem. Eng. J. 2020. V. 397. P. 125418.
Вервикишко Д.Е. Корреляция между нанопористой структурой углеродных материалов и функциональными характеристиками суперконденсаторов на их основе. Дис. … канд. физ.-мат. наук. М.: ОИВТ РАН, 2014.
Бузмаков А.В., Асадчиков В.Е., Золотов Д.А. и др. // Кристаллография. 2018. Т. 63. С. 1007. https://doi.org/10.1134/S0023476118060073
Гайнутдинов Р.В., Толстихина А.Л., Белугина Н.В. и др. // Кристаллография. 2018. Т. 63. С. 766. https://doi.org/10.1134/S0023476118050119
Gates-Rector S., Blanton T. // Powder Diffr. 2019. V. 34. № 4. P. 352.
Bergerhoff G., Brown I.D., Allen F. // International Union of Crystallography. Chester. 1987. V. 360. P. 77.
ГОСТ 4453-74. Уголь активный осветляющий древесный порошкообразный. Технические условия. Дата введения 1 января 1976 г.
Gamlen P.H., White J.W. // J. Chem. Soc. Faraday Trans. 1976. V. 72. P. 446.
Badding J.V., Scheidemantel T.J. // Solid State Commun. 2002. V. 122. № 9. P. 473.
Abdelkader A., Hussien B.M., Fawzy E.M., Ibrahim A.A. // Appl. Petrochem. Res. 2021. V. 11. № 2. P. 137.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография