

Кристаллография, 2023, T. 68, № 1, стр. 18-24
Кольцевой силикат Cs4Tm2[Si4O12](OH)2 и его аналог Cs4(Tm,Tb)2[Si4O12](OH)2 – двойники мероэдрии
Н. Б. Болотина 1, *, А. П. Топникова 2, Е. Л. Белоконева 2, О. В. Димитрова 2, А. С. Волков 2, Л. В. Зорина 3
1 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
2 Московский государственный университет им. М.В. Ломоносова
Москва, Россия
3 Институт физики твердого тела РАН
Черноголовка, Россия
* E-mail: bolotina@crys.ras.ru
Поступила в редакцию 25.08.2022
После доработки 25.08.2022
Принята к публикации 29.08.2022
- EDN: DNOCPX
- DOI: 10.31857/S0023476123010058
Аннотация
Методом гидротермального синтеза получены новые кольцевые силикаты Cs4Tm2[Si4O12](OH)2 и Cs4(Tm,Tb)2[Si4O12](OH)2. Выявлена структурная аналогия между ними и ранее изученными силикатами K4Sc2(OH)2(Si4O12) и K2Sc[Si2O6]F. Их кристаллические структуры определены как ромбические мероэдрические двойники по утраченному элементу тетрагональной симметрии. Структуры новых кольцевых силикатов родственны структурам фресноита и тетрагонального мелилита.
ВВЕДЕНИЕ
Силикаты редкоземельных элементов (REE) сочетают в себе термическую устойчивость и механическую прочность с нелинейно-оптическими, сегнетоэлектрическими, ионпроводящими, каталитическими, люминесцентными свойствами, востребованными в современных технологиях, что делает актуальным синтез новых фаз, исследование их структуры и свойств.
Структуры силикатов различаются по типу конфигураций из SiO4-тетраэдров, образующих изолированные группы, кольца, цепочки, слои и каркасы [1, 2]. В структурах известных Tm- и Tb-силикатов [1, 3] можно выделить конфигурации различного типа: ортогруппы в Tm2SiO5 [4], NaTb9(SiO4)6O2 (структурный тип апатита) [5]; диортогруппы в Tm2Si2O7 (аналог тортвейтита) [6, 7], K3TmSi2O7 [8], Tb2Si2O7 [9], K3TbSi2O7 [8] и Tb4S3Si2O7 [10]; триортогруппы в K3TbSi3O8(OH)2 [11]; орто- и триортогруппы в Tm4(Si3O10)(SiO4) [12]; тетраортогруппы в Ba2Tm2Si4O12F2 и Ba2Tb2Si4O13 [13]; шестичленные кольца в Na3Tb3Si6O18 ⋅ H2O (синтетический джеренит) [14]; цепочки – неразветвленная Rb2TbGaSi4O12 [15], волластонитовая Na2Tb1.08Ca2.92Si6O18H0.8 [16] и спиральная Na3TbSi3O9 ⋅ 3H2O [17]; слои в Cs3TmSi4O10F2 и Cs3TbSi4O10F2 [18]; пакеты в Cs3TbSi8O19 ⋅ 2H2O [19] и Na4K2Tb2Si16O38 ⋅ 10H2O [20]. Некоторые из кристаллов силикатов с Tm и Tb обладают люминесцентными свойствами.
В настоящей работе приведены условия гидротермального синтеза новых Cs,Tm- и Cs(Tm,Tb)-силикатов, выполнены структурная расшифровка двойников и кристаллохимический анализ, выявлены структурные связи между родственными силикатами.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез, состав и свойства кристаллов. Кристаллы нового силиката получены в гидротермальных условиях при температуре 280°С и давлении ~100 атм. Коэффициент заполнения автоклава выбран таким образом, чтобы давление оставалось постоянным. Массовое соотношение исходных компонентов составляло Tm2O3 : Tb2O3 : : SiO2 = 2 : 1 : 1 соответственно. Cs2CO3 был добавлен в раствор в качестве минерализатора. Отношение твердой и жидкой фаз 1 : 5. Синтез проводили в стандартном автоклаве объемом 5 см3, футерованном фторопластом. Длительность эксперимента составила 14 сут, что было необходимо для полного завершения реакции. Значение рН, измеренное после прохождения реакции, равно 9.
Под бинокулярным микроскопом обнаружены бесцветные прозрачные кристаллы тетрагонального и кубического облика, некоторые в сростках размером до 1 мм. Помимо них в опыте имелись похожие, но отличающиеся морфологией призматические бесцветные прозрачные кристаллы, наблюдавшиеся исключительно в сростках. Выход кристаллов был высоким, порядка 90%. Состав кристаллов определен с помощью рентгеноспектрального анализа в лаборатории локальных методов исследования вещества МГУ на микрозондовом комплексе на базе растрового электронного микроскопа Jeol JSM-6480LV. В состав кристаллов кубического облика входят атомы Cs, Tm, Si, O; кристаллы второй разновидности содержат Cs, Tm, Tb, Si, O. Кристаллы первой разновидности преимущественно средние и мелкие, с ребром куба менее 0.2 и 0.1 мм соответственно. Вторая разновидность представлена более крупными кристаллами от 0.15 до 1 мм.
Рентгеноструктурное исследование. Для рентгеноструктурного анализа Cs,Tm-силиката выбрали небольшой монокристалл размером 0.14 × × 0.12 × 0.11 мм, по форме близкий к кубику. Интенсивности дифракционных рефлексов измеряли в полной сфере обратного пространства на дифрактометре XCalibur S с CCD-детектором. Для анализа Cs(Tm,Yb)-силиката использовали небольшой изометричный обломок размером 0.112 × × 0.063 × 0.039 мм. Набор дифракционных данных получен в полной сфере обратного пространства на дифрактометре Oxford Diffraction Gemini R c CCD-детектором. Обработка данных в обоих случаях выполнена по программе CrysAlisPro [21]. Оба соединения кристаллизуются в тетрагональных решетках с почти одинаковыми периодами a ≃ 9.40, c ≃ 9.14 Å.
Структура Cs,Tm-силиката решена прямыми методами по программе SHELXS [22] комплекса WinGX [23] в тетрагональной группе Р42/mnm (№ 136). Установлены позиции атомов Tm, Cs, Si и атомов кислорода О1–O4 в вершинах полиэдров для Tm (октаэдры) и Si (тетраэдры). Расчет баланса валентных усилий по Полингу показал, что атом в позиции О4 принадлежит гидроксильной группе ОН, но позиция атома водорода не была локализована. Структурную модель уточняли методом наименьших квадратов по программе SHELXL [24] комплекса WinGX [23] в анизотропном приближении параметров смещений всех атомов. Фактор уточнения составил R = 2.48%, но был сильно завышен параметр атомных смещений u11 атома О4. Тщательный анализ дифракционной картины выявил сверхструктурные рефлексы (h + 0.5, k + 0.5, l), поэтому был осуществлен переход к базису a + b, –a + b, c. Параметры новой ячейки a = b = 13.2943(5), c = 9.1434(4) Å. Координаты атомов в новом базисе хорошо соответствуют ромбической группе Pbam (№ 55). Хотя у Pbam есть две тетрагональные надгруппы, P4/mbm (№ 127) и P42/mbc (№ 135), ни одна из них не подходит для данного кристалла. Структура (Cs,Tm)-силиката уточнена по программе Jana2006 [25] в группе симметрии Pbam как структура мероэдрического двойника, компоненты которого связаны поворотом на 90° вокруг оси с. Уточнение завершилось с R-фактором 3.45% и с отношением объемов компонент 0.505(3) : 0.495(3).
Структуру Cs(Tm,Tb)-силиката изучали по той же схеме в предположении равных количеств Tm и Tb в составе. Уточнение завершилось с R-фактором 2.90%; уточненные объемы компонент двойника составили 0.415(2) и 0.585(2) от объема кристалла.
Кристаллографические данные, характеристики эксперимента и результаты уточнения структур приведены в табл. 1. Табл. 2 содержит координаты атомов и параметры атомных смещений для Cs,Tm-силиката. Основные межатомные расстояния для этого соединения приведены в табл. 3. Координаты атомов и межатомные расстояния в структуре Cs(Tm,Tb)-силиката незначительно отличаются от величин в табл. 2, 3. Полная информация о двух структурах содержится в базе данных CCDC (CSD) [1], номера депозитов 2203488 и 2203489. Рисунки выполнены с использованием программ ATOMS [26] и Diamond.
Таблица 1.
Кристаллографические характеристики, данные эксперимента и результаты уточнения структур Cs4Tm2[Si4O12](OH)2 и Cs4(Tm,Tb)2[Si4O12](OH)2
| Химическая формула | Cs4Tm2[Si4O12](OH)2 | Cs4(Tm,Tb)2[Si4O12](OH)2 |
|---|---|---|
| М | 1205.8 | 1195.8 |
| Сингония, пр. гр., Z | Ромбическая, Pbam, 4 | |
| a, b, c, Å | 13.2918(5), 13.2968(5), 9.1434(4) | 13.2869(3), 13.2934(2), 9.1166(2) |
| V, Å3 | 1616.0(1) | 1610.25(6) |
| Dx, г/см3 | 4.956 | 4.933 |
| Излучение; λ, Å | МоKα; 0.71073 | |
| μ, мм–1 | 20.152 | 19.106 |
| T, К | 293(2) | 150(2) |
| Размер образца, мм | 0.14 × 0.12 × 0.11 | 0.112 × 0.063 × 0.039 |
| Дифрактометр | XCalibur S | Oxford Diffraction Gemini R |
| Тип сканирования | ω | |
| θmax, град | 30.76 | 37.8 |
| Пределы hkl | –19 ≤ h ≤ 18, –18 ≤ k ≤ 18, –13 ≤ l ≤ 12 |
–22 ≤ h ≤ 22, –22 ≤ k ≤ 21, –15 ≤ l ≤ 15 |
| Количество рефлексов: измеренных/независимых (all), Rint/с I ≥ 3 σ(I) (obs) | 26 172/2570, 0.0599/2072 | 14 449/4267, 0.0249/3780 |
| Метод уточнения | МНК по F(hkl) | |
| Весовая схема | 1/[σ2(F) + 0.0009F 2] | |
| Число параметров | 77 | 76 |
| Rall/wRall | 0.0478/0.0637 | 0.0343/0.0597 |
| Robs/wRobs | 0.0345/0.0590 | 0.0295/0.0560 |
| S | 1.25 | 1.19 |
| Δρmin/Δρmax, э/Å3 | –1.63/1.49 | –2.74/3.24 |
| Программы | Jana2006 [25] | |
Таблица 2.
Координаты атомов и эквивалентные параметры атомных смещений в структуре Cs4Tm2[Si4O12](OH)2
| Атом | Позиция Уайкова | x/a | y/b | z/c | Ueq, Å2 |
|---|---|---|---|---|---|
| Tm1 | 8i | 0.24908(3) | 0.25009(4) | 0.25081(8) | 0.0152(4) |
| Cs1 | 4g | 0.9885(2) | 0.21830(10) | 0 | 0.0241(5) |
| Cs2 | 4h | 0.0099(3) | 0.14888(11) | 0.5 | 0.0275(5) |
| Cs3 | 4g | 0.35219(10) | 0.9893(2) | 0 | 0.0283(5) |
| Cs4 | 4h | 0.28561(9) | 0.9907(3) | 0.5 | 0.0208(4) |
| Si1 | 8i | 0.1208(2) | 0.00237(16) | 0.1799(3) | 0.0048(6) |
| Si2 | 8i | 0.9978(2) | 0.3826(3) | 0.3200(4) | 0.0178(9) |
| O1 | 4e | 0 | 0 | 0.2433(11) | 0.005(2) |
| O2 | 4f | 0 | 0.5 | 0.2934(15) | 0.019(3) |
| O3 | 4g | 0.1272(8) | 0.0136(8) | 0 | 0.017(2) |
| O4 | 4h | 0.9899(9) | 0.3891(10) | 0.5 | 0.027(3) |
| O5 | 8i | 0.1733(5) | 0.0968(5) | 0.2473(7) | 0.0101(14) |
| O6 | 8i | 0.1075(8) | 0.3326(7) | 0.2663(11) | 0.032(2) |
| O7 | 8i | 0.3251(5) | 0.4021(5) | 0.2251(8) | 0.0104(14) |
| O8 | 8i | 0.3918(8) | 0.1666(7) | 0.2530(10) | 0.030(2) |
| Oh1 | 4g | 0.2339(8) | 0.2285(9) | 0 | 0.024(2) |
| Oh2 | 4h | 0.2650(9) | 0.2509(8) | 0.5 | 0.022(3) |
Таблица 3.
Основные межатомные расстояния в Cs4Tm2[Si4O12](OH)2
| Атомы | Расстояния, Å |
|---|---|
| TmO4(OH)2-октаэдр | |
| Tm1–O5 | 2.274(7) |
| Tm1–O6 | 2.183(10) |
| Tm1–O7 | 2.272(7) |
| Tm1–O8 | 2.199(10) |
| Tm1–Oh1 | 2.320(2) |
| Tm1–Oh2 | 2.288(1) |
| 〈Tm–O〉 | 2.256 |
| SiO4-тетраэдры | |
| Si1–O1 | 1.708(5) |
| Si1–O3 | 1.654(3) |
| Si1–O5 | 1.563(7) |
| Si1–O7 (iii) | 1.570(7) |
| 〈Si1–O〉 | 1.624 |
| Si2–O2 (i) | 1.580(5) |
| Si2–O4 | 1.651(4) |
| Si2–O6 (i) | 1.675(11) |
| Si2–O8 (ii) | 1.670(10) |
| 〈Si2–O〉 | 1.644 |
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
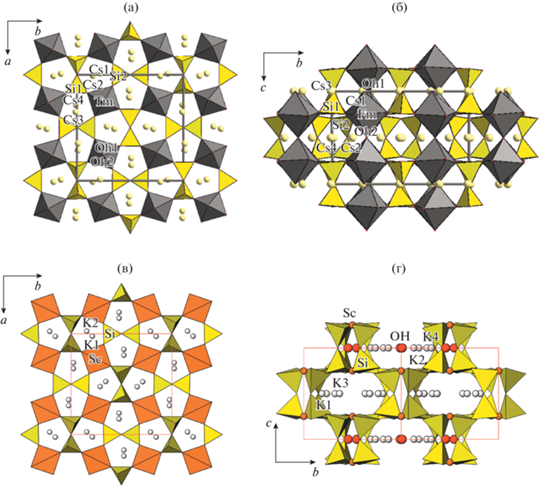
Tm- и (Tm,Tb)-силикаты относятся к кольцевым силикатам. Анионные радикалы в кристаллической структуре – изолированные четырехчленные кольца из SiO4-тетраэдров – перпендикулярны плоскости ab и ориентированы по плоскостям ac и bc. В проекции на плоскость ab (рис. 1а) кольца представлены парами тетраэдров, ориентированных перпендикулярно друг другу. Октаэдры TmO4(OH)2 и (Tm,Tb)O4(OH)2 соединяются по вершинам в колонки, вытянутые вдоль оси с (рис. 1б). Боковыми вершинами октаэдров колонки соединяются с кольцами [Si4O12] и образуют смешанный микропористый каркас, в пустотах которого находятся атомы цезия. Аналогичным образом устроена двойниковая структура (K,Sc)-силиката K4Sc2(OH)2(Si4O12), полученного в близких гидротермальных условиях. Структура была изучена в [27] в группе симметрии Pbam. Параметры ромбической элементарной ячейки составили: a = 12.725(4), b = 12.741(6), c = 8.441(3) Å. Структура представляет собой смешанный каркас с анионным радикалом из четырехчленных колец [Si4O12], скрепленных колонками Sc-октаэдров (проекции структуры (рис. 1в, 1г) построены по координатам из [27]). Отклонения симметрии этой и вновь изученных структур силикатов от тетрагональной проявляются в смещениях атомов из плоскостей симметрии, характерных для группы Р42/mnm в “малой” ячейке, разворотах SiO4-тетраэдров в кольцах и RЕE-октаэдров в колонках.
Рис. 1.
Кристаллические структуры Cs4Tm2[Si4O12](OH)2 и K4Sc2(OH)2(Si4O12) [27] в проекциях на плоскости ab (а, в) и bc (б, г). Показаны SiO4-тетраэдры, TmO4(OH)2- и ScO4(OH)2-октаэдры; атомы Tm, Sc, Cs, K и ОН-группы изображены шарами.
Скандий – элемент-аналог REE – характеризуется наименьшим среди них ионным радиусом и наиболее простым электронным строением. Вершины полиэдров и атомы Cs в Cs,Tm-силикате отстоят друг от друга дальше, чем вершины аналогичных полиэдров и атомы K в K,Sc-силикате. Последний был охарактеризован в [27] как полисинтетический двойник прорастания, в котором близкие по объему компоненты двойникуются плоскостью (1$\bar {1}$0). Для двух новых силикатов закон двойникования определен как поворот на 90° вокруг оси [001]. Оба закона в программе Jana2006 задаются матрицами для преобразования индексов дифракционных рефлексов и приводят к одному результату при уточнении структурной модели. Физически кристаллы двойникуются скорее всего по плоскости, как предполагалось в [27] для K,Sc-силиката.
Силикат K2Sc[Si2O6]F [28] обладает несоразмерно модулированной структурой. Симметрия кристалла представлена (3 + 2)D-группой симметрии P42/mnm(α,α,0)000s(–α,α,0)0000 в рамках “малой” ячейки с параметрами a = 8.9878(1), c = = 8.2694(2) Å. Группа симметрии P42/mnm базисной структуры та же, что была выбрана на первом этапе для Cs,Tm-силиката, а расхождение в параметрах ячейки обусловлено различием ионных радиусов Cs и K, а также Tm и Sc. Волновые векторы модуляции q1 = 0.2982(4)(a* + b*) и q2 = = 0.2982(4)(–a* + b*) равны по длине и ориентированы во взаимно перпендикулярных направлениях, будучи связанными четверной осью симметрии. Базисная структура, установленная в “малой” тетрагональной ячейке (рис. 2), является тетрагональным аналогом ромбических сверхструктур, рассмотренных выше. Параметры (3 + 2)D-структуры, уточненной в [28] с R-фактором 5.14%, были использованы для характеризации межатомных расстояний и координационного окружения катионов на разных участках периода модуляционной волны в реальном кристалле. По аналогии со структурами упомянутых выше силикатов не исключаем для структуры K2Sc[Si2O6]F возможность понижения симметрии до ромбической и двойникования одним из утраченных элементов тетрагональной симметрии. Не подвергая сомнению результаты [28], можно было бы попытаться упростить модель. В качестве группы симметрии для базисной структуры можно выбрать ромбическую подгруппу Pnnm (№ 58) группы P42/mnm, симметрию модулированной структуры описать группой (3 + 1)D симметрии Pnnm(α,β,0)000 с волновым вектором q = 0.2982(4)(a* + b*) и дополнить модель двойниковой матрицей, определяющей поворот на 90° вокруг оси [001].
Рис. 2.
Базисная структура несоразмерно модулированного кристалла K2Sc(Si2O6)F в проекциях на плоскости ab (а) и bc (б). Показаны SiO4-тетраэдры, ScO4F2-октаэдры; атомы Sc, K и F изображены шарами.

Рассмотрение сходных структур силикатов показывает, что проекция на плоскость ab структуры фресноита Ba2TiSi2O8 [29] (a = 8.516, c = 5.218 Å, P4bm) выглядит практически аналогично соответствующей проекции новой структуры (рис. 1а, 3а). Принципиальное различие состоит в том, что атомы Ti находятся в полуоктаэдрах, а вместо колец имеются диортогруппы, что обусловлено отсутствием зеркальной плоскости: смешанные слои остаются полярными и не соединяются в каркас (рис. 3б).
Рис. 3.
Проекция кристаллической структуры фресноита на плоскость: ab (а), bc (б). Показаны SiO4-тетраэдры, TiO5-полуоктаэдры; атомы Ba, Ti изображены шарами.

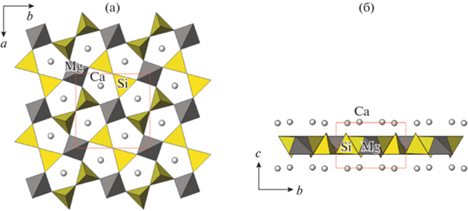
Структуры K4Sc2(OH)2(Si4O12) и тетрагонального мелилита Ca2Mg[Si2O7] [30] (a = 7.860, c = = 5.024 Å, P$\bar {4}$21m) сопоставлены в [27]. Показано, что основное различие состоит в замене крупного мелилитового MgO4-тетраэдра на еще более крупный полиэдр – октаэдр (рис. 4). Для мелилитов характерны несоразмерные модуляции.
ВЫВОДЫ
Получены и структурно изучены новые силикаты Cs4Tm2[Si4O12](OH)2 и Cs4(Tm,Tb)2[Si4O12](OH)2. Выявлена структурная аналогия между ними и ранее изученными силикатами K4Sc2(OH)2 (Si4O12) и K2Sc[Si2O6]F. Показано, что понижение симметрии новых силикатов от тетрагональной до ромбической сопровождается двойникованием по утраченному элементу тетрагональной симметрии. Структуры новых кольцевых силикатов родственны структурам фресноита и тетрагонального мелилита.
Авторы выражают благодарность Н.В. Зубковой за помощь в получении экспериментальных данных, а также сотрудникам лаборатории локальных методов исследования вещества МГУ за определение состава кристаллов.
Проведенный Н.Б. Болотиной анализ двойниковой структуры силикатов выполнен при поддержке Министерства науки и высшего образования РФ в рамках выполнения работ по Государственному заданию ФНИЦ “Кристаллография и фотоника” РАН. Получение экспериментальных данных при низких температурах Л.В. Зориной частично выполнено в рамках госзадания ИФТТ РАН.
Список литературы
The Cambridge Crystallographic Data Centre (CCDC). Inorganic Crystal Structure Data Base – ICSD. https://www.ccdc.cam.ac.uk/, http://www.fiz-karlsruhe.de
Пущаровский Д.Ю. // Структурная минералогия силикатов и их синтетических аналогов. М.: Недра, 1986. 160 с.
Crystallography Open Database. crystallography.net/cod
Mueller-Bunz H., Schleid T. // Z. Anorg. All. Chem.1999. B. 625. S. 613.
Garra W., Marchetti F., Merlino S. // J. Solid State Chem. 2009. V. 182. P. 1529.
Kahlenberg V., Aichholzer P. // Acta Cryst. E. 2014. V. 70. P. i34.
Fleet M.E., Liu X. // Am. Mineral. 2004. V. 89. P. 396.
Vidican I., Smith M.D., zur Loye H.C. // J. Solid State Chem. 2003. V. 170. P. 203.
Fleet M.E., Liu X. // Z. Krist. 2003. B. 218. S. 795.
Sieke C., Hartenbach I., Schleid T. // Z. Natur. B. 2002. B. 57. S. 1427.
Ananias D., Kostova M., Paz F.A.A. et al. // J. Am. Chem. Soc. 2004. V. 126. P. 10410.
Hartenbach I., Lissner F., Schleid T. // Z. Natur. B. 2003. B. 58. S. 925.
Fulle K., Sanjeewa L.D., McMillen C.D., Kolis J.W. // Acta Cryst. B. 2017. V. 73. P. 907.
Topnikova A.P., Belokoneva E.L., Dimitrova et al. // Crystallography Reports. 2016. V. 61. № 4. P. 566.
Lee Ch.-Sh., Liao Yu.-C., Hsu J.-T. et al. // Inorg. Chem. 2008. V. 47. P. 1910.
Bao X., Liu X., Liu X. // RSC Adv. 2017. V. 7. P. 50195.
Wang G., Li J., Yu J. et al. // Chem. Mat. 2006. V. 18. P. 5637.
Morrison G., Latshaw A.M., Spagnuolo N.R., Zur Loye H.-C. // J. Am. Chem. Soc. 2017. V. 139. № 41. P. 14743.
Zhao X., Li J., Chen P. et al. // Inorg. Chem. 2010. V. 49. P. 9833.
Ananias D., Ferreira A., Rocha J. et al. // J. Am. Chem. Soc. 2001. V. 123. P. 5735.
Agilent Technologies, CrysAlisPro Software System, version 1.171.3735. Agilent Technoligies UK Ltd. Oxford, UK, 2014.
Sheldrick G.M. // Acta Cryst. A. 2008. V. 64. P. 112.
Farrugia L.J. // J. Appl. Cryst. 2012. V. 45. P. 849.
Sheldrick G.M. // Acta Cryst. C. 2015. V. 71. P. 3.
Petříček V., Dušek M., Palatinus L. // Z. Kristallogr. 2014. B. 229. S. 345.
Dowty E. // ATOMS. Shape Software, Kingsport, Tennessee, USA, 2006.
Пятенко Ю.А., Воронков А.А., Жданова Т.А. // Докл. АН СССР. 1979. Т. 248. С. 868.
Hejny C., Kahlenberg V., Eberhard T., Krüger H. // Acta Cryst. B. 2016. V. 72. P. 209.
Masse R., Grenier J.C., Durif A. // Bull. Soc. Franc. Mineral. Crist. 1967. V. 90. P. 20.
Bindi L., Bonazzi P., Dusek M. et al. // Acta Cryst. B. 2001. V. 57. P. 739.
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография