

Кристаллография, 2023, T. 68, № 2, стр. 268-275
Исследование структурных основ субстратной специфичности пуриннуклеозидфосфорилазы из Thermus Thermophilus методами структурной биоинформатики
И. Ф. Гарипов 1, *, В. И. Тимофеев 1, 2, Е. А. Заяц 3, Ю. А. Абрамчик 3, М. А. Костромина 3, И. Д. Константинова 3, Р. С. Есипов 3
1 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
2 Национальный исследовательский центр “Курчатовский институт”
Москва, Россия
3 Институт биоорганической химии им. академиков М.М. Шемякина и Ю.А. Овчинникова РАН
Москва, Россия
* E-mail: ildar.garipov.f@gmail.com
Поступила в редакцию 22.07.2022
После доработки 05.09.2022
Принята к публикации 05.09.2022
- EDN: GAXNVC
- DOI: 10.31857/S0023476123010101
Аннотация
Проведено моделирование молекулярной динамики дикой формы белка пуриннуклеозидфосфорилазы с двумя субстратами – аденозином и гуанозином. Проведено аналогичное моделирование белка мутантной формы с теми же субстратами. Из полученных траекторий методом MM-GBSA оценено изменение свободной энергии при формировании полученных комплексов.
ВВЕДЕНИЕ
Пуриннуклеозидфосфорилазы (ПНФ) катализируют обратимый фосфоролиз пуриновых рибо- и 2'-дезоксирибонуклеозидов с образованием свободного азотистого основания и (дезокси-)рибоза-1'-фосфата (рис. 1).
Рис. 1.
Схема реакции, катализируемой пуриннукдеозидфосфорилазой из Thermus thermophilus HB27. Аденозин/аденин – R1 = NH2, R2 = H. Гуанозин/гуанин – R1 = O, R2 = NH2.

Благодаря своей каталитической активности ПНФ способны в присутствии ионов фосфата осуществлять реакцию трансгликозилирования: перенос азотистого основания с одного нуклеозида на другой [1–3]. Пуриннуклеозидфосфорилазы играют значительную роль в метаболизме нуклеиновых кислот и реутилизации пуриновых азотистых оснований, а также нашли широкое применение в каскадном синтезе модифицированных нуклеозидов [17–21].
ПНФ из различных организмов различаются по ряду параметров: масса, четвертичная структура и субстратная специфичность, на основе чего можно выделить два функциональных класса. Ферменты первого класса ПНФI, встречающиеся у всех живых организмов, представляют собой гомотримеры массой ~90 кДа и узко специфичны в отношении 6-оксопуриновых нуклеозидов. Ферменты второго класса ПНФII, характерные только для микроорганизмов, являются гомогексамерами с молекулярной массой 110–150 кДа и обладают более широкой специфичностью, взаимодействуя как с 6-оксопуринами, так и с 6-аминопуринами [3].
Известно, что геном термофильной эубактерии штамма Thermus thermophilus HB27 содержит гены ПНФ как первого, так и второго класса. Объектом данного исследования является пуриннуклеозидфосфорилаза II из Thermus thermophilus HB27 (ThPNPII). Как представитель второго класса ПНФ, TthPNPII является гексамером, способным осуществлять фосфоролиз гуанозина и аденозина [1]. Данный фермент обладает широкой субстратной специфичностью в отношении модифицированных не природных пуриновых азотистых оснований [1]. Субстратами для TthPNPII являются не только рибозиды, но и дезоксирибозиды и различные производные 2-дезоксирибозы. Стоит также отметить высокую степень гомологии TthPNPII в сравнении с ферментом человека – 40% [1, 3].
Для TthPNPII аденозин является значительно лучшим субстратом по Km и Kcat в сравнении с гуанозином, что обусловлено различием заместителя в шестом положении пурина [1]. Сравнительный анализ ПНФ из термофильных бактерий семейства Deinococcus–Thermus, проведенный в [2], показал, что определяющую роль в субстратной специфичности TthPNPII (в [2] вывод сделан для тримерного ПНФ, гомологичного исследуемому TthPNPII) играет аминокислотный остаток Asp235. Аспартат в данном положении обусловливает высокое сродство фермента к аденозину, замена его на аспарагин (мутация D235N) увеличивает активность фермента относительно гуанозина [2].
Для исследования структурных основ субстратной специфичности TthPNPI рассмотрим окружение субстрата в активном центре фермента в радиусе до 4 Å от атомов субстрата (рис. 2б): Tyr85, Ala113, Phe187, Phe192, Leu198, Ile209, Gly210, Met211, Thr234, Asp235, Ala237, His244, Val250, а также принадлежащей соседнему мономеру Phe153. Можно видеть, что O6 и N7 атомы гуанозина находятся на расстоянии, достаточном для формирования водородных связей с атомами Asp235. Аденозин отличается от гуанозина наличием в шестом положении аминогруппы вместо оксогруппы, данная аминогруппа также может образовывать водородные связи с атомами Asp235.
Рис. 2.
Пространственная структура гексамера TthPNPII, в активных центрах расположен фосфат, изображенный сферами (а). Активный центр TthPNPII, светлыми линиями показан Phe153 из полипептидной цепи соседней субъединицы, сферами – фосфат, толстыми стержнями – гуанозин, пунктирными линиями – возможные водородные связи (б).

Мутация D235N несколько меняет картину взаимодействий, так как дикарбоновая аминокислота заменяется полярно нейтральной, однако амидогруппа аспарагина (СОNH2) также может образовать водородную связь либо с кислородом в положении 6 гуанозина, либо с атомом N7 аденозина [1]. Благодаря полученной в [1] кристаллической структуре белка TthPNPII (рис. 2а) представляется возможным провести соответствующие модификации in silico и сравнить энергии связывания фермента с аденозином и гуанозином до и после мутации с последующей декомпозицией энергии по аминокислотным остаткам, чему и посвящено данное исследование.
МАТЕРИАЛЫ И МЕТОДЫ
Создание структур комплексов. Для получения необходимого комплекса в программе PyMOL [17] структуру исследуемого белка TthPNPII (PDB ID: 6TK9) совместили со структурой гомологичной ПНФ (PDB ID: 3IEX), находящейся в комплексе с гуанозином. Далее c использованием программы PyMOL гуанозин и остаток Asp 235 в белке заменили соответственно на аденозин и Asn. Тем самым сгенерировали четыре необходимые для молекулярной динамики (МД) комплекса дикого и мутантных типов ферментов отдельно с каждым из субстратов.
Моделирование молекулярной динамики. Для предварительной подготовки файлов, моделирования МД и анализа энергий системы использовали пакет программ для МД Amber [4, 5]. Использовали наборы силовых полей ff14SB [6] и GAFF (General AMBER Force Field) [7] для молекул ферментов и лигандов соответственно. Для параметризации лигандов использовали программу antechamber [4]. Для воды использовали модель TIP3P [8]. Молекулу комплекса поместили в кубическую ячейку. Минимальное расстояние от молекулы белка до граней ячейки составляло 1.5 нм. Ячейка была заполнена молекулами воды. В каждую из систем добавили несколько ионов натрия для нейтрализации заряда системы. На первом этапе для каждой из систем провели минимизацию энергии. Далее системы были уравновешены при температуре 300 К и давлении 1 бар путем моделирования динамики в NVT- и NPT-ансамблях (50 пс в каждом) соответственно. Температуру и давление в системах контролировали с использованием модифицированного термостата Ланжевена [9] и баростата Берендсена [10] с временными константами τT = 1 пс и τP = 2 пс соответственно. Продуктивное 10 нс моделирование МД для каждой из систем проводили в NPT-ансамбле с шагом интегрирования 2 фс. Дальнодействующие электростатические взаимодействия рассчитывали с использованием схемы суммирования по Эвальду в варианте PME [7, 11]. Кулоновские потенциалы и потенциалы Леннарда–Джонса были усечены до 1.4 нм, что является наиболее оптимальным для используемого силового поля [5].
Оценка энергии взаимодействия лигандов с белком. Для расчета энергии взаимодействия лигандов с рецептором использовали метод MM-GBSA (Molecular mechanics – Generalized Born surface area) [12, 13]. Расчет проводили с помощью программы MMPBSA.py [14]. Использовали 1000 фреймов для каждой из систем. Для вычисления энергии взаимодействия использовали модифицированную обобщенную модель Борна, предназначенную для макромолекул [15].
РЕЗУЛЬТАТЫ
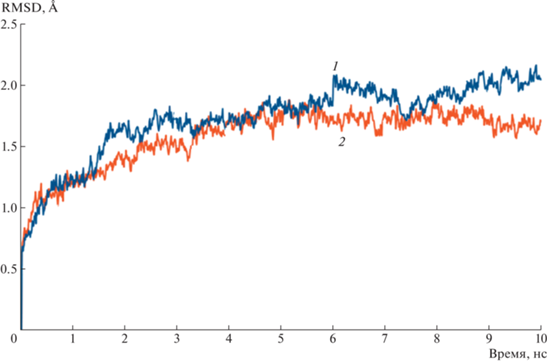
На рис. 3 представлены зависимости RMSD (Root Mean Square Deviation, среднеквадратичное отклонение) от времени для Cα-атомов дикого и мутантных форм ферментов в комплексе с гуанозином. RMSD комплекса с нативным ферментом плавно стабилизируется около значения 1.75 Å, тогда как у комплекса мутантного типа плато RMSD устанавливается при значении 2 Å нм и только после 4 нс моделирования. Кривая RMSD комплекса мутантного типа менее плавная и больше в среднем по значению. Из этого можно заключить, что комплекс гуанозина с белком мутантного типа менее стабилен, чем с нативной формой.
Рис. 3.
Графики зависимости среднеквадратичного отклонения (RMSD) атомов от времени для комплексов гуанозина с ферементом дикого (1) и мутантного типа (2).

Для аденозина кривая RMSD (рис. 4) фермента мутантного типа в отличие от вычислительного эксперимента с гуанозином показывает большую стабильность комплекса с лигандом, достигая диапазона значений 1.6–1.75 Å. Комплекс нативного фермента с аденозином имеет менее сглаженную кривую и на интервалах времени 1.5–3, 6–7.2 и 8–10 нс значения RMSD заметно больше. Из графика следует, что отклонения атомов полипептидной цепи нативного белка растут на протяжении моделирования, достигая значения 2 Å.
Рис. 4.
Графики зависимости среднеквадратичного отклонения (RMSD) атомов от времени для комплексов аденозина с ферементом дикого (1) и мутантного типа (2).
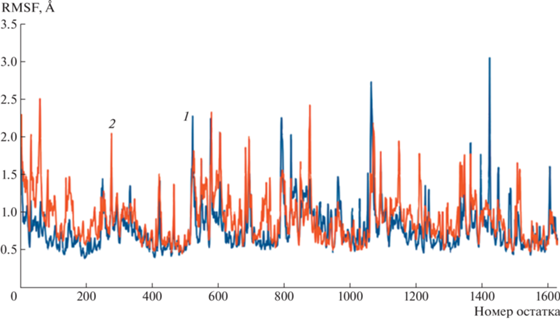
Графики RMSF (Root Mean Square Fluctuation, среднеквадратичная флуктуация) комплексов с гуанозином (рис. 5) показывают заметные изменения в подвижности участков полипетидной цепи. Для комплекса мутантного типа флуктуации большей части остова белка увеличиваются, что касается и активного центра, хотя и максимальное значение флуктуаций ~3 Å достигается для нативного фермента. Кривые RMSF показывают, что комплекс мутанта с гуанозином в целом стал более подвижным и пики флуктуаций начали приходиться на другие аминокислоты. Кривая RMSF для комплекса мутантного типа также может свидетельствовать о дестабилизации и влиянии мутации на конформационные превращения, претерпеваемые белком в растворе.
Рис. 5.
График флуктуации координат атомов, усредненные по всему времени моделирования (RMSF) комплексов гуанозина с ферментом дикого (1) и мутантного типа (2).
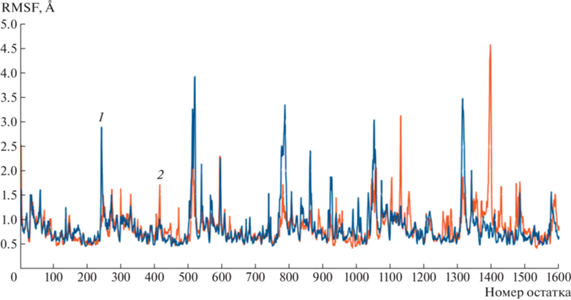
Графики RMSF комплексов с аденозином не сильно разнятся до и после мутации (рис. 6). Заметное различие в кривых RMSF наблюдается на остатках β-листов, расположенных рядом с активными центрами, в которых проводили замену D235N; пиковое значение 4.5 Å приходится на β-листы (остатки 1419–1434) мономера F. Таким образом, существенного изменения в подвижности комплекса с аденозином до и после мутации, судя по кривым, не произошло, по крайней мере картина отличается от результатов с гуанозином. Нельзя не заметить, что первый отличительный пик кривой 1 (рис. 6) приходится на His244, расположенный в активном центре. Этот пик отсутствует у того же аминокислотного остатка гистидина в мутантной форме белка, как и у обоих комплексов с гуанозином, что свидетельствует о его менее фиксированном положении в пространстве. His244, как показано на рис. 2б, способен установить водородную связь с оксогруппой гуанозина, тогда как в связывании аденозина его роль, судя по всему, становится другой либо вовсе утрачивается. Это, в свою очередь, может быть обусловлено изменением распределения частичных зарядов в активном центре после мутации и стерическим различием субстратов.
Рис. 6.
График флуктуации координат атомов, усредненный по всему времени моделирования (RMSF), комплексов аденозина с ферментом дикого (1) и мутантного типа (2).
Также рассмотрено положение субстратов в результате моделирования МД. На рис. 7 построены кривые RMSD для неводородных атомов гуанозина в комплексе с ферментами дикого (кривая 1) и мутантного типа (кривая 2). Кривая комплекса гуанозина с нативным ферментом заметно флуктуирует до 7 нс с максимальным отклонением 1 Å, что вполне ожидаемо. Интереснее выглядит кривая гуанозина в комплексе с мутантом: первые 5 нс лиганд в обоих случаях имел схожее значение отклонения от начального положения, однако после 5 нс RMSD лиганда резко увеличилось на 1 Å, достигнув более-менее стабильного положения при 2 Å от начального положения.

Аналогичные кривые RMSD для атомов аденозина в комплексе с ферментом показаны на рис. 8. Кривая комплекса с ферментом мутантного типа (кривая 2) выше, чем для комплекса с ферментом дикого типа (кривая 1), почти на всем интервале моделирования (за исключением последней наносекунды), из чего можно заключить, что мутация несколько повлияла на положение субстрата в активном центре, в среднем значения для двух случаев раличаются на 0.4 Å. Таким образом, полученные графики свидетельствуют о применимости траекторий для дальнейшего вычисления энергий. Влияние мутации зависит от субстрата. Комплекс ПНФ с аденозином стал более стабильным после замены аспарагиновой кислоты на аспарагин согласно кривой RMSD. В случае комплекса ПНФ с гуанозином замена Asp235 на Asn существенно увеличила подвижность большинства аминокислот (рис. 3) и заметно повлияла на динамику белковой глобулы.

Методом MM-GBSA из четырех траекторий рассчитаны энергии связи субстратов с белком в растворе (табл. 1–4).
Таблица 1.
Изменение компонент свободной энергии при образовании комплекса фермента дикого типа с гуанозином
| Компонента свободной энергии | Среднее значение, ккал/моль | Стандартное отклонение, ккал/моль | Ошибка среднего, ккал/моль |
|---|---|---|---|
| ΔEVdW | –34.5 | 2.95 | 0.01 |
| ΔEel | –39.49 | 4.64 | 0.15 |
| ΔGpolar | 52.05 | 2.83 | 0.08 |
| ΔGnonpolar | –4.00 | 0.15 | 0.01 |
| ΔGgas | –73.99 | 3.88 | 0.13 |
| ΔGsolv | 50.05 | 2.81 | 0.09 |
| ΔGbind | –23.95 | 3.43 | 0.11 |
Таблица 2.
Изменение компонент свободной энергии при образовании комплекса фермента мутантного типа с гуанозином
| Компонента свободной энергии | Среднее значение, ккал/моль | Стандартное отклонение, ккал/моль | Ошибка среднего, ккал/моль |
|---|---|---|---|
| ΔEVdW | –41.24 | 3.49 | 0.11 |
| ΔEel | –38.56 | 9.95 | 0.32 |
| ΔGpolar | 46.31 | 5.33 | 0.17 |
| ΔGnonpolar | –4.74 | 0.15 | 0.01 |
| ΔGgas | –79.80 | 9.45 | 0.31 |
| ΔGsolv | 41.57 | 5.27 | 0.17 |
| ΔGbind | –38.23 | 5.66 | 0.19 |
Таблица 3.
Изменение компонент свободной энергии при образовании комплекса фермента дикого типа с аденозином
| Компонента свободной энергии | Среднее значение, ккал/моль | Стандартное отклонение, ккал/моль | Ошибка среднего, ккал/моль |
|---|---|---|---|
| ΔEVdW | –25.79 | 3.25 | 0.10 |
| ΔEel | –140.93 | 21.39 | 0.68 |
| ΔGpolar | 156.73 | 20.30 | 0.64 |
| ΔGnonpolar | –3.47 | 0.32 | 0.01 |
| ΔGgas | –166.72 | 21.52 | 0.69 |
| ΔGsolv | 153.27 | 20.19 | 0.64 |
| ΔGbind | –13.45 | 3.45 | 0.11 |
Таблица 4.
Изменение компонент свободной энергии при образовании комплекса фермента мутантного типа с аденозином
| Компонента свободной энергии | Среднее значение, ккал/моль | Стандартное отклонение, ккал/моль | Ошибка среднего, ккал/моль |
|---|---|---|---|
| ΔEVdW | –34.43 | 2.43 | 0.08 |
| Δ Eel | –56.19 | 8.32 | 0.26 |
| ΔGpolar | 86.07 | 8.67 | 0.27 |
| ΔGnonpolar | –3.93 | 0.23 | 0.01 |
| ΔGgas | –90.62 | 8.65 | 0.27 |
| ΔGsolv | 82.14 | 8.64 | 0.27 |
| ΔGbind | –8.48 | 4.06 | 0.13 |
Из рассчитанных значений следует понижение энергии связи ∆∆Gbind = –14.3 ккал/моль для комплекса с гуанозином, тогда как для аденозина произошло изменение в обратную сторону и ∆∆Gbind = 4.97 ккал/моль. Так как ∆Gbind < 0 во всех случаях, комплекс с любым субстратом термодинамически выгоден. Изменение энергий связи в противоположном направлении для аденозина и гуанозина свидетельствует об увеличении специфичности фермента к гуанозину в результате мутации D235N, что согласуется с данными [1, 2].
Проведенный вычислительный эксперимент показал, что мутация D235N сказывается на субстратной специфичности и динамике фермента, причем характер влияния мутации на динамику зависит от природы субстрата: комплекс мутанта с гуанозином становится более подвижным и менее стабильным, а с аденозином – более устойчивым, с меньшими отклонениями Cα-атомов пептидной цепи. TthPNPII, в нативной форме специфичная к аденозину и способная расщеплять гуанозин [1], после замены остатка аспарагиновой кислоты на аспарагин начала формировать, согласно результатам расчета методом MM-PBSA, термодинамически более выгодный комплекс с гуанозином на ∆∆Gbind = –14.3 ккал/моль. Для аденозина энергетический выигрыш связи с субстратом наоборот уменьшился на ∆∆Gbind = = 4.97 ккал/моль.
Исследование выполнено при финансовой поддержке Российского научного фонда (проект № 21-13-00429) в рамках расчета МД и при поддержке Министерства науки и высшего образования РФ в рамках выполнения работ по Государственному заданию ФНИЦ “Кристаллография и фотоника” РАН в части анализа результатов молекулярного моделирования.
Список литературы
Timofeev V.I., Fateev I.V., Kostromina M.A. et al. // J. Biomol. Struct. Dyn. 2020. V. 40. P. 1. https://doi.org/10.1080/07391102.2020.1848628
Tomoike F., Kuramitsu S., Masui R. // Extremophiles. 2013. V. 17. P. 505. https://doi.org/10.1007/s00792-013-0535-7
Погосян Л.Г., Акопян Ж.И. // Биомедицинская химия. 2013. Т. 59. № 5. С. 483. https://doi.org/10.18097/pbmc20135905483
Salomon-Ferrer R., Case D.A., Walker R.C. // WIREs Comput. Mol. Sci. 2013. V. 3. P. 198. https://doi.org/10.1002/wcms.1121
Case D.A., Cheatham T.E., III, Darden T. et al. // J. Comput. Chem. 2005. V. 26. P. 1668. https://doi.org/10.1002/jcc.20290
Maier J.A., Martinez C., Kasavajhala K. et al. // J. Chem. Theory Comput. 2015. V. 11. P. 3696. https://doi.org/10.1021/acs.jctc.5b00255
Salomon-Ferrer R., Goetz A.W., Poole D. et al. // J. Chem. Theory Comput. 2013. V. 9. P. 3878. https://doi.org/10.1021/ct400314y
Jorgensen W. L., Chandrasekhar J., Madura J.D. et al. // J. Chem. Phys. 1983. V. 79. P. 926. https://doi.org/10.1063/1.445869
Allen M.P., Tildesley D.J. Computer simulation of liquids. New York: Oxford university press, 1991. https://doi.org/10.2307/2938686
Berendsen H.J.C., Postma J.P.M., van Gunsteren W.F. et al. // J. Chem. Phys. 1984. V. 81. P. 3684. https://doi.org/10.1063/1.448118
Darden T., York D., Pedersen L. // J. Chem. Phys. 1993. V. 98. P. 10089. https://doi.org/10.1063/1.464397
Kollman P.A., Massova I., Reyes C. et al. // Acc. Chem. Res. 2000. V. 33. P. 889. https://doi.org/10.1021/ar000033j
Srinivasan J., Trevathan M.W., Beroza P. et al. // Theor. Chem. Acc. 1999. V. 101. P. 426. https://doi.org/10.1007/s002140050460
Miller B.R., McGee T.D., Swails J.M. et al. // J. Chemical Theory and Computation. 2012. V. 8. P. 3314. https://doi.org/10.1021/ct300418h
Onufriev A., Bashford D., Case D.A. // Proteins. 2004. V. 55. P. 383. https://doi.org/10.1002/prot.20033
Schrödinger L.L.C. The PyMOL Molecular Graphics System, Version 2.0
Mikhailopulo I.A., Miroshnikov A.I. // Acta Naturae. 2010. V. 2. P. 36. https://doi.org/10.32607/20758251-2017-9-2-47-58
Fateev I.V., Kostromina M.A., Abramchik Y.A. et al. // Biomolecules. 2021. V. 11. P. 586. https://doi.org/10.3390/biom11040586
Roy B., Depaix A., Périgaud C. et al. // Chem. Rev. 2016. V. 116. P. 7854. https://doi.org/10.1021/acs.chemrev.6b00174
Almendros M., Berenguer J., Sinisterra J.V. // Appl. Environmental Microbiology. 2012. V. 78. P. 3128. https://doi.org/10.1128/AEM.07605-11
Fateev I.V., Kharitonova M.I., Antonov K.V. et al. // Chemistry. 2015. V. 21. P. 13401. https://doi.org/10.1002/chem.201501334
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография