

Кристаллография, 2023, T. 68, № 2, стр. 298-305
Высокоемкие частицы карбоната кальция как основа pН-чувствительных контейнеров для доксорубицина
Т. Н. Паллаева 1, А. В. Михеев 1, Д. Н. Хмеленин 1, Д. А. Еуров 2, Д. А. Курдюков 2, В. К. Попова 3, Е. В. Дмитриенко 3, Д. Б. Трушина 1, 4, *
1 Институт кристаллографии им. А.В. Шубникова ФНИЦ “Кристаллография и фотоника” РАН
Москва, Россия
2 Физико-технический институт им. А.Ф. Иоффе
Санкт-Петербург, Россия
3 Институт химической биологии и фундаментальной медицины СО РАН
Новосибирск, Россия
4 Первый Московский государственный медицинский университет им. И.М. Сеченова
Москва, Россия
* E-mail: trushina.d@mail.ru
Поступила в редакцию 23.09.2022
После доработки 03.10.2022
Принята к публикации 03.10.2022
- EDN: BSBCCT
- DOI: 10.31857/S0023476123020121
Аннотация
Наноструктурированные субмикронные частицы карбоната кальция размерами 500 ± 90 и 172 ± 75 нм синтезированы в ходе массовой кристаллизации в водных растворах с добавлением глицерина, а также смеси полиэтиленгликоля, полисорбата и клеточной среды. Наночастицы CaCO3 : Si : Fe размером 65 ± 15 нм получены методом темплатного синтеза в порах частиц кремнезема. Изучены кристаллическая структура и полиморфизм полученных частиц, определено влияние размера и структуры частиц на эффективность их загрузки противораковым соединением, а также его высвобождение в модельных условиях при разных рН.
ВВЕДЕНИЕ
Положительные результаты терапии широкого спектра заболеваний в значительной степени зависят от возможности осуществлять доставку препаратов в целевой орган, ткань или клетку. Несмотря на длительные исследования в области адресной доставки лекарств, в настоящее время бόльшая часть лекарственных препаратов не обладает адресностью и распределяется по всему организму, попадая преимущественно в клетки макрофагов печени и селезенки. Развитие побочных эффектов и снижение эффективности удельной дозы препарата особенно критичны при доставке химиотерапевтических препаратов с выраженными токсическими свойствами. Для устранения недостатков традиционной терапии и уменьшения побочных эффектов особую привлекательность приобрела идея, основанная на использовании сосудистых аномалий опухолей для обеспечения доступа к ним лекарственных препаратов с избежанием проникновения в нормальные ткани [1]. Особенности сосудов опухоли, в частности повышенная проницаемость и возможность удерживать введенные препараты и частицы, получили название EPR-эффекта [2]. С момента открытия и до сих пор EPR-эффект считается наиболее важной концепцией при разработке систем доставки противоопухолевых препаратов [1, 3–5]. Однако медленная диффузия лекарственных препаратов в опухолевую ткань в настоящее время признана ограничивающим фактором, серьезно снижающим лекарственную эффективность в клетках-мишенях. Помимо этого, особенности пор в кровеносных капиллярах опухоли могут сильно варьироваться в зависимости от природы опухолей, стадии развития, места положения и других факторов, а последние исследования в этой области показывают, что EPR-эффект гораздо сильнее выражен у грызунов, чем у людей [6–9]. Для более существенного увеличения эффективности терапии перспективной стратегией представляется использование особенностей микроокружения опухоли в дополнение к EPR-эффекту, в частности слабокислого рН [1, 4, 10]. Это делает перспективным разработку рН-чувствительных систем доставки, которые высвобождают инкапсулированное содержимое при понижении рН среды. Среди чувствительных к рН высоко пористых частиц благодаря хорошей биосовместимости и низкой токсичности выделяются частицы СаСО3 [11–15]. Одна из полиморфных модификаций СаСО3, ватерит, характеризуется значительной сорбционной емкостью, что позволяет сорбировать различные вещества с эффективностью до 10–15% от массы самих частиц [16]. В настоящее время для доставки in vitro в основном используют частицы ватерита среднего диаметра 2–5 мкм [17, 18]. Частицы СаСО3 диаметром менее 1 мкм не так распространены из-за сложности их синтеза [19]. Известно, что частицы ватерита быстро высвобождают загруженные в них противораковые вещества (камптотецин, доксорубицин и доксициклин) при pH от 4 до 6 (до 90%), в то время как при pH = 7.4 высвобождение ничтожно мало [12, 13, 17, 20, 21].
В одной из первых работ по изучению влияния незагруженных частиц СаСО3 размером 30–200 нм на рост опухоли у грызунов было показано, что частицы CaCО3 избирательно накапливаются во внеклеточной области опухолей, повышая и поддерживая рН опухоли на уровне ∼7.4 благодаря высокой буферной способности [22]. В [23] продемонстрировано, что непрерывная инфузия 100 ± 9 нм частиц CaCO3 способна ингибировать метастазирование опухоли в модели агрессивно метастазирующего ортотопического рака молочной железы. В [24] выявлено, что при совместном культивировании клеток рака молочной железы и фибробластов добавление наночастиц CaCO3 (120 ± 30 нм) привело к избирательному ингибированию роста и замедлению миграции раковых клеток без влияния на фибробласты. Этот эффект связывают с тем, что растворение частиц CaCO3 в слабокислой среде, характерной для окружения раковых клеток, обеспечивает хорошую буферизацию pH в пределах нормального физиологического диапазона, что приводит к метаболическому перепрограммированию раковых клеток и может уменьшить их агрессивность, не влияя на рост и поведение окружающих нормальных клеток.
Степень накопления частиц в опухолевой ткани определяется диаметром частиц, их формой, зарядом и природой поверхности [4, 9]. Благодаря возможности получать частицы CaCО3 с требуемыми характеристиками с помощью варьирования условий их синтеза, которая была изучена ранее [19, 25, 26], в настоящей работе синтезирована серия частиц CaCО3 в диапазоне размеров от 50 до 500 нм. Изучено влияние размера и структуры частиц СаСО3 на эффективность их загрузки доксорубицином, а также его высвобождение в модельных условиях при нейтральном и кислом рН.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Материалы и реактивы. В ходе работы использовали хлорид кальция (CaCl2⋅2H2O и CaCl2⋅ ⋅6H2O), карбонат натрия (Na2CO3), гидрокарбонат натрия (NaHCO3), карбонат аммония ((NH4)2 CO3), гидроксид калия (KOH), хлорид железа (III) (FeCl3), фосфатно-солевой буфер (PBS), хлорид магния (MgCl2), глицерин, Твин 20 и гидрохлорид доксорубицина производства Sigma-Aldrich (Германия), полиэтиленгликоль (ПЭГ 2000 Да) производства Carl Roth (Германия), DMEM (Dulbecco’s modified Eagle medium) производства GIBCO Life Technologies (США). Для проведения экспериментов воду очищали с помощью системы Milli-Q Plus.
Получение частиц CaCO3. Сферические субмикронные частицы CaCO3 были получены по методике, описанной в [19]. Для этого к перемешиваемой на магнитной мешалке смеси глицерина с 0.33 М водного раствора CaCl2 быстро добавляли равный объем смеси глицерина с эквимолярным водным раствором Na2CO3. Реакцию проводили в смеси глицерин : вода при содержании глицерина 80%. Реакционную смесь перемешивали со скоростью 500 об./мин в течение трех часов, затем суспензию полученных частиц CaCO3 трижды промывали от ионов Na+ и Cl– дистиллированной водой. Для предотвращения агрегации частиц суспензию периодически подвергали ультразвуковому воздействию. Все образцы высушивали, частицы карбоната кальция хранили в виде порошка.
Частицы СaCO3 получали также методом соосаждения [25]. Для этого под действием ультразвука к 1 мл водного раствора, содержащего 0.10 М NaHCO3, 0.1 мг/мл ПЭГ, 0.1% об. Твин 20 и DMEM, добавляли 100 мкл водного раствора, содержащего 0.1 М СaCl2 и MgCl2, 0.1% об. DMEM. Частицы отделяли центрифугированием (10 мин, 13.400 об./мин) и хранили в дистиллированной воде.
Синтез наночастиц состава CaCO3 : Si : Fe проводили с помощью темплатного метода, описанного в [26]. Для этого использовали монодисперсные сферические частицы кремнезема (mSiO2) с внешним диаметром 200 нм, содержащие цилиндрические наноканалы диаметром 3 нм [27]. Для подготовки мезопористых частиц mSiO2 к синтезу осуществляли их капиллярную пропитку 1 М водным раствором CaCl2 в течение 24 ч. Затем частицы высушивали при 60°С, полученный порошок выдерживали в 2 М водном растворе (NH4)2CO3. После этого частицы редиспергировали в деионизованной воде, центрифугировали и высушивали на воздухе. Описанную процедуру синтеза CaCO3 повторяли 6 раз. Далее проводили травление материала темплата (аморфного SiO2) с использованием 3 М водного раствора КОН в течение 5 ч при температуре 70°С. Затем частицы центрифугировали и редиспергировали в деионизованной воде. Для получения агрегативно устойчивого гидрозоля к водной суспензии наночастиц добавляли 0.1 М раствор FeCl3. Полученную суспензию выдерживали в течение суток, после чего частицы CaCO3 : Si : Fe многократно центрифугировали и промывали деионизованной водой.
Загрузка частиц CaCO3 доксорубицином. Для загрузки частиц CaCO3 и CaCO3 : Si : Fe доксорубицином проводили адсорбцию из раствора доксорубицина на предварительно сформированные нано- и субмикрочастицы. Для этого навески частиц определенной массы помещали в раствор доксорубицина (0.15 мг/мл) и инкубировали на шейкере в течение двух часов, затем центрифугировали и промывали 1 раз деионизованной водой. Для определения концентрации доксорубицина супернатанты исследовали спектрофотометрически на длине волны поглощения доксорубицина (480 нм).
Высвобождение доксорубицина при различных рН. Высвобождение доксорубицина из частиц изучали в буферных растворах с различным рН. Для этого 6 мг частиц ресуспендировали в 1.5 мл фосфатного буфера с рН = 4 или 7 и непрерывно перемешивали на шейкере. Через определенные промежутки времени (15, 30 мин, 1, 2, 6, 24 ч) образцы центрифугировали, и супернатант добавляли к 1.5 мл свежего буферного раствора с соответствующим рН. Супернатант анализировали спектрофотометрически при длине волны 480 нм для определения концентрации доксорубицина.
Физико-химические методы исследования. Анализ размера, формы, морфологии поверхности и структуры частиц проводили на сканирующем электронном микроскопе Jeol 7401F при напряжении 5 кВ.
Исследования частиц методом просвечивающей электронной микроскопии (ПЭМ) выполняли на микроскопе Tecnai Osiris (FEI, США) с ускоряющим напряжением 200 кВ, широкоугловым детектором темного поля и EDX-спектрометром Bruker SuperX, а также на микроскопе Jeol JEM-2100F (JEOL, США), оборудованном EDX-спектрометром INCA (Oxford Instruments, Великобритания).
Гидродинамический размер частиц и ζ-потенциал их поверхности в водной суспензии определяли с помощью автоматического анализатора Zetasizer Nano-ZS (Malvern, Великобритания).
Порошковые рентгеновские дифрактограммы субмикронных частиц снимали на лабораторных дифрактометрах Rigaku Miniflex 600 и D2 Phaser (Bruker, Германия) с использованием источника CuKα (λ = 1.5406 Å, 40 кВ, 15 мА) в режиме съемки с шагом 0.02° и со скоростью 1 шаг/с в интервале углов 2θ 18°–75°. Определение фазового состава и расчет среднего размера области когерентного рассеяния (ОКР) проводили методом Ритвельда при помощи программы FullProf.
Эффективность загрузки частиц CaCO3 и CaCO3 : Si : Fe доксорубицином оценивали спектрофотометрически с помощью двухлучевого сканирующего спектрофотометра Lambda-С650 (Perkin Elmer, США) с диапазоном длин волн 190–900 нм. Оптическую плотность раствора регистрировали на длине волны 480 нм, соответствующей максимуму поглощения доксорубицина. Количество вещества, инкорпорированного в частицы CaCO3 и CaCO3 : Si : Fe, определяли с помощью предварительно построенных калибровочных прямых по разнице концентрации раствора вещества после адсорбции по отношению к исходному раствору. Эффективность капсулирования (ЭК) определяли по формуле
где Kд – количество вещества (концентрация), добавленного к частицам, Kсуп – количество вещества в супернатанте после адсорбции. Загрузку частиц доксорубицином в массовых процентах (мас. %) рассчитывали как отношение массы включенного вещества к массе частиц. Для изучения высвобождения доксорубицина из частиц определяли содержание вещества в супернатантах. Данные, представленные на графиках, являются усредненными значениями серии экспериментов (3–5), проведенных в одинаковых условиях, и среднеквадратичными отклонениями серии измерений.РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
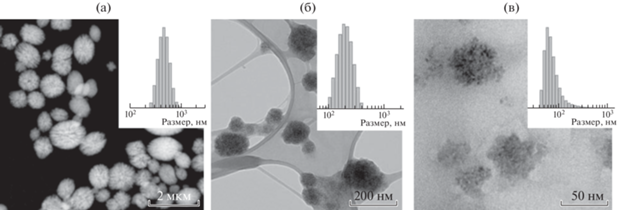
Частицы карбоната кальция, полученные в результате смешивания водных растворов солей, представляют собой не агрегированные сферические частицы с развитой поверхностью (рис. 1). При выбранном составе кристаллизационной смеси получены субмикрочастицы карбоната кальция, средний гидродинамический размер которых составляет 500 ± 90 нм (СаСО3–500) и 172 ± 75 нм (СаСО3–200) (рис. 1а, 1б). Гидродинамический диаметр наночастиц CaCO3 : Si : Fe, синтезированных в порах частиц кремнезема, составляет 65 ± 15 нм (CaCO3 : Si : Fe–50). Распределения по размерам частиц СаСО3 и CaCO3 : Si : Fe в водной суспензии приведены на вставках рис. 1.
Рис. 1.
ПЭМ-изображения частиц СаСО3–500 (а), СаСО3–200 (б) и CaCO3 : Si : Fe–50 (в). На вставках – результаты динамического светорассеяния суспензий частиц в воде.
На рис. 2 представлены энергодисперсионные рентгеновские спектры для трех образцов частиц и распределения химических элементов для отдельных частиц в этих образцах. Энергодисперсионный анализ частиц CaCO3–500 показывает значительное преобладание кальция, углерода и кислорода, которые распределены равномерно. Для частиц СаСО3–200 идентифицируются магний и фосфор, колокализованные с кальцием, поскольку частицы синтезировали в питательной клеточной среде, богатой этими элементами. В образце наночастиц CaCO3 : Si : Fe–50 определяются остаточный кремний (после растворения частиц кремнезема) и железо, которые применяли для стабилизации наночастиц. На карте распределения элементов видно, что железо колокализуется не только одновременно с кальцием, но и в свободном виде, что говорит о его избыточном содержании для улучшения стабильности наночастиц.
Рис. 2.
Энергодисперсионные рентгеновские спектры и карты распределения химических элементов для частиц СаСО3–500 (синяя линия), СаСО3–200 (зеленая линия) и CaCO3 : Si : Fe–50 (красная линия).

Структуру и фазовый состав субмикро- и наночастиц СаСО3 изучали методом порошковой рентгеновской дифракции. Для анализа фазового состава частиц использовали модель структуры ватерита из [28, 29] (пр. гр. P63/mmc, параметры гексагональной ячейки a = b = 4.131, c = 8.492 Å). Отметим, что структура поликристаллического ватерита до сих пор обсуждается и нет единой модели, которая описывала бы все рефлексы на дифрактограммах независимо от способа получения частиц. Структура кальцита, определенная в 1914 г. [30], была одной из первых структур, изученных с помощью рентгеновских лучей. Характеристические пики на полученных дифрактограммах (рис. 3а) при углах 2θ, равных 20.77°, 24.70°, 26.90°, 32.60°, 38.65°, 43.65°, 48.75°, 49.70°, 55.50°, соответствуют кристаллографическим плоскостям ватерита (004), (110), (112), (114), (211), (300), (304), (118) и (224). Пики при углах 2θ, равных 23.03°, 29.34°, 32.16°, 35.98°, 39.38°, 43.11°, 47.51°, 48.52° и 57.30°, соответствуют кристаллографическим плоскостям кальцита (012), (104), (006), (110), (113), (103), (202), (016), (018) и (122).
Рис. 3.
Порошковые рентгеновские дифрактограммы для частиц СаСО3–500 и CaCO3 : Si : Fe–50 (а), ПЭМ-изображение отдельной частицы СаСО3–200, ее увеличенное изображение и соответствующая картина электронной дифракции (б).

Все обнаруженные пики для образца СаСО3–500 относятся к двум фазам ватерита и кальцита, полнопрофильный анализ методом Ритвельда подтвердил, что кристаллы СаСО3–500 состоят на 99.4% из фазы ватерита с 0.6% включением кальцита. На дифракционной кривой образца наночастиц CaCO3 : Si : Fe–50 (рис. 2а) наблюдается набор рефлексов, соответствующих кальциту, примесных кристаллических фаз не обнаружено. Рассчитанный методом Ритвельда средний размер ОКР для наночастиц CaCO3 : Si : Fe–50 составил ∼45 нм. Достаточно большое значение ОКР может быть связано с тем, что образец предварительно сушили при температуре 60°С, что способствует перекристаллизации наночастиц карбоната кальция в субмикронные частицы [26]. Как было определено ранее, для субмикронных поликристаллов СаСО3–500 ОКР может принимать форму эллипсоида с длиной главных осей ∼120 и 50 нм [31].
На рис. 3б представлены ПЭМ-изображение отдельной частицы СаСО3–200 и картина электронной дифракции, полученная с этой частицы. Отсутствие четких пиков интенсивности свидетельствует о том, что частицы СаСО3–200 имеют аморфную структуру. Для сравнения загрузочных емкостей полученных частиц, имеющих разные средние размеры и кристаллическую структуру, одинаковые навески частиц инкубировали в растворе доксорубицина при одинаковых условиях.
Для расчета концентрации доксорубицина в растворах была построена калибровочная прямая (рис. 4а), с использованием которой рассчитана загрузка частиц СаСО3 препаратом. Показано, что загрузка составила 4 мас. % для частиц CaCO 3 : Si : Fe–50, 4.8 мас. % для частиц СаСО3–200 и 6.5 мас. % для частиц СаСО3–500, что соответствует 27, 32 и 44% эффективности инкапсулирования от исходного раствора доксорубицина. Синтезированные двумя методами частицы CaCO3 имеют различный размер, кристаллическую структуру и загрузочную емкость для противоракового препарата, которые приведены в табл. 1. С учетом того, что погрешности для определения загрузочной емкости частиц составляют ∼1 мас. %, можно считать, что три полученных образца CaCO3 имеют сравнимые загрузочные емкости для доксорубицина.
Рис. 4.
Калибровочная прямая для доксорубицина (а), высвобождение доксорубицина из частиц СаСО3–500 и CaCO3 : Si : Fe–50 в фосфатный буфер при различных рН (б).

Таблица 1.
Характеристики синтезированных частиц CaCO3
| Образец | Гидродинамический диаметр, нм | Полиморфный состав | Загрузка доксорубицином, мас. % |
|---|---|---|---|
| СаСО3–500 | 500 ± 90 | 99.4% ватерита | 6.5 |
| СаСО3–200 | 172 ± 75 | аморфная структура | 4.8 |
| CaCO3 : Si : Fe–50 | 65 ± 15 | 100% кальцита | 4 |
В качестве модельной среды для изучения высвобождения доксорубицина был выбран натрий-фосфатный буфер, поскольку осмолярность и концентрации ионов в нем соответствуют значениям в крови, тканевых жидкостях и тканях организма человека. На рис. 4б представлены результаты высвобождения доксорубицина из кристаллических частиц СаСО3 двух размеров при их инкубации в фосфатном буфере с рН 4 и 7. Из кривых на рис. 4б видно, что для всех образцов высвобождение доксорубицина осуществляется в два этапа. Начальный этап характеризуется резким увеличением концентрации препарата в растворе, затем происходит постепенный выход оставшегося в частицах доксорубицина. Данное обстоятельство указывает на то, что на начальном этапе наблюдали выход соединения с поверхности и из пор частиц за счет процесса десорбции, а также вследствие первичного растворения карбонатной матрицы в поверхностных слоях по сравнению с объемом частицы. Последующее замедление данного процесса может быть связано с постепенным растворением карбонатной матрицы при кислотных значениях рН. Обычно интенсивный выход характеризует высвобождение иммобилизованных веществ из высокопористых частиц, которое можно замедлить с помощью покрытия частиц полимерными оболочками [32, 33]. При сравнении высвобождения доксорубицина при нейтральных (рН = 7) и более кислотных условиях (рН = 4) наиболее интенсивное высвобождение препарата наблюдается из частиц СаСО3–500 при рН = 4, что однозначно связано с растворением карбонатной матрицы при кислотных значениях рН. При увеличении рН наблюдается замедление высвобождения препарата, что объясняется большей стабильностью карбоната кальция при нейтральном значении рН. Доксорубицин из частиц CaCO3 : Si : Fe–50 высвобождается практически одинаково при разных рН, что связано со стабильной кристаллической структурой наночастиц, которые представляют собой кальцит, а также с присутствием в их составе Si и Fe, которые могут влиять на скорость растворения.
ЗАКЛЮЧЕНИЕ
В процессе массовой кристаллизации при соосаждении с добавлением в реакционный объем глицерина или смеси ПЭГ, Твин 20 и DMEM синтезированы субмикронные частицы СаСО3, имеющие средние гидродинамические диаметры 500 ± 90 и 172 ± 75 нм соответственно. С помощью темплатного синтеза в мезопористых частицах кремнезема получены наночастицы СаСО3 : Si : Fe диаметром 65 ± 15 нм. Исследование образцов методами порошковой рентгеновской дифракции и электронной дифракции показало, что структура частиц различна, и наночастицы, стабилизированные ионами железа, представляют собой стабильную модификацию кальцита. Субмикронные частицы СаСО3, полученные соосаждением, имеют структуру ватерита или аморфны. Несмотря на различие структур синтезированных частиц эффективности их загрузки противораковым соединением сопоставимы и составляют 4–6.5 мас. %. При инкубации наночастиц кальцита при рН = 4 и 7 высвобождение иммобилизованного доксорубицина происходит одинаково. Стабильность наночастиц кальцита в кислом рН обусловлена не только наиболее термодинамически стабильной полиморфной модификацией, но и наличием в их составе кремния и железа. Наиболее интенсивное высвобождение доксорубицина происходит из субмикронных метастабильных частиц ватерита при рН = 4, что объясняется растворением частиц при кислотных значениях рН. Замедление высвобождения инкапсулированного вещества со временем связано с постепенным ростом рН в результате растворения частиц. Таким образом, частицы CaCO3 и CaCO3 : Si : Fe могут служить контейнерами для доксорубицина с возможностью выбрать такие параметры частиц, чтобы вещество либо длительное время не высвобождалось, либо высвобождалось в кислой среде.
Д.А. Еуров и Д.А. Курдюков выражают благодарность М.А. Яговкиной и Д.А. Кириленко за исследования частиц CaCO3 : Si : Fe методами рентгеновской дифракции и ПЭМ.
Работа в части получения и характеризации частиц CaCO3–500 выполнена в рамках государственного задания ФНИЦ “Кристаллография и фотоника” РАН с использованием оборудования ЦКП ФНИЦ “Кристаллография и фотоника” РАН. Синтез наночастиц CaCO3 : Si : Fe выполнен в рамках государственного задания 0040-2019-0012. Синтез частиц CaCO3–200 выполнен в рамках государственного задания ИХБФМ СО РАН 121031300042-1. Работы по загрузке и высвобождению доксорубицина выполнены при поддержке Российского научного фонда (грант № 21-74-10058).
Список литературы
Danhier F., Feron O., Préat V. // J. Control. Release. 2010. V. 148. № 2. P. 135. https://doi.org/10.1016/j.jconrel.2010.08.027
Matsumura Y., Maeda H. // Cancer Res. 1986. V. 46. P. 6387.
Pérez-Herrero E., Fernández-Medarde A. // Eur. J. Pharm. Biopharm. 2015. V. 93. P. 52. https://doi.org/10.1016/j.ejpb.2015.03.018
Rodrigues C.F., Alves C.G., Lima-Sousa R. et al. // Advances and Avenues in the Development of Novel Carriers for Bioactives and Biological Agents. Elsevier. 2020. P. 283. https://doi.org/10.1016/B978-0-12-819666-3.00010-9
Parra Nieto J., Del Cid M.A.G., de Cárcer I.A. et al. // Biotechnol. J. 2021. V. 16. № 2. P. 2000150. https://doi.org/10.1002/biot.202000150
Danhier F. // J. Control. Release. 2016. V. 244. P. 108. https://doi.org/10.1016/j.jconrel.2016.11.015
Rosenblum D., Joshi N., Tao W. et al. // Nat. Commun. 2018. V. 9. № 1. P. 1. https://doi.org/10.1038/s41467-018-03705-y
Nichols J.W., Bae Y.H. // J. Control. Release. 2014. V. 190. P. 451. https://doi.org/10.1016/j.jconrel.2014.03.057
Wilhelm S., Tavares A.J., Dai Q. et al. // Nat. Rev. Mater. 2016. V. 1. P. 1. https://doi.org/10.1038/natrevmats.2016.14
Reshetnyak Y.K. // Clin. Cancer Res. 2015. V. 21. № 20. P. 4502. https://doi.org/10.1158/1078-0432.CCR-15-1502
Nakamura J., Poologasundarampillai G., Jones J.R. et al. // J. Mater. Chem. B. 2013. V. 1. № 35. P. 4446. https://doi.org/10.1039/C3TB20589D
Maleki Dizaj S., Sharifi S., Ahmadian E. et al. // Expert Opin. Drug Deliv. 2019. V. 16. № 4. P. 331. https://doi.org/10.1080/17425247.2019.1587408
Zhang Y., Cai L., Li D. et al. // Nano Res. 2018. V. 11. № 9. P. 4806. https://doi.org/10.1007/s12274-018-2066-0
Sudareva N.N., Popryadukhin P.V., Saprykina N.N. et al. // Cell. Ther. Transplant. 2020. V. 9. № 2. P. 13. https://doi.org/10.18620/ctt-1866-8836-2020-9-2-13-19
Fu J., Leo C.P., Show P.L. // Biochem. Eng. J. 2022. P. 108446. https://doi.org/10.1016/j.bej.2022.108446
Trushina D.B., Borodina T.N., Belyakov S. et al. // Mater. Today Adv. 2022. V. 14. № 2022. P. 100214. https://doi.org/10.1016/j.mtadv.2022.100214
Qiu N., Yin H., Ji B. et al. // Mater. Sci. Eng. C. 2012. V. 32. № 8. P. 2634. https://doi.org/10.1016/j.msec.2012.08.026
Liu S.S., Liu L.J., Xiao L.Y. et al. // J. Mater. Chem. B. 2015. V. 3. № 42. P. 8314. https://doi.org/10.1039/C5TB01692D
Trushina D.B., Bukreeva T.V., Antipina M.N. // Cryst. Growth Des. 2016. V. 16. № 3. P. 1311. https://doi.org/10.1021/acs.cgd.5b01422
Wang A., Yang Y., Zhang X. et al. // Chempluschem. 2016. V. 81. № 2. P. 194. https://doi.org/10.1002/cplu.201500515
Choukrani G., Maharjan B., Park C.H. et al. // Mater. Sci. Eng. C. 2020. V. 106. P. 110226. https://doi.org/10.1016/j.msec.2019.110226
Som A., Raliya R., Tian L. et al. // Nanoscale. Royal Soc. Chem. 2016. V. 8. № 25. P. 12639. https://doi.org/10.1039/C5NR06162H
Som A., Raliya R., Paranandi K. et al. // Nanomedicine. 2019. V. 14. № 2. P. 169. https://doi.org/10.2217/nnm-2018-0302
Lam S.F., Bishop K.W., Mintz R. et al. // Sci. Rep. 2021. V. 11. № 1. P. 9246. https://doi.org/10.1038/s41598-021-88687-6
Popova V., Poletaeva Y., Pyshnaya I. et al. // Nanomaterials. 2021. V. 11. № 11. P. 2794. https://doi.org/10.3390/nano11112794
Eurov D.A., Kurdyukov D.A., Boitsov V.M. et al. // Microporous Mesoporous Mater. 2022. V. 333. P. 111762. https://doi.org/10.1016/j.micromeso.2022.111762
Trofimova E.Y., Kurdyukov D.A., Yakovlev S.A. et al. // Nanotechnology. 2013. V. 24. № 15. P. 155601. https://doi.org/10.1088/0957-4484/24/15/155601
Kamhi S.R. // Acta Cryst. 1963. V. 16. № 8. P. 770. https://doi.org/10.1107/S0365110X63002000
Pokroy B., Kabalah-Amitai L., Polishchuk I. et al. // Chem. Mater. 2015. V. 27. № 19. P. 6516. https://doi.org/10.1021/acs.chemmater.5b01542
Bragg W.L. // Proc. R. Soc. London. A. 1914. V. 89. № 613. P. 468. https://doi.org/10.1098/rspa.1914.0015
Трушина Д.Б., Бородина Т.Н., Сульянов С.Н. и др. // Кристаллография. 2018. Т. 63. № 6. С. 956. https://doi.org/10.1134/S0023476118060309
Borodina T., Marchenko I., Trushina D. et al. // J. Pharm. Pharmacol. 2018. V. 70. P. 1164. https://doi.org/10.1111/jphp.12958
Borodina T.N., Trushina D.B., Marchenko I.V. et al. // BioNanoSci. 2016. V. 6. № 3. P. 261. https://doi.org/10.1007/s12668-016-0212-2
Дополнительные материалы отсутствуют.
Инструменты
Кристаллография