

Мембраны и мембранные технологии, 2023, T. 13, № 1, стр. 42-55
Метод жидкофазного фторирования полимеров как способ увеличения селективности разделения O2/N2
Д. А. Сырцова a, *, А. Ю. Алентьев a, С. В. Чирков a, Д. А. Безгин a, Р. Ю. Никифоров a, И. С. Левин a, Н. А. Белов a
a Федеральное государственное бюджетное учреждение науки Ордена Трудового Красного Знамени
Институт нефтехимического синтеза им. А.В. Топчиева РАН
119991 Москва, Ленинский просп., 29, Россия
* E-mail: syrtsova@ips.ac.ru
Поступила в редакцию 16.08.2022
После доработки 03.10.2022
Принята к публикации 07.10.2022
- EDN: HISSUE
- DOI: 10.31857/S2218117223010066
Аннотация
В работе проведена односторонняя модификация гомогенных полимерных пленок из поливинилтриметилсилана (ПВТМС), поли(2,6-диметилфениленоксида-1,4) (ПФО) и полибензодиоксана (PIM-1) методом жидкофазного фторирования фтор-азотной смесью в перфтордекалине. Время фторирования составляло от 10 до 60 мин. Для образцов ПФО методом рентгенофазового анализа показано, что исходные образцы включают, помимо аморфной, β-нанокристаллическую фазу (48%) и установлено, что фторирование не оказывает существенного влияния на степень кристалличности исследуемых пленок. Изучено влияние времени фторирования на эффективные коэффициенты проницаемости, диффузии и растворимости кислорода и азота. Установлено, что модификация приводит к снижению как эффективных коэффициентов диффузии, так и эффективных коэффициентов растворимости газов, при этом результирующий рост селективности проницаемости составил от 30% для ПВТМС до увеличения в 2 раза в случае ПФО и PIM-1. Установлено, что такое улучшение показателей по селективности проницаемости достигается преимущественно за счет увеличения селективности растворимости. Получены значения эффективных коэффициентов проницаемости газов для смеси O2/N2. Найдено, что достигнутые значения факторов разделения для модифицированных образцов близки к идеальной селективности пленок. Полученные результаты демонстрируют возможности эффективного применения данного метода не только для модификации гомогенных полимерных пленок исследуемых полимеров, но и мембран с селективным непористым слоем на их основе.
ВВЕДЕНИЕ
Задача разделения компонентов воздуха, и в первую очередь, разделение смеси кислород/азот, была одной из первых задач, при решении которых мембранные процессы успешно зарекомендовали себя еще в 80-х гг. 20 в. как конкурентные традиционным разделительным методам, например, адсорбционному, абсорбционному и криогенному [1]. Были разработаны промышленные полимерные мембраны с непористым селективным слоем, например, асимметричная плоская мембрана из поливинилтриметилсилана (ПВТМС) (ИНХС РАН, НПО “Кусково”, СССР), половолоконная мембрана на основе полиимида Upilex-R (Ube Industries Япония), половолоконная мембрана из полиметилпентена (ПМП) (Dow Chemicals, Япония), композиционная половолоконная мембрана на основе полисульфона Monsanto (США) и др., обеспечивающие требуемые в то время показатели. На сегодняшний день мембранная технология в случае многотоннажного производства по-прежнему во многом уступает криогенному методу и короткоцикловой адсорбции (КЦА), однако проведенные технико-экономические расчеты [2] показывают, что для малотоннажных процессов, например, медицины, самолётостроения и др. именно применение мембран оказывается наиболее эффективным. Отметим, при разделении компонентов воздуха коммерческий интерес представляет как получение смесей, обогащенных кислородом, так и смесей, содержащих преимущественно азот [1–3]. Список современных коммерческих мембран для решения таких задач достаточно широк (табл. 1), но при этом необходимость дальнейшего роста экономической эффективности стимулирует как поиск новых мембранных материалов (полимерных, неорганических, материалов со смешанными матрицами [2]), так и использование различных методов модификации существующих полимеров и полимерных мембран, позволяющих улучшить их показатели для газоразделительных процессов.
Таблица 1.
Современные мембраны для разделения компонентов воздуха
| Мембрана | Полимер | Производитель | Тип мембраны | Ссылки |
|---|---|---|---|---|
| SEPAREL | Полиметилпентен | DIC Corp | ПВ, AC | [4] |
| Generon | Тетрабромполикарбонат | Generon | ПВ, АС | [5] |
| Parker | Полифениленоксид | Parker-Hannifin | ПВ | [6] |
| Sepuran | Полиимид P84 | Evonik | ПВ | [7] |
| Upilex-R | Полиимид BPDA-pp'ODA | Ube | ПВ | [8] |
| AF2400 | Teflon AF2400 | Biogeneral | ПВ, АС | [9] |
| PRIZM | Полисульфон | Air products | ПВ | [10] |
Таблица 2.
Газотранспортные и разделительные свойства мембран и мембранных материалов на основе различных полимеров для разделения компонентов воздуха
| Полимер | Коэффициент проницаемости, Баррер* | Селективность O2/N2 |
Ссылки |
|---|---|---|---|
| ПСФ | 1.1 | 6.2 | [11] |
| ПИ(Matrimid) | 1.95 | 6.5 | [12] |
| ПИ(Matrimid 5218) | 1.5 | 6.6 | [13] |
| PIM-1 | 580 | 3.2 | [14] |
| PIM-1 | 1300 | 2.7 | [3] |
| ПМП | 32.3 | 3.8 | [15] |
| ПВТМС | 30 | 4.0 | [16] |
| ПФО | 15.1 | 4.5 | [17] |
| ПФО | 17 | 3.8 | [18] |
| ПФО с 20% SBS | 18.5 | 3.80 | [19] |
| ПИ 6FDA/BATFM | 27.1 | 3.80 | [20] |
| ПИ 6FDA/PPDA/CF3 | 30 | 4.30 | [21] |
| ПФО с 1.0% H40 | 32.2 | 14.60 | [22] |
| Углеродная мембрана на основе ПИ | 812.0 | 7.50 | [23] |
| Полимер | Проницаемость, GPU** | Селективность O2/N2 | Ссылки |
| ПК | 36.0 | 5.3 – | [24] |
| ПИ (6FDA-IPDA) | 65.0 | 6.3 | [24] |
| ПИ(6FDA-6FAP) | 794 | 5.3 | [25] |
| ПЭС | 9.3 | 5.8 | [26] |
| ПСФ | 7.8–21.6 | 5.4–6.7 | [27] |
| ПСФ | 6.9 | 7.3 | [28] |
Одним из наиболее перспективных направлений модификации полимеров является обработка поверхности, позволяющая изменять такие свойства поверхностного и приповерхностных слоев, как, например, гидрофильность поверхности, плотность полимерной матрицы, газопроницаемость, при этом сохраняя исходные объемные свойства полимерной пленки. К эффективным современным методам поверхностной модификации полимерных пленок и мембран можно отнести обработку поверхности в плазме, в том числе, низкотемпературной, в различных средах [29–31], метод радиационной прививки [32], галогенирование, в том числе, фторирование, как газофазное, которое может включать: прямое фторирование, которое проводится газовой смесью F2/N2 или F2/He [33–44]; фторирование в плазме [32, 45] и обработка дихлордифторметаном под воздействием γ-излучения и искрового разряда [46], так и жидкофазное [47]. Несмотря на то, что к настоящему моменту основное внимание уделяется применению поверхностного фторирования как эффективному способу повышения селективности разделения метан-содержащих смесей (Не/СН4, Н2/СН4, СО2/СН4) [37, 41–49], было показано, что данный метод обработки поверхности мембран позволяет также добиться повышения селективности О2/N2, например, для политриметилсилилпропина (ПТМСП), для которого абсолютные значения модифицированных пленок хоть и проигрывали по уровню селективности целому ряду полимеров, тем не менее, превышали исходные значения более чем в 2 раза [37, 42].
В данной работе представлены результаты исследований влияния условий односторонней модификации новым методом жидкофазного фторирования (фторирование элементным фтором в среде перфторированной жидкости [47, 50, 57]) на газоразделительные и газотранспортные свойства полимеров различной химической структуры, уже применяемых и перспективных для разделения компонентов воздуха. Выбор данного метода обусловлен тем, что жидкофазное фторирование [47, 50, 57] в случае модификации газоразделительных полимерных пленок и мембран имеет ряд преимуществ по сравнению с модификацией путем газофазного фторирования [35–46, 50]. Так, например, более мягкие условия проведения процесса [47, 57] позволяют существенно снизить вклад деструкции полимера в процессе обработки, что влияет на химическую структуру фторированного слоя, и, следовательно, на газотранспортные и селективные свойства модифицированных мембран [50, 57].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Приготовление полимерных пленок
В работе исследованы исходные гомогенные полимерные пленки из ПВТМС, полученные поливом 3 мас. % раствор полимера (в случае ПФО и PIM-1 растворителем являлся хлороформ, в случае ПВТМС – толуол) на целлофан. Структурные формулы полимеров представлены на рис. 1. Образцы сушили при комнатной температуре. Полученные пленки отделяли от подложки и вакуумировали до постоянной массы в эксикаторе (остаточное давление при вакуумировании не превышало 6 мбар). Толщина пленок составляла 40–45 мкм в случае ПФО и ПВТМС, 85–95 мкм в случае PIM-1 и отдельно были получены пленки PIM-1 толщиной 120 мкм для определения коэффициентов диффузии исходного образца.
Электронная микроскопия и элементный анализ полимерных образцов проводили с использованием 3D-сканирующего электронного микроскопа Quanta 200 (FEI, США) с системой определения характеристик материалов Pegasus (EDAX, США). Исследование химического состава поверхности образцов проводили на рентгеновском фотоэлектронном спектрометре FLEXPS (Specs, Германия). Спектрометр оборудован полусферическим анализатором электронов Phoibos 150 и детектором электронов c многоканальной пластиной на 150 каналов и линией задержки 1-DLD.
Определение степени кристалличности пленок ПФО
Определение степени кристалличности пленок ПФО до и после фторирования проводили на дифрактометре Rigaku, Япония. Экспериментальные дифрактограммы были получены с помощью рентгеновского источника с вращающимся медным анодом Rotaflex RU-200, режим работы источника 50 кВ–100 мА. Источник был оснащен горизонтальным широкоугольным гониометром Rigaku D/Max-RC и вторичным графитовым монохроматором (длина волны λ монохроматизированного излучения составляла 1.542 ангстрем). Диапазон измерения углов дифракции – 3–40 град по 2θ, измерение велось в режиме непрерывного сканирования со скоростью 2 град/мин и шагом 0.04 град. θ –2θ сканирование производили по схеме Брэгга–Брентано. Образцы пленок закрепляли на алюминиевых рамках, в этом случае сканирование вели в геометрии “на отражение”. Следует отметить, что при использовавшейся длине волны рентгеновского излучения в геометрии на отражение пучок сканировал всю толщину пленки.
Полученные дифрактограммы обрабатывали с помощью программы Fityk [51]: вычитали фон, затем дифрактограмму методом деконволюции аппроксимировали суммой семи гауссовских пиков, два из которых (с угловыми положениями около 14 и 23 град), согласно работе [52] относили к аморфной фазе при расчете степени кристалличности. Идентификацию кристаллических пиков и индексов Миллера hkl проводили в соответствие с данными работы [53]. По угловому положению кристаллического пика 2θ по формуле Вульфа–Брэгга
вычисляли межплоскостные расстояния dhkl. Степень кристалличности СI образцов рассчитывали соласно методу Руланда [54] по формуле где Acr – сумма интегральных интенсивностей (площадей) пиков, соответствующих кристаллической фазе, Asum – суммарная площадь всех пиков, которыми аппроксимировали дифрактограмму.Размер кристаллитов Dhkl вдоль кристаллографического направления hkl для каждого кристаллического пика оценивали по формуле Шеррера (Scherrer equation):
где λ – длина волны рентгеновского излучения, K = 0.94 – постоянная Шеррера для сферических частиц, FWHM – ширина на полувысоте пика с угловым положением 2θhkl.Методика жидкофазного фторирования
В работе проводили одностороннюю поверхностную модификацию полимерных пленок методом жидкофазного фторирования. Схема реактора представлена на рис. 2. Предварительно полимерную пленку для сравнения (холостой опыт) готовили в реакторе 1 без применения фторирующей смеси. Для этого, в держатель для мембран 2 устанавливали пленку 3, закрепляли держатель в реакторе, в котором уже находился перфтордекалин (ПФД). В течение 30 мин перемешивали ПФД с помощью магнитной мешалки 4. Таким образом, поверхностный слой пленки насыщался ПФД. По истечении 30 мин держатель мембраны извлекали из стакана и доставали пленку. Остатки ПФД на поверхности пленки аккуратно стряхивали, а затем пленку размещали между двух дисков из фильтровальной бумаги.
Рис. 2.
Принципиальная схема установки для поверхностного фторирования полимерных пленок и мембран: 1 – фторопластовый стакан (реактор); 2 – держатель для мембран; 3 – полимерная пленка; 4 – якорь магнитной мешалки; 5 – регулятор расхода фтор-азотной смеси.
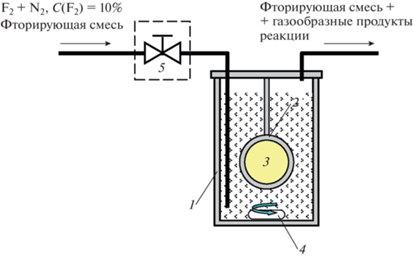
Поверхностное фторирование проводили по следующей методике: через фторопластовый стакан с налитым ПФД в течение 60 мин барботировали смесь газов: 10% фтора и 90% азота с расходом смеси 0.5 мл/с. Данный расход устанавливали с помощью расходомера. Включали магнитную мешалку и, по истечению заданного времени, закрывали подачу газовой смеси. Далее приступали к обработке мембран в ПФД, насыщенным фтором. Образец помещали в держатель и устанавливали его в реактор с ПФД таким образом, что поверхностное жидкофазное фторирование проходило с одной стороны. Для предотвращения попадания фторирующей смеси на обратную сторону пленки, необрабатываемую сторону образца закрывали специальным зажимом из тефлона. Включали подачу фторазотной смеси в реактор и одновременно начинали перемешивание жидкой фазы. Расход газа составлял 0.5 мл/с, температура реактора – 20–21°С. Через 15 мин обработки закрывали подачу газа и выключали магнитную мешалку. Образец из реактора просушивали с помощью фильтровальной бумаги. Эксперимент по поверхностному фторированию фтор-азотной смесью проводили в течение 10, 15, 30, 45 и 60 мин. Далее все образцы размещали в эксикаторе и вакуумировали в течение 15 мин.
Методика измерения параметров газопроницаемости
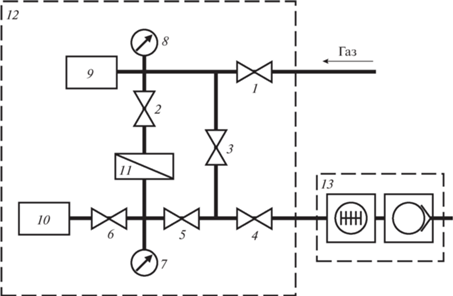
Эффективные коэффициенты проницаемости P [см3 (н.у.) см/см2 с см рт. ст.] и диффузии D [см2/с] для He, H2, O2, N2, CO2, CH4 (чистота 99.99%) определяли на барометрической установке “Баротрон” (рис. 3) при температуре 22 ± 2°С. Давление над мембраной варьировали в интервале 1–3 бар, давление под мембраной не превышало ∼16 мбар. В ходе эксперимента мембрану герметично закрепляли в мембранной ячейке 5. С помощью турбомолекулярного и форвакуумного мембранного насосов 13 подмембранное и надмебранное пространство откачивали до давления 10–2 мбар. Затем с помощью датчиков давления проводили определение фонового сигнала изменения давления за время t в калиброванном объеме 9, который регистрировали с помощью АЦП. После этого в надмембранное пространство ячейки подавали исследуемый газ и определяли Δp/Δt для исследуемого газа Измерение стационарного потока через полимерную пленку осуществляли при временах (4–6) × θ, где θ – время запаздывания. Значение эффективного коэффициента проницаемости при н.у. рассчитывали по формуле:
(4)
$P = \frac{{Vl}}{{St\Delta p}} = \frac{{\Delta p}}{{\Delta t}}\frac{{{{{{V}_{{\text{к}}}}{{T}_{0}}} \mathord{\left/ {\vphantom {{{{V}_{{\text{к}}}}{{T}_{0}}} {\left( {{{T}_{0}} + T} \right)}}} \right. \kern-0em} {\left( {{{T}_{0}} + T} \right)}}}}{{{{p}_{{{\text{atm}}}}}}}\frac{l}{{S\left( {{{p}_{f}} - {{p}_{p}}} \right)}},$Рис. 3.
Принципиальная схема барометрической установки Баротрон: 1–6 – клапаны; 7, 8 – датчики давления; 9, 10 – емкости с калиброванными объемами; 11 – мембранная ячейка; 12 – воздушный термостат; 13 – система вакуумирования.
Для расчета эффективного коэффициента диффузии использовали метод Дейнеса–Баррера:
где θ – время запаздывания, с. Эффективные коэффициенты растворимости газов S [${{{\text{см}}_{{\text{г}}}^{{\text{3}}}} \mathord{\left/ {\vphantom {{{\text{см}}_{{\text{г}}}^{{\text{3}}}} {{\text{см}}_{{\text{п}}}^{{\text{3}}}}}} \right. \kern-0em} {{\text{см}}_{{\text{п}}}^{{\text{3}}}}}$ см рт. ст.] рассчитывали как P/D.Селективность проницаемости, диффузии и растворимости газов определяли как:
(6a)
$\alpha = {{{{P}_{i}}} \mathord{\left/ {\vphantom {{{{P}_{i}}} {{{P}_{j}}}}} \right. \kern-0em} {{{P}_{j}}}},$(6b)
${{\alpha }^{D}} = {{{{D}_{i}}} \mathord{\left/ {\vphantom {{{{D}_{i}}} {{{D}_{j}}}}} \right. \kern-0em} {{{D}_{j}}}},$(6c)
${{\alpha }^{S}} = {{{{S}_{i}}} \mathord{\left/ {\vphantom {{{{S}_{i}}} {{{S}_{j}}}}} \right. \kern-0em} {{{S}_{j}}}},$Измерения газоразделительных свойств мембран для смеси O2/N2 состава 30/70 об. % проводили при t = 22–23°C с возможностью определения их концентрации в потоке ретентата и пермеата дифференциальным методом на газоразделительной установке с хроматографическим детектированием [55]. В качестве газа-носителя использовали Не.
Коэффициент проницаемости компонента А в смеси находили по формуле:
(7)
${{P}_{A}} = \frac{l}{{\Delta p}}\frac{{273\,\,~{\text{K}}}}{{TS}}\frac{{{{p}_{{{\text{атм}}}}}}}{{76~}}\left( {{{\omega }_{А}}\frac{{\Delta V}}{{\Delta t}}~} \right),$Фактор разделения f рассчитывали следующим образом:
(8)
$f = \frac{{{{{{Y}_{{{{H}_{2}}}}}} \mathord{\left/ {\vphantom {{{{Y}_{{{{H}_{2}}}}}} {{{Y}_{{{{C}_{x}}}}}}}} \right. \kern-0em} {{{Y}_{{{{C}_{x}}}}}}}}}{{{{{{X}_{{{{H}_{2}}}}}} \mathord{\left/ {\vphantom {{{{X}_{{{{H}_{2}}}}}} {{{X}_{{{{C}_{x}}}}}}}} \right. \kern-0em} {{{X}_{{{{C}_{x}}}}}}}}},$Относительная погрешность определения коэффициентов проницаемости составляла 5–7%, коэффициентов диффузии – 10%.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Влияние модификации на степень кристалличности пленок ПФО
Типичные дифрактограммы исходных и модифицированных образцов пленок ПФО представлены на рис. 4. В табл. 3 представлены значения индексов Миллера hkl в соответствии с данными работы [53], угловые положения “кристаллических” дифракционных максимумов 2θ, рассчитанные для них межплоскостные расстояния dhkl, относительные интенсивности дифракционных максимумов Ihkl, размеры кристаллитов Dhkl, рассчитанные по формуле Шеррера, и степени кристалличности CI, рассчитанные для исследованных образцов.
Рис. 4.
Дифрактограммы (а) исходной (0 мин) и фторированных в течение (б) 30 мин и (в) 60 мин пленок ПФО.
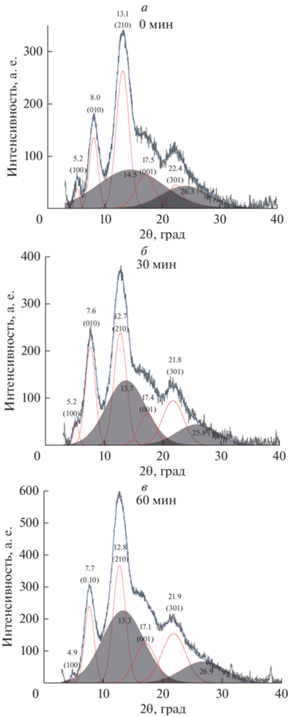
Таблица 3.
Угловые положения дифракционных максимумов 2θ, межплоскостные расстояния dhkl, относительные интенсивности дифракционных максимумов Ihkl, размеры кристаллитов Dhkl и степени кристалличности CI для исходной и фторированных пленок ПФО
| Время фторирования, мин | 0 (исходная) | 30 | 60 |
|---|---|---|---|
| hkl | 2θ, град | ||
| 100 010 210 001 301 |
5.2 8.0 13.1 17.5 22.4 |
5.2 7.6 12.7 17.4 21.8 |
4.9 7.7 12.8 17.1 21.9 |
| hkl | dhkl, Å | ||
| 100 010 210 001 301 |
17.1 11.1 6.8 5.1 4.0 |
17.1 11.6 6.9 5.1 4.1 |
18.1 11.6 6.9 5.1 4.1 |
| hkl | Ihkl, % | ||
| 100 010 210 001 301 |
2.3 9.8 18.2 3.7 13.6 |
1.7 13.4 19.1 7.1 12.8 |
0.1 8.8 17.9 10.3 14.5 |
| hkl | Dhkl, nm | ||
| 100 010 210 001 301 |
6.9 4.2 3.4 3.2 1.6 |
7.9 4.1 3.3 2.6 2.0 |
23.9 4.5 3.4 2.0 1.8 |
| CI, % | 48 | 54 | 52 |
Анализ полученных дифрактограмм показывает, что модификация пленок ПФО не приводит к изменению угловых положений дифракционных максимумов и межплоскостных расстояний. Незначительные изменения относительных интенсивностей дифракционных максимумов и, соответственно, размеров кристаллитов не приводит к изменению степени кристалличности (табл. 3). Так, средняя степень кристалличности исходных образцов пленок ПФО составила 48 ± 2%, в то время как после фторирования степень кристалличности в пределах ошибки не изменилась и составила 51 ± 3%.
Влияние модификации на химический состав приповерхностного слоя полимерных пленок
Химический состав приповерхностного слоя толщиной ~1 нм фторированных полимерных пленок изучали методом рентгеновской фотоэлектронной спектроскопии (РФЭС) для образцов, фторированных в течение 60 мин. Атомные концентрации элементов, представленные в табл. 4, для исходных полимерных пленок были близки их содержанию в мономерном звене полимеров: для ПФО отношение C : O соответствует 8.0 : 1.2; для ПВТМС отношение C : Si – 5.0 : 1.2; для PIM‑1 отношение C : O : N – 29 : 11 : 1.4. Присутствие малой концентрации атомов фтора в поверхностном слое пленки PIM-1 может быть следствием остаточных концевых групп макроцепей, поскольку сомономером PIM-1 является 1,4‑дициано-2,3,5,6-тетрафторбензол.
Таблица 4.
Атомные концентрации (%) C, O, F, N и Si в приповерхностном слое исходных и фторированных полимеров (${{t}_{{{{{\text{F}}}_{2}}}}}$ = 60 мин)
| Полимер | C | O | N | F | Si |
|---|---|---|---|---|---|
| ПФО (исходный) | 87.2 | 12.7 | – | 0 | – |
| ПФО (F2) | 54.3 | 19.7 | – | 22.2 | – |
| ПВТМС (исходный) | 77.9 | 2.84 | – | 0 | 19.2 |
| ПВТМС (F2) | 49.9 | 17.9 | – | 26.9 | 3.67 |
| PIM-1 (исходный) | 82.3 | 13.5 | 3.9 | 0.2 | – |
| PIM-1 (F2) | 56.3 | 21.3 | 2.4 | 20.0 | – |
Проведение жидкофазного фторирования в среде ПФД в течение 60 мин приводит во всех случаях к возрастанию содержания атомов фтора в химической структуре полимера до 20–27 ат. %. Фтор может входить в структуру полимерной цепи путем взаимодействия с функциональными группами полимерной цепи через замещение атомов водорода в алифатических и в ароматических структурах, насыщение конденсированных связей [47, 50, 56]. Уменьшение концентрации атомов Si и N в приповерхностном слое пленок ПВТМС и PIM-1 после фторирования может быть связано с частичным разрывом связей C–Si, C–N. Ранее было показано, что простая эфирная связь C–O и одинарная С–С связь при условиях жидкофазного фторирования оказываются весьма стабильными [56, 57], что, вероятно, выполняется и в случае поверхностного фторирования исследуемых в работе полимерных пленок.
В предыдущих работах также было продемонстрировано, что процесс фторирования полимеров сопровождается их окислением, что приводит к появлению во фторированном слое кислородсодержащих групп [33, 35, 37, 39]. Аналогичные закономерности характерны и для жидкофазного фторирования исследуемых полимерных образцов: во всех случаях наблюдается увеличение концентрации атомов кислорода в приповерхностном слое полимерных пленок (табл. 4). Вероятными источниками кислорода в процессе фторирования могут выступать недостаточная очистка от кислорода и влаги, содержащихся в объеме ПФД, полимерной пленке и материалах установки, взаимодействие фтора с остаточной влагой и оксидами металлов в объеме установки с выделением кислорода, а также формированием долгоживущих радикалов во фторированном слое полимерной матрицы [58–60] и их последующим взаимодействием с кислородом воздуха. Таким образом, как правило, в ходе поверхностного фторирования образуются композиционные мембраны с тонким фторированным слоем. Ранее для случая жидкофазного фторирования полинорборнена и ацетата целлюлозы было показано, что химический состав обработанного материала изменяется от поверхности по толщине пленки, причем фторированный слой имел контраст, отличный от необработанного полимера, что позволило оценить глубину фторирования [57]. Однако особенностью изучаемых в текущей работе полимерных пленок является отсутствие контраста на микрофотографиях сканирующей электронной спектроскопии поперечного среза фторированных полимерных пленок. Более того, в случае ПВТМС и ПФО атомы фтора не детектировались методом энерго-дисперсионной спектроскопии. Это факт может быть следствием (а) либо низкой концентрации атомов фтора во всем объеме фторированного слоя, (б) либо высокой его концентрацией в узком интервале толщины фторированного слоя. В обоих случаях концентрация атомов фтора в локальном объеме образца, в котором происходит возбуждение атомов полимерных цепей рентгеновским излучением (“зона генерации”), оказывается недостаточной для детектирования. В случае PIM-1 методом ЭДС поперечных срезов были получены профили концентрации атомов фтора от поверхности вдоль перпендикуляра по толщине пленки. Все полученные зависимости от времени обработки фтором были аппроксимированы следующим выражением:
где C(F) – атомная концентрация фтора (ат. %), tF – время фторирования (мин), L – глубина фторирования (мкм), A, B, C – константы, равные 0.4586, 0.0347 и 0.0125 соответственно. Анализ этой зависимости позволяет рассчитать глубину фторирования L0, в которой производная концентрации фтора равна нулю. При всех временах фторирования толщина фторированного слоя не превышала 16 мкм, что, по-видимому, может быть связано с глубиной проникновения ПФД в высокопроницаемую полимерную матрицу PIM‑1. Таким образом, оценка толщины фторированного слоя обработанной полимерной пленки сопровождается рядом экспериментальных трудностей, преодоление которых требует дополнительных исследований.Исследование влияния модификации на параметры газопереноса через полимерные пленки
Одним из основных параметров процесса модификации влияющим на структуру и свойства полимерных мембран в случае поверхностного фторирования как газофазного, так и жидкофазного, является время фторирования [36, 38, 43, 44]. В работе изучено влияние времени фторирования в диапазоне 5–60 мин на эффективные коэффициенты проницаемости исследуемых полимерных пленок. Полученные результаты представлены на рис. 5.
Рис. 5.
Влияние времени фторирования на эффективные коэффициенты проницаемости кислорода (a) и азота (б) для исследованных образцов полимерных пленок.
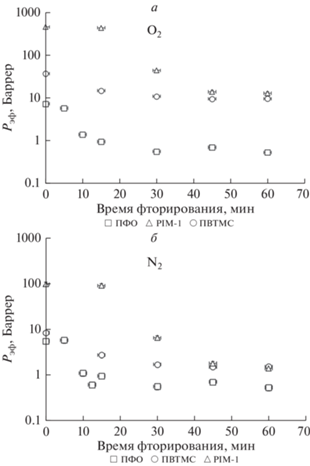
Видно, что c увеличением времени обработки как для кислорода, так и для азота наблюдается ожидаемое уменьшение эффективных коэффициентов проницаемости, как и в случае газофазного фторирования. Однако, отметим, что характер зависимостей существенным образом зависит от полимера. Так, в случае PIM-1 проведение “холостого” опыта с перфтордекалином без фторирующей фтор-азотной смеси показало, что в этом случае уровень газопроницаемости и для кислорода, и для азота падает в несколько раз относительно исходного образца, так, проницаемость О2 снизилась с 1590 Баррер до 460, а по азоту – с 610 до 97 Баррер. Для двух других полимеров эффекта заметного изменения газопроницаемости после аналогичных экспериментов с растворителем не наблюдается. Это объясняется повышенным свободным объемом PIM-1, размер элемента которого для свежеполученной пленки полимера составляет более 10 Ǻ [14, 61] и оказывается доступным для диффузии молекул ПФД размером 7.43 Ǻ. Уровень исходной газопроницаемости (холостой опыт) для PIM-1 сохраняется вплоть до 15 мин обработки F2, а затем идет монотонное снижение показателей, тогда как в случае же ПВТМС и ПФО отмечено снижение эффективных коэффициентов проницаемости уже после 5 мин модификации, а после 30 мин кривые выходят на плато. Для исследования факторов, приводящих к таким отличиям, в работе исследовано влияние времени фторирования на эффективные коэффициенты диффузии и рассчитаны эффективные коэффициенты растворимости газов в модифицированных полимерных пленках. Полученные данные представлены на рис. 6.
Рис. 6.
Влияние времени фторирования на эффективные коэффициенты диффузии кислорода (a) и азота (б) для исследованных образцов полимерных пленок.
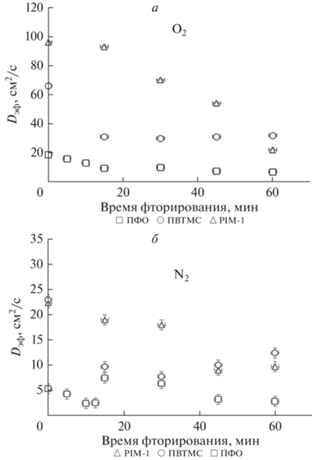
Рис. 7.
Влияние времени фторирования на эффективные коэффициенты растворимости кислорода (a) и азота (б) для исследованных образцов полимерных пленок.
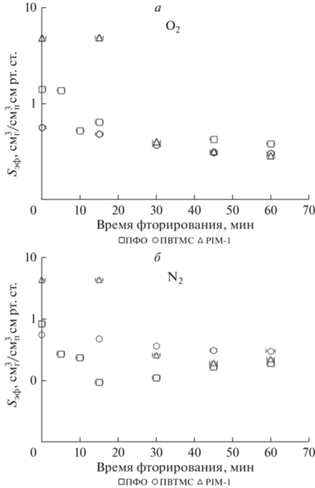
Из сравнения данных, представленных на рис. 5 и 6 видно, что для PIM-1 характер зависимости эффективных коэффициентов проницаемости и для О2 и для N2 от времени обработки соответствует характеру аналогичных зависимостей, полученных для эффективных коэффициентов диффузии газов. При этом эффективные коэффициенты растворимости (рис. 5) также сохраняются на уровне начальных значений при временах до 15 мин модификации, а затем снижаются на порядок, сохраняя этот уровень значений до 60 мин фторирования. Такое изменение газотранспортных параметров преимущественно связано с уменьшением свободного объема полимера в результате введения объемного атома фтора в структуру полимерных цепей. В случае ПВТМС наблюдается снижение и эффективных коэффициентов диффузии и эффективных коэффициентов растворимости обоих газов до 30 мин обработки и в дальнейшем, уровень этих параметров сохраняется на достигнутом уровне. Совпадение характера зависимостей этих параметров газопереноса указывает на то, что основное влияние на снижение проницаемости в случае ПВТМС, как и в случае PIM-1, оказывает уменьшение свободного объема во фторированном слое полимерной матрицы, причем, за 30 мин степень фторирования ПВТМС достигает предельных значений в результате возникающих диффузионных ограничений для фтора.
Анализ полученных экспериментальных данных показал, что для модифицированных пленок частично-кристаллического ПФО при общем снижении уровня газопроницаемости, характер зависимостей от времени фторирования для эффективных коэффициентов проницаемости, диффузии и растворимости различен. Так, уровень эффективных коэффициентов диффузии кислорода снижается со временем фторирования, тогда как для азота, при общей тенденции к снижению значений относительно исходных, в интервале 15–30 мин отмечено возрастание эффективного коэффициента диффузии до уровня начальных показателей после которого значения этого параметра уменьшились почти в 2 раза. Отмечено существенное снижение эффективного коэффициента растворимости обоих газов относительно исходного уровня для этого временного интервала обработки. Расчет селективностей проницаемости, диффузии и растворимости (табл. 5) показывает, что на этом участке наблюдается максимум селективности проницаемости (селективность O2/N2 возрастает почти в 2 раза относительно исходной) при неожиданном минимуме селективности диффузии и скачке селективности растворимости, которая при 15 мин фторирования выше исходной почти в 3 раза. Такие изменения не могут быть объяснены снижением свободного объема аналогично аморфным ПВТМС и PIM-1. С учетом того, что по данным РСА, степень кристалличности ПФО в процессе фторирования практически не изменяется, скачок селективности диффузии и растворимости, по-видимому, связан с процессом фторирования аморфной фазы ПФО, приводящему на определенном этапе фторирования к “разрыхлению” полимерных цепей за счет водородзаместительного фторирования и увеличения длины связи С–F относительно связи С–Н. Высокие значения селективности растворимости для ПФО на этом временном интервале обработки можно объяснить тем, что хотя в общем случае, косвенная оценка эффективных коэффициентов растворимости оправдана, в случае модификации частично-кристаллических полимеров этот подход может иметь ряд ограничений.
Таблица 5.
Влияние времени фторирования на селективность проницаемости, диффузии и растворимости газов (${{c}_{{{{{\text{F}}}_{2}}}}}$ = 10 об. %)
| Полимер | Время обработки, мин | α | αD | αS |
|---|---|---|---|---|
| ПФО | 0 | 4.1 | 3.2 | 1.3 |
| 5 | 4.4 | 3.1 | 1.9 | |
| 10 | 5.9 | 3.2 | 1.6 | |
| 15 | 8.1 | 1.6 | 5.4 | |
| 30 | 7.0 | 1.6 | 4.4 | |
| 45 | 5.4 | 2.3 | 2.4 | |
| 60 | 6.0 | 2.5 | 2.5 | |
| ПВТМС | 0 | 4.0 | 2.9 | 1.5 |
| 15 | 5.5 | 3.1 | 1.8 | |
| 30 | 5.7 | 2.9 | 2.0 | |
| 45 | 6.4 | 3.1 | 2.1 | |
| 60 | 6.3 | 2.6 | 2.5 | |
| PIM-1 | 0 | 4.8 | 4.3 | 1.1 |
| 15 | 5.2 | 4.5 | 1.2 | |
| 30 | 9.2 | 4.1 | 2.2 | |
| 45 | 8.9 | 4.4 | 2.0 | |
| 60 | 8.0 | 4.3 | 1.8 |
Как показывают данные, представленные в табл. 5, для всех изученных полимеров жидкофазное фторирование позволяет достичь повышенных значений селективности проницаемости O2/N2. Так, для ПФО и PIM-1 селективность проницаемости возрастает почти в 2 раза относительно исходных значений, для ПВТМС рост составил около 30%. Сравнительный анализ данных, представленных в табл. 5 показывает, что существенный вклад в увеличение селективности проницаемости в том числе и для высокопроницаемого полимера, вносит возрастание селективности растворимости, тогда как для таких пар как Не/СН4, Н2/СН4, СО2/СН4 основным фактором, приводящим к росту селективности проницаемости, вносит селективность диффузии [48].
Известно, что идеальная селективность даже в случае непористых газоразделительных мембран может отличаться от фактора разделения [62–66]. Как правило, такой эффект наблюдается для СО2-содержащих и углеводород-содержащих смесей, но для проверки сходимости данных, полученных для индивидуальных газов с данными для смеси O2/N2, в работе получены значения факторов разделения для смеси O2/N2 для исследованных исходных и фторированных полимеров (табл. 6) при повышенном давлении над мембраной. Из представленных данных видно, что значения этого параметра оказываются близки к значениям идеальной селективности, небольшое снижение отмечено только для ПФО, при этом уровень селективности и для этого полимера все равно оказывается существенно выше исходной.
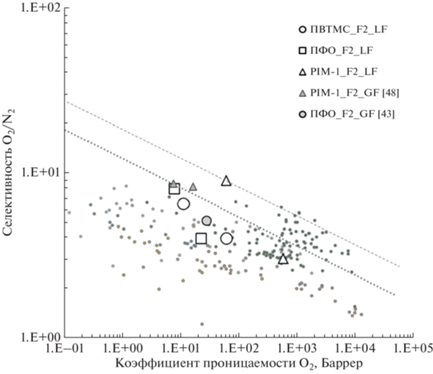
Проведенное сравнение достигнутых соотношений проницаемость/селективность (рис. 8) демонстрирует, что обработка поверхности и приповерхностных слоев полимерных пленок методом жидкофазного фторирования с более мягкими условиями проведения модификации относительно метода прямого газофазного фторирования с точки зрения снижения локального перегрева в процессе фторирования, приводит, по-видимому, к снижению уровня сшивок полимерных фрагментов, связанного с возможными разрывами полимерной цепи и отрывом боковых групп вследствие реакций рекомбинации и диспропорционирования долгоживущих радикалов во фторированном слое [58–60]. Это позволяет сохранить более высокий уровень проницаемости газов относительно исходного по сравнению с газофазной обработкой пленок. При этом, повышение селективности как диффузии, так и растворимости вследствие введения фтора в полимерную цепь приводит к возрастанию селективности разделения, в частности, пары O2/N2. Установлено, что максимальные значения селективности, в частности, α = 8 для ПФО, 6.4 для ПВТМС и 9 для PIM-1, существенно превышающие исходные значения, достигаются при подборе оптимальных условий, в том числе, времени фторирования, отличающего для различных полимеров.
Рис. 8.
Соотношение эффективных коэффициентов проницаемости О2 и идеальной селективности O2/N2 для исходных полимеров, полимеров, модифицированных методом жидкофазного фторирования (LF) и полимеров, модифицированных методом газофазного фторирования (GF) [43, 48]. На диаграмме представлены верхние границы распределения Робсона 2008 г. [67] и 2015 г. [68].
ЗАКЛЮЧЕНИЕ
Рассмотренный в работе метод жидкофазного фторирования продемонстрировал высокую эффективность применения предложенного подхода для существенного улучшения газоразделительных полимерных пленок из полимеров различной химической структуры, в том числе, частично кристаллических, на примере ПФО, ПВТМС и PIM-1 не только для метан-содержащих газовых смесей, но также для смеси O2/N2 при сохранении высокого уровня потока по кислороду относительно исходных пленок. Рост селективности составил от 30% для ПВТМС до увеличения в 2 раза для ПФО и PIM-1. Установлено, что такое улучшение показателей по селективности проницаемости достигается преимущественно за счет увеличения селективности растворимости, в отличие от метан-содержащих смесей, в случае которых наибольших вклад вносит селективность диффузии. Отметим, что для пары O2/N2 метод жидкофазного фторирования демонстрирует более высокие результаты, как по достигаемым значениям селективности, так и по соотношению эффективный коэффициент проницаемости/селективность относительно метода прямого газофазного фторирования. При этом достигнутое соотношение для PIM-1 находится на уровне границы 2015 г. диаграммы Робсона. [68]. Показано, что в случае жидкофазного фторирования высокопроницаемых полимеров с большим доступным объемом следует учитывать влияние перфтордекалина как растворителя и установлено, что, как и в случае газофазного фторирования, для достижения максимальных значений селективности требуется подбор оптимальных условий модификации, в частности, времени фторирования. Установлено, что полученные факторы разделения смеси O2/N2 близки к идеальным селективностям исследуемых газов, что позволит в дальнейшем использовать данный метод для модификации не только гомогенных полимерных пленок исследуемых полимеров, но и мембран с селективным непористым слоем на их основе.
Список литературы
Membrane technology and applications // Richard W. Baker. 3rd ed. 1. Membranes (Technology) 583 p.
Chong K.C., Lai O., Thiam S., Teoh C., Heng L. // J. Engineering Science and Technology. 2016. V. 11. № 7. P. 1016.
Thomas S., Pinnau I., Du N., Guiver M.D. // J. Membrane Science. 2009. V. 333(1–2). P. 125.
https://www.separel.com/en/about/.
https://www.generon.com/product_type/membrane-modules/.
https://www.parker.com/Literature/Hiross%20Zander %20Division/PDF%20Files/PIS/K3.1.333_Technology_ overview_membrane_modules_for_nitrogen_and_oxygen_systems_EN.pdf.
https://www.membrane-separation.com/en/nitrogen-generation-with-sepuran-n2/product-overview.
https://www.ube.com/contents/en/chemical/separation/n2_separator.html.
https://www.biogeneral.com/teflon-af/.
http://www.airproducts.ru/supply-modes/prism-membranes.
Pinnau I., Koros W.J. // J. Appl. Polym. Sci. 1991. V. 43. P. 1491.
Shishatskiy S., Nistor C., Popa M., Pereira Nunes S., Peinemann K.V. // Adv. Eng. Mater. 2006. V. 8. № 5. P. 390.
Zhang Y., Musselman I.H., Ferraris J.P., Balkus K.J., Jr. // J. Membrane. Sci. 2008. V. 313. P. 170.
Budd P.M., McKeown N.B., Ghanem B.S., Msayib K.J., Fritsch D., Starannikova L., Belov N., Sanfirova O., Yampolskii Y., Shantarovich V. // J. Membrane. Sci. 2008. V. 325. P. 851.
Lopez C., Wilkes G.L., Stricklen P.M., White S.A. // J. Macromolecular Science, Part C. 1992. P. 301.
Тепляков В.В., Дургарьян С.Г. // Высокомолекулярные соединения. 1984. Т. 26. № 7. С. 1498.
Wright T., Paul D.R. // J. Appl. Polym. Sci. 1998. V. 67. P. 875.
Polotskaya G.A., Agranova S.A., Gazdina N.V., Kuznetsov Yu.P., Nesterov V.V. // J. Appl. Polymer Science. 1996. V. 62. P. 2215.
Song K.W., Ka K.R., Kim C.K. // Industrial and Engineering Chemistry Research. 2010. V. 49. P. 6587.
Tong H., Hu C., Yang S., Ma Y., Guo H., Fan L. // Polymer. 2015. V. 69. P. 138.
Luo S., Liu Q., Zhang B., Wiegand J.R., Freeman B.D., Guo R. // J. Membrane Sci. 2015. V. 480. P. 20.
Sterescu D.M., Stamatialis D.F., Mendes E., Kruse J., Ra1tzke K., Faupel F., Wessling M. // Macromolecules. 2007. V. 40. P. 5400.
Singh-Ghosal A., Koros W.J. // J. Membrane Sci. 2000. V. 174. P. 177.
Pinnau I., Koros W.J. // Ind. Eng. Chem. Res. 1991. P. 30.
Niwa M., Kawakami H., Nagaoka S., Kanamori T., Shinbo T. // J. Membrane Sci. 2000. V. 17. P. 253.
Chung T.S., Teoh S.K., Hu X. // J. Membrane Sci. 1997. V. 133. P. 161.
Pesek S.C., Koros W.J. // J. Membrane Sci. 1993. V. 81. P. 71.
Ismail A.F., Dunkin I.R., Gallivan S.L., Shilton S.J. // Polymer. 1999. V. 40. P. 6499.
Zarshenas K., Raisi A., Aroujalian A. // RSC Adv. 2015. V. 5. P. 19760.
Lin X., Chen J., Xu J. // J. Membrane Sci. 1994. V. 90(1–2). P. 81.
Matsuyama H., Teramoto M., Hirai K. // J. Membrane Sci. 1995. V. 99(2). P. 139.
.Borisov S., Khotimskiy V.S., Rebrov A.I., Rykov S.V., Slovetskiy D.I., Pashunin Yu.M. // J. Membrane Sci. 1997. V. 125. P. 319.
Tressaud A., Durand E., Labrugère C., Kharitonov A.P., Kharitonova L.N. // J. Fluorine Chemistry. 2007. V. 128(4). P. 378.
Susanta B. Handbook of Specialty Fluorinated Polymers: Preparation, Properties, and Application // Elsevier Inc. 2015. 329 p.
Peyroux J., Duboi M., Tomasella E., Batisse N., Kharitonov A.P., Flahaut D., Romana L., Thomas F. // Surface & Coatings Technology. 2016. V. 292. P. 144.
Le Roux J.D., Paul D.R., Kampa J., Lagow R.J. // J. Membrane Science. 1994. V. 90(1–2). P. 21.
Kharitonov A.P., Kharitonova L.N. // Pure Appl. Chem. 2009. V. 81. № 3. P. 451.
Le Roux J.D., Paul D.R., Arendt M.F., Yuan Y., Cabasso I. // J. Membrane Sci. 1994. V. (1–2). P. 37.
Kharitonov A.P., Moskvin Yu.L., Teplyakov V.V., Le Roux J.D. // J. Fluorine Chemistry. 1999. V. 93. P. 129.
Langsam M. // Patent CA 1320804 C. 1993.
Le Roux J.D., Teplyakov V.V., Paul D.R. // J. Membrane Sci. 1994. V. 90. P. 55.
Langsam M., Anand M., Karwacki E.J. // Gas Separation and Purification. 1988. V. 2(4). P. 162.
Kharitonov P., Taege R., Ferrier G., Teplyakov V.V., Syrtsova D.A., Koops G.-H. // J. Fluorine Chem. 2005. V. 126(2). P. 251.
Сырцова Д.А. Автореферат диссертации на соискание степени к. х. н. 2001.
Srobel M., Corn S., Lyons C.S., Korba G.A. // J. Polym. Sci.: Part A: Polymer Chem. 1987. V. 25. P. 1295.
Das P.S., Adhikari B., Maiti S. // Macromleculare Chemie. 1991. V. 188. P. 27.
Blinov I.A., Mukhortov D.A., Yampolskii Yu.P., Belov N.A., Alentiev A.Yu., Chirkov S.V., Bondarenko G.N., Kostina Yu.V., Legkov S.A., Perepuchov A.M., Kambur M.P., Kambur P.S., Kapustin V.V., Vozniuk O.N., Kurapova E.S. // J. Fluorine Chemistry. 2020. V. 234. P. 109526.
Ma X., Li K., Zhu Zh., Dong H., Lv J., Wang Y., Pinnau I., Li J., Chen B., Han Y. // J. Mater. Chem. A. 2021. V. 9. P. 18 313.
Fuoco A., Satilmis B., Uyar T., Monteleone M., Esposito E., Muzzi C., Tocci E., Longo M., Santo M, Lanč M., Friess K, Vopička O., Izak P., Jansen J. // J. Membrane Sci. 2019. V. 594. P. 117460.
Belov N.A., Pashkevich D.S., Alentiev A.Y., Tressaud A. // Membranes. 2021. V. 11(9). P. 713.
Wojdyr M. // J. Appl. Crystallogr. 2010. V. 43(5). P. 1126.
Alentiev A.Yu., Levin I.S., Buzin M.I., Belov N.A., Nikiforov R.Yu., Chirkov S.V., Blagodatskikh I.V., Kechekyan A.S., Kechekyan P.A., Bekeshev V.G., Ryzhikh V.E., Yampolskii Yu.P // Polymer. 2021. V. 226. P. 123804.
Nagendra B., Cozzolino A., Daniel C., Rizzo P., Guerra G., De Rosa C., D’Alterio M., Tarallo O., Nuzzo A. // Macromolecules. 2019. V. 52. P. 9646.
Ruland W. // Acta Crystallographica. 1961. V. 14(11). P. 1180.
Безгин Д.А., Никифоров Р.Ю., Белов Н.А., Миленин С., Ямпольский Ю.П., Музафаров А.М. // Мембраны и мембранные технологии. 2021. V. 11. № 1. P. 3.
Sievert A.C., Tong W.R., Nappa M.J. // J. Fluorine Chemistry. 1991. V. 53(3). P. 397.
Белов Н.А., Блинов И.А., Суворов А.В., Никифоров Р.Ю., Чирков С.В., Алентьев А.Ю., Камбур М.П., Костина Ю.В., Левин И.С., Шапагин А.В., Ямпольский Ю.П. // Мембраны и мембранные технологии. 2021. Т. 11. № 2. С. 127.
Назаров В.Г. // Рос. хим. журн. 2006. Т. 50. С. 77.
Харитонов А.П. // Изв. Рос. Акад. наук. Энергетика. 2008. № 2. С. 149.
Peyroux J., Dubois M., Tomasella E., Petit E., Flahaut D. // Appl. Surf. Sci. 2014. V. 315. P. 426.
Starannikova L.E., Alentiev A.Yu., Nikiforov R.Yu., Ponomarev I.I., Blagodatskikh I.V., Nikolaev A.Yu., Shantarovich V.P., Yampolskii Yu.P. // Polymer. 2021. V. 212. P. 123 371.
Schultz J., Peinemann K.V. // J. Membrane Science. 1996. V. 110. № 1. P. 37.
Sridhar S., Veerapur R.S., Patil M.B., Gudasi K.B., Aminabhavi T.M. // J. Appl. Polymer Sci. 2007. V. 106. № 3. P. 1585.
Donohue M.D., Minhas B.S., Lee S.Y. // J. Membrane Sci. 1989. V. 42. № 3. P. 197.
Yeom C.K., Lee S.H., Lee J.M. // J. Appl. Polymer Science. 2000. V. 78. № 1. P. 179.
Falbo F., Tasselli F., Brunetti A., Drioli E., Barbieri G. // J. Chemical Engineering. 2014. V. 31. P. 4.
Robeson L.M. // J. Membrane. Sci. 2008. V. 320(1–2), 390.
Comesana-Gandara B., Chen J., Bezzu C.G., Carta M., Rose I., Ferrari M.-C., Esposito E., Fuoco A., Jansen J.C., McKeown N.B. // Energy Environ. Sci. 2019. V. 12. P. 2733.
Дополнительные материалы отсутствуют.
Инструменты
Мембраны и мембранные технологии