

Микология и фитопатология, 2022, T. 56, № 2, стр. 114-126
De Novo Transcriptome Assembly and Annotation of a New Plant Pathogenic Corinectria sp. Strain in Siberia
V. V. Biriukov 1, 2, *, I. N. Pavlov 3, 4, **, Yu. A. Litovka 3, 4, ***, N. V. Oreshkova 1, 2, 3, 5, ****, V. V. Sharov 1, 2, *****, E. P. Simonov 6, ******, D. A. Kuzmin 1, *******, K. V. Krutovsky 1, 5, 7, 8, ********
1 Siberian Federal University
660041 Krasnoyarsk, Russia
2 Krasnoyarsk Science Center of the Siberian Branch of the Russian Academy of Sciences
660036 Krasnoyarsk, Russia
3 V.N. Sukachev Institute of Forest of the Siberian Branch of the Russian Academy of Sciences
660036 Krasnoyarsk, Russia
4 Reshetnev Siberian State University of Science and Technology
660037 Krasnoyarsk, Russia
5 G.F. Morozov Voronezh State University of Forestry and Technologies
394087 Voronezh, Russia
6 University of Tyumen
625003 Tyumen, Russia
7 Georg-August University of Göttingen
37077 Göttingen, Germany
8 N.I. Vavilov Institute of General Genetics of the Russian Academy of Sciences
119333 Moscow, Russia
* E-mail: vladislav.v.biriukov@gmail.com
** E-mail: forester24@mail.ru
*** E-mail: litovkajul@rambler.ru
**** E-mail: oreshkova@ksc.krasn.ru
***** E-mail: vsharov@sfu-kras.ru
****** E-mail: ev.simonov@gmail.com
******* E-mail: dm.kuzmin@gmail.com
******** E-mail: konstantin.krutovsky@forst.uni-goettingen.de
Поступила в редакцию 28.06.2021
После доработки 10.11.2021
Принята к публикации 23.12.2021
- EDN: KEBQNM
- DOI: 10.31857/S0026364822020052
Аннотация
A new fungal disease affecting Siberian fir (Abies sibirica) and similar to canker in conifers caused by the fungus Corinectria fuckeliana has been observed in Central Siberia since 2006. Despite the similarity of symptoms related to Corinectria sp., the morphology and origin of this new fungal disease remained unknown. The aim of the study was de novo transcriptome sequencing and annotation of the causal agent of the disease and its phylogenetic comparison with the most closely related species for further study of the pathogenicity of that species for Abies sibirica. A pure culture of the anticipated fungus was isolated, and its transcriptome was sequenced and annotated. De novo transcriptome assemblies were generated using different software (Trinity, CLC Genomic Workbench Assembler, and RNASpades), and 57% of 14 120 transcripts assembled using the CLC Genomic Workbench Assembler were completely annotated. Phylogenetic analysis based on four genes encoded 28S ribosomal RNA, α-actin, β-tubulin, and translation elongation factor 1 alpha, respectively, demonstrated that the discovered strain is very similar to Corinectria fuckeliana species, but likely represents a new species or very different strain confirming the assumption that the species or strain found in Siberia is a new one that has not been studied previously.
INTRODUCTION
Siberian fir (Abies sibirica Ledeb.) is one of the most widespread evergreen conifer species in Russia. This species occupies a huge geographic territory, including the Northeastern part of European Russia, the Ural Mountains, the major part of the Siberia taiga zone, the Russian Far East region, as well as a great part of the mountain forests in Southern Siberia, and forms mainly stands mixed with conifers and other tree species and thus plays a very important ecological role in ecosystem functioning.
In the last decades, the extensive dieback of natural conifer stands caused mainly by root rot disease pathogens has been observed in Siberia and the Far East. It is likely that the dieback is also promoted by a combination of abnormal climatic conditions (including extreme droughts), decreased biological resistance of conifers against pathogens and other factors favorable for pathogenic organisms (Pavlov, 2015). Siberian fir is one of the most economically important tree species; its timber is extensively used in the construction, pulp and paper industry, and the essential oil extracted from needles is used in medicine. Therefore, the massive conifer forests dieback due to the aforementioned biotic and abiotic factors can lead to huge economic losses.
Corinectria fuckeliana (C. Booth) C. González et P. Chaverri is widely known as a wound parasite and an endophytic colonizer of conifer sapwood found in northern European countries, Canada, Chile, and New Zealand, where it parasitizes mainly on different spruce, fir, larch, and pine species (Schultz, 1990; Crane et al., 2009; Dick, Crane, 2009; Morales, 2009; González, Chaverri, 2017). A new Siberian fir disease, associated likely with this fungus, has been observed in the East Sayan Mountains region since 2006. The disease has been exhibiting symptoms that are similar to those caused by the fungus C. fuckeliana. They include massive stem-canker lesions of branches and twigs, their deformation and dieback, and the appearance of multiple elongated wounds of various shapes with resin flow. The affected trees with dieback branches are subject to windbreak and gradual biodegradation with a sharp deterioration in the wood quality. In subsequent years, the disease has spread 450 km northwards from the area where it was first reported. Despite the similarity of symptoms to Corinectria, little was known about the basic biology, morphology, and origin of the causal agent of the disease. Pavlov and his colleagues (Pavlov et al., 2020) isolated 47 different morphotypes of pure cultures of fungi from affected trees, and the morphotype representing Acremonium-like anamorph was the most frequently isolated fungus. Moreover, they conducted a phylogenetic analysis of four isolates using nucleotide sequences of three protein-coding genes, α-actin (Act), β-tubulin (Btub) and translation elongation factor 1 alpha (Tef1α) genes, and the complete internal transcribed spacer of rDNA (ITS) and demonstrated that these Acremonium-like Siberian isolates formed a separate subclade within a clade composed of previously described Corinectria genus species (Pavlov et al., 2020).
There are 221 genomes representing 15 different genera of the Nectriaceae family (which also includes the Corinectria genus) available in the NCBI Genome Database (accessed on March 2021), including 187 genomes of Fusarium species, 13 genomes for Calonectria genus, but only six for Neonectria genus, two per each of Ilyonectria, Dactylonectria and Pseudonectria genera, and only one genome per each of Aquanectria, Coccinonectria, Corinectria, Cylindrocarpon, Cylindrodendrum, Nectria, Stylonectria, Thelonectria and Xenoacremonium genera. Moreover, the transcriptome data are less available for these species. Only two transcriptome shotgun assembly (TSA) entries for the Nectriaceae family are available in the NCBI database (for Fusarium virguliforme and F. acuminatum species, respectively).
The main aim of the present study was de novo transcriptome sequencing and annotation of the most pathogenic isolate previously determined and described in Pavlov et al. (2020) (hereinafter, Siberian isolate or strain N1/NfP5.7), and its phylogenetic comparison with the most closely related species. De novo transcriptome assemblies were generated using different software to further select the best assembly for annotation. Phylogenetic analysis demonstrated that the discovered isolate was similar to Corinectria fuckeliana, but may represent a new strain or even species confirming that the Siberia’s isolates have not been studied previously. Thereby, the obtained data (both TSA and SRA) make possible further studies of the pathogenicity for Abies sibirica and other conifers of a previously undescribed Corinectria strain or species. Moreover, the results may provide some additional insight into the Corinectria genus.
MATERIAL AND METHODS
Cultures and media. The strain N1/NfP5.7 was isolated into a pure culture from affected areas of A. sibirica branches (Central Siberia, Krasnoyarsk, 56°01′41.46″N; 92°34′39.30″E) (Pavlov et al., 2020). The morphological and cultural features of Corinectria strain N1/NfP5.7 were studied on potato-dextrose agar (PDA), carrot agar (CA), and slow nutrient agar (SNA) for 3–4 weeks at 22 ± 1°C. Colony growth was measured on 90 mm diameter Petri dishes containing PDA, 5 mm deep, inoculated with a 10 mm diameter mycelial plug after 14 days. Microconidia, mycelium and conidiophores were observed using an Olympus CX41 microscope (Olympus Co., Tokyo, Japan) and a scanning electron microscope Hitachi SU3500 (Hitachi, Tokyo, Japan).
RNA isolation and sequencing. Total RNA was isolated from the mycelium of the 21-day-old culture of the strain N1/NfP5.7 using the Plant/Fungi Total RNA Purification Kit (Norgen Biotec Corp., Thorold, Ontario, Canada). Quality of the extracted RNA was evaluated using Agilent Bioanalyzer 2100 and RNA 6000 Nano Kit assay (Agilent Technologies, Santa Clara, California, USA). The NEBNext Poly(A) mRNA Magnetic Isolation Module was used to separate intact poly(A) + RNA from the total extracted RNA. The TruSeq Stranded Total RNA with Ribo-Zero Plant kit (Illumina, Inc., San Diego, California, USA) was used to remove ribosomal RNA (rRNA), convert mRNA to complementary DNA (cDNA), and generate the paired-end sequencing library that was then sequenced on an Illumina MiSeq with 2 × 75 cycles using the MiSeq Reagent Kit v3 for 150 cycles (Illumina, Inc., San Diego, California, USA).
Transcriptome assembly and annotation. FastQC software v.0.11.5 was used to evaluate the quality of sequencing data. The pre-processing of sequencing data, which included the removal of adapters and low-quality sequences, was carried out using the Trimmomatic program v0.36 (Bolger, 2014) with a minimum quality cut-off threshold of Q = 19 and a minimum length of 30 bp. Then, SortMeRNA version 2.1 and the SILVA rRNA datasets were used to remove the rRNA fraction from our data (Kopylova et al., 2012; Quast et al., 2013).
De novo assemblers Trinity v2.8.2, RNASpades (SPAdes v. 3.12.0), and the CLC Genomic Workbench Assembler v. 5.1.1 were used to assemble the transcriptome (Haas et al., 2013; Bushmanova et al., 2019). Trinity software was used twice: without and with normalization of the reads (maximum coverage 200X), respectively. Moreover, the RNASpades output included three sets of transcripts, depending on the filtration level (soft, normal, and hard filtration). Benchmarking Universal Single-Copy Orthologs (BUSCO) software v3 was used to evaluate the completeness of the obtained transcriptomes based on the ascomycetes orthologous genes dataset (ascomycota_odb9) (Seppey et al., 2019). OmicsBox platform was used to carry out transcriptome annotation (Götz et al., 2008). Assembled transcriptome sequences were aligned to the non-redundant fungi nucleotide database with a BLASTx e-value of 1e-05 to search for homology with the most closely related species. In addition, the InterProScan program implemented into OmicsBox was used to identify known conservative active sites, domains, and repeats in the obtained transcripts (Jones et al., 2014). Gene ontology (GO) mapping and annotation were also performed using OmicsBox.
Phylogenetic analysis. In order to verify the generic taxonomic status of the discovered N1/Npf5.7 strain (presumably Corinectria sp.) in the Nectriaceae family, a multi-gene alignment, including sequences of 28S ribosomal RNA (28S rRNA) gene, and three protein-encoding genes Act, Btub and Tef1α taken from the NCBI GenBank was generated and used for the phylogenetic analysis. The NCBI GenBank accession numbers for each gene and nucleotide sequence used in the study are presented in Table 1. The nucleotide sequences for protein-encoded genes representing Siberian isolate N1/NfP5.7 were extracted from the assembled transcriptome data using the Blast program. The 28S rRNA sequence was extracted using RNAmmer software version 1.2 (Lagesen et al., 2007). Multiple sequence alignment (MSA) was generated using the MAFFT v7.407 program and the iterative refinement method with the weighted sum of pairs score (L-INS-i) (Katoh et al., 2005; Katoh, Standley, 2013). Poorly aligned regions were trimmed using the GBlocks server considering reading frame position and then manually finalized. Concaterpillar v. 1.7.2 was used to perform the topological congruence test and branch length compatibility assessment with the GTR substitution model (Leigh et al., 2008). The resulting MSAs were concatenated to make a supermatrix partitioned based on a combination of genes (28S rRNA gene and three different codon positions for each protein-encoding gene). Partition Finder v2.2.1 was subsequently used to find the proper nucleotide substitution models for the partition scheme. Phylogenetic trees were reconstructed using IQ-TREE v1.5.6 using the maximum-likelihood method (Nguyen et al., 2015). Node support was inferred with ultrafast bootstrap approximation with 100 000 replicates. In addition, Bayesian phylogenetic inference was conducted using MrBayes v3.2.7a with its default settings using the substitution model found with PartitionFinder (Huelsenbeck, Ronquist, 2001).
Table 1.
Genera, species and genes used for phylogenetic analysis and their NCBI GenBank accession numbers
| *Genus | Species name | Strain | Gene | GenBnk ID | |
|---|---|---|---|---|---|
| MycoBank | NCBI GenBank | ||||
| Cinnamomeonectria | Cinnamomeonectria cinnamomea | Neonectria cinnamomea | IMI 325256 | 28S | KJ022074.1 |
| IMI 325256 | ACT | KJ022288.1 | |||
| IMI 325256 | TEF1 | KJ022395.1 | |||
| IMI 325256 | TUB | KJ022343.1 | |||
| Corinectria | Corinectria fuckeliana | Neonectria fuckeliana | GJS02-67 | 28S | HM364320.1 |
| GJS02-67 | ACT | HM352886.1 | |||
| GJS02-67 | TEF1 | HM364354.1 | |||
| GJS02-67 | TUB | HM352867.1 | |||
| Corinectria fuckeliana | IMI 342668 | 28S | KJ022070.1 | ||
| IMI 342668 | ACT | KJ022285.1 | |||
| IMI 342668 | TEF1 | KJ022404.1 | |||
| IMI 342668 | TUB | KJ022340.1 | |||
| Nectria | Nectria antarctica | N. antarctica | A.R. 2767 | 28S | HM484560.1 |
| A.R. 2767 | ACT | HM484501.1 | |||
| A.R. 2767 | TEF1 | HM484516.2 | |||
| A.R. 2767 | TUB | HM484601.1 | |||
| CBS 308.34 | 28S | MH867042.1 | |||
| CBS 308.34 | ACT | JF832482.1 | |||
| CBS 308.34 | TEF1 | JF832519.1 | |||
| CBS 308.34 | TUB | JF832886.1 | |||
| N. balansae | N. balansae | CBS 124070 | 28S | JF832710.1 | |
| CBS 124070 | ACT | JF832484.1 | |||
| CBS 124070 | TEF1 | JF832521.1 | |||
| CBS 124070 | TUB | JF832907.1 | |||
| N. berberidicola | N. berberidicola | XJAU 2433-1 | 28S | MH793632.1 | |
| XJAU 2433-1 | ACT | MH793662.1 | |||
| XJAU 2433-1 | TEF1 | MH793617.1 | |||
| XJAU 2433-1 | TUB | MH818840.1 | |||
| N. cinnabarina | N. cinnabarina | A.R. 4302 | 28S | HM484736.1 | |
| A.R. 4302 | ACT | HM484627.1 | |||
| A.R. 4302 | TEF1 | HM484654.1 | |||
| A.R. 4302 | TUB | HM484820.1 | |||
| N. dematiosa | N. dematiosa | XJAU 2025-4 | 28S | MH793625.1 | |
| XJAU 2025-4 | ACT | MH793655.1 | |||
| XJAU 2025-4 | TEF1 | MH793610.1 | |||
| XJAU 2025-4 | TUB | MH818833.1 | |||
| N. magnispora | N. magnispora | MAFF 241418 | 28S | JF832686.1 | |
| MAFF 241418 | ACT | JF832498.1 | |||
| MAFF 241418 | TEF1 | JF832541.1 | |||
| MAFF 241418 | TUB | JF832898.1 | |||
| N. mariae | N. mariae | A.R. 4274 | 28S | JF832684.1 | |
| A.R. 4274 | ACT | JF832499.1 | |||
| A.R. 4274 | TEF1 | JF832542.1 | |||
| A.R. 4274 | TUB | JF832899.1 | |||
| N. nigrescens | N. nigrescens | XJAU 2255-4 | 28S | MH793628.1 | |
| XJAU 2255-4 | ACT | MH793658.1 | |||
| XJAU 2255-4 | TEF1 | MH793613.1 | |||
| XJAU 2255-4 | TUB | MH818836.1 | |||
| N. pseudotrichia | N. polythalama | ICMP 2505 | 28S | JF832696.1 | |
| ICMP 2505 | ACT | JF832500.1 | |||
| ICMP 2505 | TEF1 | JF832524.1 | |||
| ICMP 2505 | TUB | JF832901.1 | |||
| N. pseudocinnabarina | N. pseudocinnabarina | G.J.S. 09-1358 | 28S | JF832700.1 | |
| G.J.S. 09-1358 | ACT | JF832503.1 | |||
| G.J.S. 09-1358 | TEF1 | JF832536.1 | |||
| G.J.S. 09-1358 | TUB | JF832904.1 | |||
| N. pseudotrichia | N. pseudotrichia | G.J.S. 09-1329 | 28S | JN939827.1 | |
| G.J.S. 09-1329 | ACT | JF832506.1 | |||
| G.J.S. 09-1329 | TEF1 | JF832530.1 | |||
| G.J.S. 09-1329 | TUB | JF832902.1 | |||
| N. pulcherrima | N. pulcherrima | IMI 325242 | 28S | KJ022071.1 | |
| IMI 325242 | ACT | KJ022290.1 | |||
| IMI 325242 | TEF1 | KJ022392.1 | |||
| IMI 325242 | TUB | KJ022345.1 | |||
| Thyronectria | Thyronectria zanthoxyli | Nectria zanthoxyli | A.R. 4280 | 28S | HM484571.1 |
| A.R. 4280 | ACT | HM484513.1 | |||
| A.R. 4280 | TEF1 | HM484523.2 | |||
| A.R. 4280 | TUB | HM484599.1 | |||
| Nectriopsis | Nectriopsis rexiana | N. exigua | G.J.S. 98-32 | ACT | GQ505979.1 |
| G.J.S. 98-32 | TEF1 | HM484852.1 | |||
| G.J.S. 98-32 | TUB | HM484883.1 | |||
| G.J.S. 98-32 | 28S | GQ505986.1 | |||
| Neonectria | Neonectria candida | N. ramulariae | ATCC 16237 | 28S | HM364310.1 |
| ATCC 16237 | ACT | HM352879.1 | |||
| ATCC 16237 | TEF1 | HM364349.1 | |||
| ATCC 16237 | TUB | DQ789857.1 | |||
| N. ditissima | N. ditissima | CBS 100.316 | 28S | HM364311.1 | |
| CBS 100.316 | ACT | HM352880.1 | |||
| CBS 100.316 | TEF1 | HM364350.1 | |||
| CBS 100.316 | TUB | HM352864.1 | |||
| N. faginata | N. faginata | CBS 134246 | 28S | KC660600.1 | |
| CBS 134246 | ACT | KC660409.1 | |||
| CBS 134246 | TEF1 | KC660457.1 | |||
| CBS 134246 | TUB | KC660743.1 | |||
| N. hederae | N. hederae | CBS 125175 | 28S | KC660615.1 | |
| CBS 125175 | ACT | KC660428.1 | |||
| CBS 125175 | TEF1 | KC660459.1 | |||
| CBS 125175 | TUB | KC660760.1 | |||
| N. microconidia | N. microconidia | MAFF 241522 | 28S | KC660596.1 | |
| MAFF 241522 | ACT | KC660427.1 | |||
| MAFF 241522 | TEF1 | KC660477.1 | |||
| MAFF 241522 | TUB | KC660759.1 | |||
| N. punicea | N. punicea | CBS 119525 | 28S | KC660592.1 | |
| CBS 119525 | ACT | KC660360.1 | |||
| CBS 119525 | TEF1 | KC660432.1 | |||
| CBS 119525 | TUB | KC660696.1 | |||
| N. shennongjiana | N. shennongjiana | HMAS 173254 | 28S | KJ022076.1 | |
| HMAS 173254 | ACT | KJ022291.1 | |||
| HMAS 173254 | TEF1 | KJ022406.1 | |||
| HMAS 173254 | TUB | KJ022346.1 | |||
| Thyronectria | Thyronectria balsamea | Nectria balsamea | A.R. 4478 | 28S | HM484567.1 |
| A.R. 4478 | ACT | HM484508.1 | |||
| A.R. 4478 | TEF1 | HM484528.2 | |||
| A.R. 4478 | TUB | HM484591.1 | |||
RESULTS
Characteristics in culture and microscopy. Colonies of Corinectria strain N1/NfP5.7 on PDA were slow-growing (1.9 mm/day at 22 ± 1°C) and irregular rounded with a mealy appearance (Fig. 1, a). Aerial mycelium was sometimes sparse, with white/creamy yellow/buff color, sometimes rust near the inoculum block, reverse with diffuse pigment from the center to the margin of the colony.
Fig. 1.
The colony of the Corinectria strain N1/NfP5.7 on PDA (a), carrot-agar (b), MEA (c), and SNA (d).
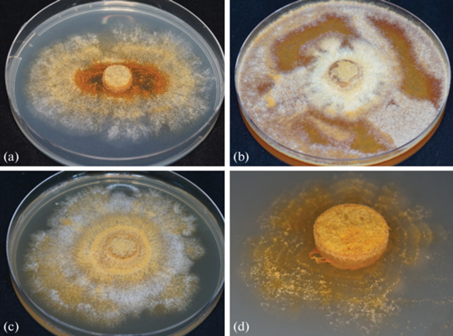
The morphology of the strain colony on PDA differed from originally described immediately after isolating the strain (Pavlov et al., 2020), which is probably due to the high variability of this trait during storage and regular subcultures of the culture, as well as the composition of the nutrient medium and cultivation conditions (24°C for 14 days, without illumination).
On CA colony was irregular rounded with concentric circles (Fig. 1, b), with abundant, white/yellow/orange aerial mycelium and the growth rate of 2.7 mm/day. On 2% malt extract agar mycelium was abundant, fluffy, white, with time light to dark yellow and ocher (Fig. 1, c). The growth rate on MEA was 2.3 mm/day. On SNA aerial mycelium was sparse (Fig. 1, d), but with abundant sporulation (Fig. 2) in slimy droplets and the growth rate of 0.5 mm/day.
Fig. 2.
Scanning electron microscopy of microconidia in false heads, mycelium and conidiophores (magnification ×470–4700) of the Acremonium-like anamorph fungal strain N1/NfP5.7.

Perithecia in situ formed clusters on wood, often with both developing and mature perithecia in the same cluster; the number of perithecia in a cluster varied. Color was from red to dark red; uniformly red in 3% KOH and yellow in 100% lactic acid. The surface was smooth and shiny, sometimes scurfy and subglobose.
Microconidia on SNA were abundant, hyaline, smooth, non-septate, ellipsoid with a pointed end, pear-shaped, formed on Acremonium-like conidiophores, often in false heads (Fig. 2). The average size of microconidia was 4.2 × 2.4 µm. Macroconidia and chlamydospores were not observed. Conidiophores were hyaline, often branching at right angles, variable in length, up to 110 µm long and up to 5 µm wide at the base, tapering towards the apex, single or sparingly branched, septate. The average length of conidiophores was 59.7 ± 2.3 µm.
Transcriptome sequencing and assembly. As a result of the primary data processing, ~27 Mbp of high-quality paired-end transcript reads in the length range of 30–60 bp with Q > 30 were obtained (i.e., the sequencing error probability was no more than 0.1%). The RNA sequencing data before and after processing are presented in Table 2.
Table 2.
RNA-seq data
| Parameter | Before processing | After processing | After SortMeRNA |
|---|---|---|---|
| Number of reads | 31 925 258 | 26 896 380 | 26 441 532 |
| Read length range, bp | 35–76 | 30–60 | 30–60 |
According to the SortMeRNA results, the total rRNA content in the sequencing data was 1.51%. Information on the rRNA content is presented in Table 3. The filtered and cleaned RNA-seq data have been deposited at the NCBI Sequence Read Archive database under the SRA study accession number SRR12783070.
Table 3.
rRNA percentage in the sequencing data
| Representative database | rRNA, % |
|---|---|
| silva-bac-16s-id90 | 0.40 |
| silva-bac-23s-id98 | 0.21 |
| silva-arc-23s-id98 | 0.00 |
| silva-arc-16s-id95 | 0.01 |
| silva-euk-18s-id95 | 0.13 |
| silva-euk-28s-id98 | 0.71 |
| rfam-5s-id98 | 0.00 |
| rfam-5.8s-id98 | 0.00 |
| Total | 1.51 |
The summary information about the assembled Siberian isolate N1/NfP5.7 transcriptomes is presented in Table 4. The longest transcriptome (24 813 047 bp) was obtained using the Trinity assembler with normalization of reads, while the shortest one, consisting of 18 697 543 bp, was obtained using the de novo assembler implemented into the CLC Genomic Workbench. Moreover, the assembly obtained with this assembler contained the smallest number of transcripts – 14 129, while the transcriptome assembled using RNASpades (with soft filtering parameter) contained the largest number of transcripts (27 040). Despite such a variation in the total length of the obtained transcriptomes and the number of transcripts composing them, the N50 values of these assemblies were very similar and ranged from 2211 to 2334 bp.
Table 4.
Main parameters of the Siberian isolate N1/NfP5.7 (Corinectria sp.) transcriptome assemblies
| Assembler | Number of contigs | Total length, bp | The longest transcript, bp | N50, bp | N90, bp |
|---|---|---|---|---|---|
| Trinity without normalization | 19 295 | 24 561 211 | 15 246 | 2226 | 521 |
| Trinity with normalization | 19 359 | 24 813 047 | 15 246 | 2235 | 526 |
| CLC | 14 120 | 18 697 543 | 16 323 | 2211 | 552 |
| RNASpades, normal filtration | 20 517 | 23 418 981 | 16 108 | 2286 | 482 |
| RNASpades, soft filtration | 27 040 | 24 166 325 | 16 108 | 2226 | 358 |
| RNASpades, hard filtration | 17 379 | 22 932 680 | 16 108 | 2334 | 570 |
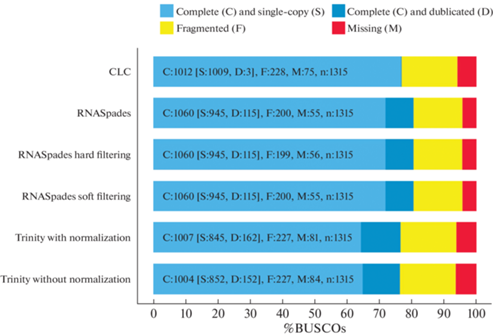
The BUSCO program results are presented in Fig. 3. The reference ascomycota_odb9 database used for the analysis contained information on 1315 orthologous genes. The BUSCO program results indicated that the most completed transcriptomes were assembled using the RNASpades program (80.6% of completeness). The percentage of duplicates in these transcriptomes was relatively high (8.7%), but less than in assemblies generated by the Trinity program (11.5% and 12.3% of duplicates among transcripts assembled without and with normalization, respectively). The least percentage of duplicates (0.2%) was in the transcriptome generated by the CLC Genomic Workbench program also with relatively high completeness (76.9%). Thus, this assembly of the N1/NfP5.7 strain transcriptome was used for further annotation. The transcriptome assemble has been deposited at DDBJ/EMBL/GenBank under the accession GIXD00000000.
Fig. 3.
Assessment of transcriptome completeness based on the orthologous ascomycete genes dataset (ascomycota_odb9).
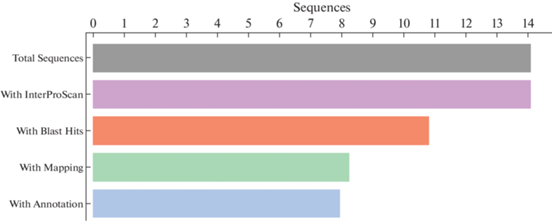
Transcriptome annotation. Summary of the transcriptome annotation of the Siberian isolate N1/NfP5.7 is shown in Fig. 4. A total of 14 120 transcripts were analyzed, for each of which a match in databases containing information about conservative domains and repeats was found. According to the BLAST results, 10 853 (~77%) sequences have homologs in the non-redundant fungi nucleotide database. For 8296 transcripts (~59%), the GO terms were retrieved. Finally, ~57% of the data (7993 transcripts) were annotated.
According to the InterProScan results, the most common repeats in the studied transcripts were WD40 (represented in 88 sequences) as well as ankyrin repeats (represented in 49 transcripts). The complete list of repeats and their abundance are presented in the Supplementary Fig. S1 (Biriukov et al., 2021).
BLAST analysis demonstrated that the largest number of nucleotide homologs for the obtained transcriptome sequences were found in the previously studied ascomycetes Neonectria ditissima (9628 homologs), Fusarium fujikuroi (5904), Nectria haematococca (5294), Fusarium verticillioides (3948), and F. oxysporum f. sp. pisi HDV247 (3828) homologs. Other species in which homologs of the obtained transcripts were found are presented in the Supplementary Fig. S2 (Biriukov et al., 2021). As a result of GO mapping, 608 469 GO terms associated with the BLAST search hits were retrieved. The databases used for GO terms search and the number of terms found in them are presented in the supplementary materials to our previous paper (Biriukov et al., 2021).
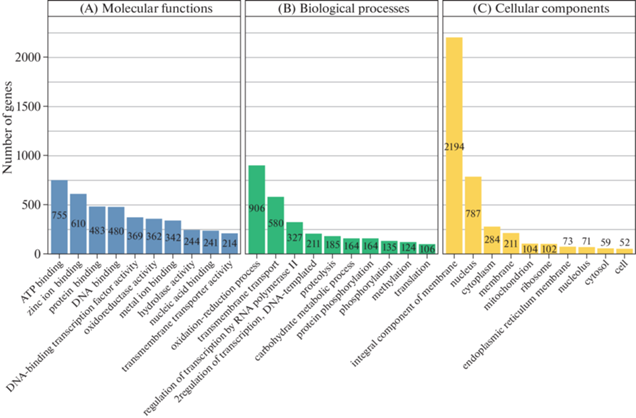
Fig. 5, A demonstrates the classification of the retrieved GO terms by the specific functions of the gene product at the molecular level. The most enriched molecular functions were ATP binding (755 terms), the zinc ion binding (610 terms), and the protein and DNA binding (483 and 480 terms, respectively). The classification of the found GO terms by biological functions showed that 906 of the studied transcripts are presumably involved in oxidation-reduction processes, 580 transcripts are involved in transmembrane transport, and 327 transcripts regulate RNA polymerase II transcription. The top-10 represented biological functions are shown in Fig. 5, B. The classification of retrieved GO terms by cellular components showed that the predominant number of transcripts studied corresponds to the integral components of the membrane (2194 transcripts), while 787 and 284 transcripts were identified as nuclear and cytoplasmic components, respectively. Other top-10 cellular components and their representation in our transcriptome data are presented in Fig. 5, C.
Fig. 5.
Top-10 GO terms in each domain: molecular functions (A), biological processes (B), and cellular components (C).
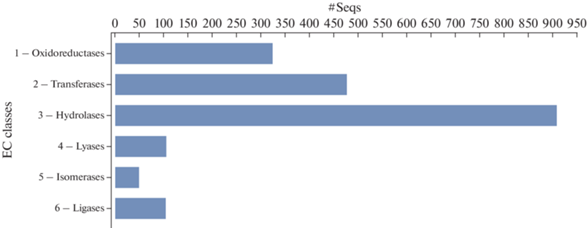
In addition, enzyme annotation through the direct GO to Enzyme Code (EC) mapping was carried out. It has been shown that the predominant enzyme classes in our dataset were hydrolases involved in intracellular processes and destruction of host cell wall composed mainly of cellulose and lignin, as well as in biomass degradation (907 transcripts), transferases (477), and oxidoreductases (323). The distribution of data on other classes of enzymes is shown in Fig. 6.
Phylogenetic analysis. Phylogenetic analysis was performed using the supermatrix comprised of nucleotide sequences of four genes of 27 fungal taxa in a MSA with a total length of 2152 bp with 331 parsimony-informative sites and 477 variable parsimony-uninformative sites. The best evolutionary nucleotide substitution models selected for tree interference are presented in Table 5. The resulting supermatrix as well as the partition scheme is available as a Supplementary Nexus File S1 at figshare (Biriukov et al., 2021).
Table 5.
Selected nucleotide substitution models for tree inference
| Genes and codons | Selected model |
|---|---|
| Btub_c1, 28S rRNA, Act_c1 | GTR + I + G |
| Tef_c2, Act_c2 | K81UF + I |
| Btub_c3, Tef1_c3 | TVM + G |
| Tef1_c1, Tub_c2 | F81 + I |
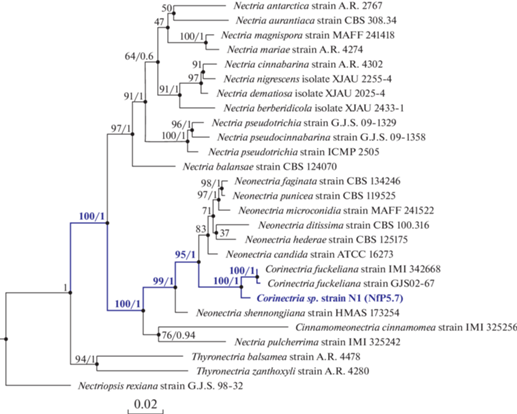
Both the maximum likelihood (ML) (Fig. 7) and the Bayesian phylogenetic trees have shown a clear separation of the Neonectria + Corinectria from other genera (100/1 based on ML ultrafast bootstrap and Bayesian posterior probabilities, respectively). The Bayes consensus tree inferred in our study is also available as Supplementary Fig. S4 at figshare (Biriukov et al., 2021). Inferred phylogenetic trees also demonstrated that Corinectria species form a monophyletic clade. The analysis showed also that the isolated Siberian N1/NfP5.7 strain belongs to the genus Corinectria and is the most similar to Сorinectria fuckeliana, but it is still well separated from this (100/1) and other species in the genus Corinectria and may represent a new strain or even species confirming that the Siberia’s isolates have not been studied previously. Moreover, it confirmed the earlier phylogenetic analysis conducted in (Pavlov et al., 2020) in which the supertree approach was used based on the same three protein-coding genes, but instead of the 28S rRNA gene used in the current study the internal transcribed spacer (ITS) marker was used in (Pavlov et al. 2020). Constructed tree from that study also showed a strong support for the clade comprised of four Siberian isolates.
Fig. 7.
Maximum Likelihood (ML) phylogenetic consensus tree based on the concatenated nucleotide sequences of four genes. The tree is rooted to Nectriopsis rexiana (N. exigua G.J.S. 98–32 in the NCBI GenBank). Values at branches indicate ML ultrafast bootstrap. For the clades, whose structure was identical to the Bayesian consensus tree, the Bayesian posterior probabilities are also indicated. Taxa names are given according to the current names specified in the MycoBank database, while strain identifiers correspond to the NCBI GenBank entries.
DISCUSSION
Very little genomic and transcriptomic data are currently available for the Nectriaceae family, especially for the Corinectria and Neonectria genera. In this work, we performed sequencing and assembly of the transcriptome for a strain of the likely new species exhibiting Acremonium-like anamorphs and causing massive cancerous disease of Abies sibirica in Central Siberia that leads to cambium necrosis and dieback. The main purpose of transcriptome sequencing in this study was to provide additional genomic resource for developing genetic markers for phylogenetic and population analyses and for prospective comparisons with other fungal transcriptomes, which are very limited for now and do not allow to use current transcriptome data for species classification. The transcriptome assembly was carried using different assemblers with the aim of further selection of a more suitable assembly for annotation. As shown in the study, assembly using Trinity and RNASpades yielded the longest transcriptomes; however, the duplicates percentage in those assemblies was high (this may be associated with program algorithms that depend on the error number in data, as well as the number of gene isoforms). For annotation purposes, the assembly generated by the CLC assembler was selected since it has the smallest number of duplications. Most transcripts (57%) were functionally annotated, and their main biological processes, cellular localization, and molecular functions were determined.
The annotation results showed that the isolated strain produces a wide range of enzymes, including enzymes involved in oxidation-reduction processes, transmembrane transport, and ATP binding. This fact explains a large number of detected transcripts, the protein products of which are part of an integral component of the membrane. In addition, the enriched enzyme classes were determined. For instance, the most abundant enzyme class was hydrolases involved in intracellular processes and destruction of host cell wall composed mainly of cellulose and lignin, as well as in biomass degradation that may be an important survival strategy for this species.
Annotation results demonstrated that obtained transcripts are enriched in WD40 repeats. These repeats consist of approximately 40–60 amino acid residues flanked by a conserved glycine-histidine sequence near its N-terminus (strand D) and another conserved tryptophan-aspartic acid dipeptide at its C-terminus (strand C). WD40 repeats-containing proteins are widely distributed among eukaryotes and perform diverse cellular functions: they participate in such important processes as G-protein-mediated signal transduction, cell division, RNA processing, transcription regulation (Mylona et al., 2006), ubiquitin-dependent protein degradation (Higa et al., 2006), chromatin modifications (Ruthenburg et al., 2006), and other processes (Smith, 2008; Stirnimann et al., 2010). In addition, they mediate the formation and regulation of dynamic multi-protein functional complexes through protein scaffolding based on protein-protein interactions (Stirnimann et al., 2010; Jain, Pandey, 2018). Interestingly, it was also demonstrated that WD40 repeats might regulate fungi fruiting body formation by controlling essential steps in eukaryotic cell differentiation (Pöggeler, Kück, 2004).
The transcriptome data obtained in this work also provided an invaluable resource in phylogenetic analysis and molecular identification of the isolated species. Phylogenetic analysis was mainly aimed at determining the taxonomic status of the pathogen genus rather than species. Phylogeny results confirm the hypothesis that the discovered Siberian isolate N1/NfP5.7 may represent a new species within the genus Corinectria due to the well-supported separation in both ML and Bayesian phylogenetic trees from other Corinectria species included in the phylogenetic study. Moreover, it confirmed the earlier phylogenetic analysis by Pavlov et al. (2020), where the constructed trees showed strong support for the clade comprised of four Siberian isolates separated from other Corinectria species. Pavlov et al. (2020) used the supertree approach and the same three protein coding genes used in the current study plus internal transcribed spacer (ITS) instead of the 28S rRNA gene used in our study. Moreover, in Pavlov et al. (2020), only a single model of nucleotide substitutions was used for all genes to construct a phylogenetic tree. In our research, we determined the best substitution model for each gene separately and, moreover, even for each codon position for protein-coding genes. This approach is more accurate and better reflects the evolutionary processes in organisms. Therefore, our research complements their study. However, to confirm a unique species status of this isolate and to provide a new scientific name for this species, more isolates must be described, and a large-scale phylogenetic validation analysis is required.
Sequencing and analysis of the transcriptome allowed us to simultaneously address two tasks - to identify phylogenetically the pathogenic strain that causes the disease of Siberian fir, and also to study the quantitative representation of transcripts of different genes, possibly associated with the pathogenicity mechanism of this strain. Transcriptome analysis is the best approach for studying the expression of different genes at the genome-wide level. Transcriptome assembly obtained with CLC Genomic Workbench in this study provides also additional genomic resources for the Nectriaceae family and can support further genetic investigations of the pathogenicity of the discovered species to A. sibirica and other conifers as well as its distribution. We also hope that our results can help other similar studies, which will advance our understanding of the host-fungi interaction mechanisms.
The authors are grateful to the two anonymous reviewers for carefully reading the manuscript and making valuable comments that helped us improve it. This study was funded by the Research Grant N 14.Y26.31.0004 from the Government of the Russian Federation and was carried out also within scientific projects № 0287-2021-0011 and № FWES-2022-0003 funded by the Ministry of Science and Higher Education of the Russian Federation.
Список литературы
Biriukov V.V., Pavlov I.N., Litovka Y.A. et al. De novo transcriptome assembly and annotation of a new plant pathogenic Corinectria sp. strain in Siberia. Figshare. 2021. https://doi.org/10.6084/m9.figshare.c.5335229
Bolger A.M., Lohse M., Usadel B. Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics. 2014. V. 30 (15). P. 2114–2120. https://doi.org/10.1093/bioinformatics/btu170
Bushmanova E., Antipov D., Lapidus A. et al. rnaSPAdes: a de novo transcriptome assembler and its application to RNA-Seq data. GigaScience. 2019. V. 8 (9). https://doi.org/10.1093/gigascience/giz100
Crane P.E., Hopkins A.J.M., Dick M.A. et al. Behaviour of Neonectria fuckeliana causing a pine canker disease in New Zealand. Can. J. Forest Res. 2009. V. 39 (11). P. 2119–2128. https://doi.org/10.1139/X09-133
Dick M.A., Crane P.E. Neonectria fuckeliana is pathogenic to Pinus radiata in New Zealand. Australasian Plant Disease Notes. 2009. V. 4 (1). P. 12–14. https://doi.org/10.1071/DN09005
González C. D., Chaverri P. Corinectria, a new genus to accommodate Neonectria fuckeliana and C. constricta sp. nov. from Pinus radiata in Chile. Mycol. Progress. 2017. V. 16 (11–12). P. 1015–1027. https://doi.org/10.1007/s11557-017-1343-8
Götz S., García-Gómez J.M., Terol J. et al. High-throughput functional annotation and data mining with the Blast2GO suite. Nucleic Acids Research. 2008. V. 36 (10). P. 3420–3435. https://doi.org/10.1093/nar/gkn176
Haas B.J., Papanicolaou A., Yassour M. et al. De novo transcript sequence reconstruction from RNA-Seq: reference generation and analysis with Trinity. Nature Protocols. 2013. V. 8 (8). P. 1494–1512. https://doi.org/10.1038/nprot.2013.084
Higa L.A., Wu M., Ye T. et al. CUL4-DDB1 ubiquitin ligase interacts with multiple WD40-repeat proteins and regulates histone methylation. Nature Cell Biology. 2006. V. 8 (11). P. 1277–1283. https://doi.org/10.1038/ncb1490
Huelsenbeck J.P., Ronquist F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics. 2001. V. 17 (8). P. 754–755. https://doi.org/10.1093/bioinformatics/17.8.754
Jain B.P., Pandey S. WD40 Repeat proteins: signalling scaffold with diverse functions. The Protein Journal. 2018. V. 37 (5). P. 391–406. https://doi.org/10.1007/s10930-018-9785-7
Jones P., Binns D., Chang H.-Y. et al. InterProScan 5: genome-scale protein function classification. Bioinformatics. 2014. V. 30 (9). P. 1236–1240. https://doi.org/10.1093/bioinformatics/btu031
Katoh K., Kuma K., Toh H. et al. MAFFT version 5: improvement in accuracy of multiple sequence alignment. Nucleic Acids Research. 2005. V. 33 (2). P. 511–518. https://doi.org/10.1093/nar/gki198
Katoh K., Standley D.M. MAFFT Multiple sequence alignment software version 7: Improvements in performance and usability. Molec. Biol. Evol. 2013. V. 30 (4). P. 772–780. https://doi.org/10.1093/molbev/mst010
Kopylova E., Noé L., Touzet H. SortMeRNA: fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics. 2012. V. 28 (24). P. 3211–3217. https://doi.org/10.1093/bioinformatics/bts611
Lagesen K., Hallin P., Rødland E.A. et al. RNAmmer: consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Research. 2007. V. 35 (9). P. 3100–3108. https://doi.org/10.1093/nar/gkm160
Leigh J.W., Susko E., Baumgartner M. et al. Testing congruence in phylogenomic analysis. Systematic Biology. 2008. V. 57 (1). P. 104–115. https://doi.org/10.1080/10635150801910436
Morales R.R. Detection of Neonectria fuckeliana in Chile associated to stem cankers and malformations in Pinus radiata plantations. Bosque (Valdivia). 2009. V. 30 (2). P. 106–110. https://doi.org/10.4067/S0717-92002009000200007
Mylona A., Fernández-Tornero C., Legrand P. et al. Structure of the tau60/Delta tau91 subcomplex of yeast transcription factor IIIC: insights into preinitiation complex assembly. Molecular Cell. 2006. V. 24 (2). P. 221–232. https://doi.org/10.1016/j.molcel.2006.08.013
Nguyen L.-T., Schmidt H.A., Haeseler A. et al. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015. V. 32 (1). P. 268–274. https://doi.org/10.1093/molbev/msu300
Pavlov I.N. Biotic and abiotic factors as causes of coniferous forests dieback in Siberia and Far East. Contemporary Problems of Ecology. 2015. V. 8 (4). P. 440–456. https://doi.org/10.1134/S1995425515040125
Pavlov I.N., Vasaitis R., Litovka Y.A. et al. Occurrence and pathogenicity of Corinectria spp. – an emerging canker disease of Abies sibirica in Central Siberia. Scientific Reports. 2020. V. 10 (1). P. 5597. https://doi.org/10.1038/s41598-020-62566-y
Pöggeler S., Kück U. A WD40 Repeat protein regulates fungal cell differentiation and can be replaced functionally by the mammalian homologue striatin. Eukaryotic Cell. 2004. V. 3 (1). P. 232–240. https://doi.org/10.1128/EC.3.1.232-240.2004
Quast C., Pruesse E., Yilmaz P. et al. The Silva ribosomal RNA gene database project: improved data processing and web-based tools. Nucleic Acids Research. 2013. V. 41 (D1). P. D590–D596. https://doi.org/10.1093/nar/gks1219
Ruthenburg A.J., Wang W., Graybosch D.M. et al. Histone H3 recognition and presentation by the WDR5 module of the MLL1 complex. Nature Structural and Molecular Biology. 2006. V. 13 (8). P. 704–712. https://doi.org/10.1038/nsmb1119
Schultz M.E. A Canker disease of Abies concolor caused by Nectria fuckeliana. Plant Disease. 1990. V. 74 (2). P. 178–180. https://doi.org/10.1094/PD-74-0178
Seppey M., Manni M., Zdobnov E.M. Busco: Assessing genome assembly and annotation completeness. Methods in Molecular Biology. 2019. V. 1962. P. 227–245. https://doi.org/10.1007/978-1-4939-9173-0_14
Smith T.F. Diversity of WD-Repeat proteins. In: C.S. Clemen, L. Eichinger, V. Rybakin (eds). The coronin family of proteins: subcellular biochemistry. Springer, N.Y., 2008. P. 20–30. https://doi.org/10.1007/978-0-387-09595-0_3
Stirnimann C.U., Petsalaki E., Russell R.B. et al. WD40 proteins propel cellular networks. Trends in Biochemical Sciences. 2010. V. 35 (10). P. 565–574. https://doi.org/10.1016/j.tibs.2010.04.003
Дополнительные материалы отсутствуют.
Инструменты
Микология и фитопатология