

Молекулярная биология, 2019, T. 53, № 1, стр. 91-100
Система CRISPR/Cas9, экспрессируемая с плазмиды, подавляет репликацию вируса простого герпеса первого типа в культуре клеток Vero
Д. С. Карпов a, b, *, В. Л. Карпов a, Р. Р. Климова c, Н. А. Демидова c, А. А. Кущ c
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
b Научно-исследовательский институт биомедицинской химии им. В.Н. Ореховича
119991 Москва, Россия
c Национальный исследовательский центр эпидемиологии и микробиологии
им. почетного академика Н.Ф. Гамалеи Минздрава России
123098 Москва, Россия
* E-mail: aleom@yandex.ru
Поступила в редакцию 28.08.2018
После доработки 29.08.2018
Принята к публикации 17.09.2018
Аннотация
Герпесвирусы – одни из самых распространенных вирусов человека. Так, вирусом простого герпеса первого типа (ВПГ1) инфицировано более 3.7 млрд человек. У большинства первично инфицированных людей вирус переходит в латентную форму, устойчивую к действию всех противовирусных препаратов. Кроме того, появляются лекарственно устойчивые штаммы герпесвирусов. Это стимулирует поиск альтернативных подходов к лечению герпесвирусных инфекций. Один из таких подходов может быть реализован благодаря технологии редактирования геномов живых организмов с помощью прокариотических систем CRISPR/Cas. Система CRISPR/Cas9 из Streptococcus piogenes эффективно подавляет ВПГ-инфекцию в составе лентивирусных векторов, встраиваемых в геном; однако тут же встают вопросы, связанные с безопасностью такого способа доставки компонентов системы. В представленной работе система CRISPR/Cas9, нацеленная против генов UL52 и UL29 праймазо-геликазного комплекса ВПГ1, введена в клетки линии Vero в виде плазмиды. Показано отсутствие значимого цитотоксичного действия системы и полное подавление инфекции ВПГ1 в клетках Vero в течение двух суток. Полученные результаты указывают на возможность использования системы CRISPR/Cas9, кодируемой плазмидой, в поиске важных для жизненного цикла ВПГ1 генов и при разработке новых стратегий борьбы с герпесвирусными инфекциями.
ВВЕДЕНИЕ
Вирусы семейства Herpesviridae – одни из самых распространенных патогенов в человеческой популяции. Так, антитела к вирусам простого герпеса 1 и 2 типов (ВПГ1 и ВПГ2) выявляют у 80–90% людей. По данным на 2012 год, ВПГ1 инфицировано более 3.7 млрд. человек в возрасте до 50 лет, или 67% населения [1]. Особенно опасна ВПГ-инфекция для лиц со сниженным иммунитетом: беременных женщин, новорожденных, ВИЧ-инфицированных, реципиентов трансплантатов. У новорожденных детей ВПГ-инфекция может вызвать хориоретиниты, микроцефалию, респираторные дисфункции, задержку развития, инвалидность и смерть [2]. Герпесвирусные инфекции (ГВИ) урогенитального тракта считаются одной из причин бесплодия [3]. Первичная инфекция может протекать как в относительно легкой (лабиальный герпес), так и в клинически тяжелой форме (рекуррентный генитальный герпес, энцефалиты), вплоть до смертельного исхода. У большинства людей первичная ГВИ переходит в латентную форму, при которой вирус находится в клетках в неактивном состоянии. Под влиянием многих факторов различной природы происходит реактивация ВПГ, приводящая к репликации вируса, к появлению клинически выраженных заболеваний, часто – в рекуррентной форме. Одна из основных проблем контроля над ГВИ состоит в том, что большинство патентованных препаратов, использующихся в терапии этой инфекции, направлено на лечение острой клинически выраженной формы, но не элиминирует латентный вирус, который может пожизненно присутствовать в организме. Другая проблема заключается в появлении штаммов, устойчивых к химическим препаратам, используемым в противовирусной терапии (например, к ацикловиру) [4, 5]. В связи с этим активно проводятся поиски новых терапевтических подходов. Один из них разработан сравнительно недавно – это технология редактирования геномов с использованием системы CRISPR/Cas9 [6–8]. На примере инфекции клеток вирусом Эпштейна–Барр продемонстрирована принципиальная возможность и перспективность использования системы CRISPR/Cas9 в отношении ДНК-геномных вирусов [9]. Недавно показано, что система CRISPR/Cas9, встроенная в геном клеток млекопитающих в составе лентивирусных конструкций, эффективно подавляет инфекцию ВПГ1 [10–13]. Несмотря на то, что система CRISPR/Cas9 в составе интегрированных в геном конструкций способна эффективно подавлять ГВИ, встает важный вопрос о безопасности этого способа доставки целевых генов в клетки человека. Возможными последствиями применения лентивирусных векторов может быть опухолевая трансформация клеток при встраивании этих векторов поблизости от протоонкогенов [14, 15], инсерционный мутагенез высокоэкспрессируемых генов [16, 17] и подавление экспрессии вносимых конструкцией генов [18]. Более безопасными генетическими векторами считаются плазмиды эписомного типа. В отличие от лентивирусных векторов, плазмиды не встраиваются в геном или встраиваются с крайне низкой вероятностью (<10–5 на клетку) [19]. Плазмиды легче и дешевле получать, транспортировать и хранить, а также можно использовать в повторных инъекциях [20]. Однако плазмиды можно ввести внутрь клеток только с помощью вспомогательных средств, например, липосом и биодеградируемых полимерных микро- и наночастиц, которые к тому же могут обладать свойствами адьювантов [20].
Цель работы состояла в оценке способности системы CRISPR/Cas9, гены которой экспрессируются с плазмиды, подавлять инфекцию ВПГ1. В качестве генов-мишеней системы выбраны вирусные гены UL8 и UL52 геликаза-праймазного комплекса (UL5-UL8-UL52), а также ген UL29, кодирующий белок ICP8, связывающийся с одноцепочечной ДНК и образующий комплекс с ДНК-полимеразой и белком – продуктом гена UL42 ВПГ1. Белки UL8, UL52 и ICP8 необходимы для ориджинзависимой репликации ВПГ1. Эти белки накапливаются в участках репликации ВПГ1 вместе с белком, связывающим ориджин (UL9), и вирусной ДНК-полимеразой (UL30/42) и образуют единый комплекс после сигнала инициации репликации [21]. В ранее проведенных исследованиях показано, что использование этих генов в качестве мишеней системы CRISPR/Cas значительно подавляет способность ВПГ1 к размножению в клетках млекопитающих [11].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Клетки. Перевиваемые клетки линии Vero получены из коллекции клеточных культур ФГБУ “НИЦЭМ им. Н.Ф. Гамалеи” Минздрава России, Москва, РФ. Для культивирования использовали среду Игла MEM с добавлением 10% эмбриональной телячьей сыворотки (FBS), 2 мМ L-глутамина, 50 мкг/мл гентамицина. Перечисленные реагенты приобретены в ООО “ПанЭко” (Россия). Клетки культивировали в атмосфере 5% СО2. В молекулярном клонировании использовали хемокомпетентные клетки Escherichia coli, штамм XL1Blue (“Евроген”, Россия).
Вирусы. Штамм F ВПГ1 получен из Государственной Коллекции вирусов ФГБУ “НИЦЭМ им. Н.Ф. Гамалеи” Минздрава России. Вирус размножали в клетках Vero, используя стандартные методы культивирования. Инфекционный титр вируса определяли модифицированным методом бляшек. Готовили серии разведений образцов вируса и вносили в 96-луночные планшеты с монослоем клеток, инкубировали при 37°С в течение 7 суток в присутствии 5% СО2. Очаги инфицированных клеток (бляшки) выявляли и подсчитывали с использованием инвертированного микроскопа Primovert (“Zeiss”, Германия). Титр вируса определяли по формуле: А = ab/$v$, где А – число бляшкообразующих единиц на 1 клетку (БОЕ/кл); a – среднее число бляшек на одну лунку; b – разведение вируса; $v$ – объем внесенного вируссодержащего материала. Инфекционный титр вируса составлял 3.3 × 106 БОЕ/кл.
Выделение геномной ДНК ВПГ1. Культуру клеток Vero, выращенную до монослоя, инфицировали ВПГ1 (штамм F). Через 48 ч собирали вируссодержащую культуральную жидкость, добавляли равный объем смеси фенол : хлороформ : изоамиловый спирт (25 : 24 : 1) и встряхивали. Фазы разделяли центрифугированием. Водную фазу смешивали с гликогеном до конечной концентрации 10 мкг/мл, добавляли 1/10 объема 3 М раствора ацетата натрия (pH 5.2) и 3 объема 96%-ного спирта и инкубировали при –20°С в течение ночи. ДНК осаждали центрифугированием и ресуспендировали в деионизованной воде (mQ).
Дизайн направляющих РНК системы CRISPR/ Cas9 против генов ВПГ1. Гены-мишени системы CRISPR/Cas9 выбраны в соответствии с результатами ранее проведенных исследований [11, 13]. Участки генов UL8, UL29 и UL52 амплифицировали с помощью пар праймеров HSV1-UL8-ch-F/HSV1-UL8-ch-R, HSV1-UL29-ch-F/HSV1-UL29-ch-R и HSV1-UL52-ch-F/HSV1-UL52-ch-R соответственно, используя в качестве матрицы геномную ДНК ВПГ1. ПЦР-продукты секвенировали и направляющие РНК против генов UL8, UL29 и UL52 подбирали с помощью программы CRISPOR [22] с параметрами по умолчанию.
Получение плазмид системы CRISPR/Cas9. Плазмиды UL8-UL29 и UL52-UL29, кодирующие по две направляющие РНК против соответствующих генов ВПГ1, получали путем клонирования в плазмиду PX458 ([23]; “Addgene” (www.addgene.org), кат. номер 48138; рис. 1а) по сайтам BbsI ПЦР-фрагментов, полученных с помощью соответствующих пар праймеров: PX458-sgRNA-UL8-F/PX458-sgRNA-UL29-R и PX458-sgRNA-UL52-F/PX458-sgRNA-UL29-R (табл. 1), – используя в качестве матрицы плазмиду AIO-mCherry ([24]; “Addgene”, кат. номер 74120). Отбор колоний E. coli проводили путем ПЦР с олигонуклеотидами U6-ch-F и PX458-sgRNA-UL29-ch-R. Корректность клонированных спейсеров направляющих РНК подтверждали секвенированием с олигонуклеотидом U6-ch-F.
Рис. 1.
Сравнительный анализ эффективности трансфекции клеток Vero плазмидами, кодирующими систему CRISPR/Cas9. а – Схема расположения основных элементов системы CRISPR/Cas9 в плазмиде PX458. Аббревиатуры: sgRNA (single guide RNA) – химерная направляющая РНК; CMV (cytomegalovirus) –цитомегаловирус; SpyCas9 – Streptococcus pyogenes Cas9; NLS (nuclear localization signal) – сигнал ядерной локализации; poly(A) ‒ сигнал полиаденилирования мРНК; AmpR – ген устойчивости к ампициллину; ori – ориджин репликации. б – Содержание клеток Vero (в %), продуцирующих репортерный белок GFP, в культурах, трансфицированных указанными плазмидами или вектором PX458 (плазмидой, кодирующей систему CRISPR/Cas9 без спейсеров против вирусных генов), через 48 ч после трансфекции. Результаты представлены как среднее значение ± стандартное отклонение (M ± SD). в – Влияние CRISPR/Cas9-плазмид на жизнеспособность трансфицированных клеток Vero. Результаты представлены как процентное содержание жизнеспособных клеток (не окрашенных трипановым синим) в клеточной популяции; M ± SD. В качестве контроля взяты интактные нетрансфицированные клетки Vero; вектор – плазмида PX458, кодирующая систему CRISPR/Cas9 без спейсеров против вирусных генов; трансфекция плазмидами, кодирующими компоненты CRISPR/Cas9, нацеленные против обозначенных вирусных генов. г – Флуоресценция GFP в клетках: нетрансфицированных (1), трансфицированных пустым вектором PX458 (2), плазмидой UL8-UL29-1a (3), плазмидой UL52-UL29-9a (4), плазмидой UL8-UL29-1b (5), плазмидой UL52-UL29-9b (6). Увеличение 400×.
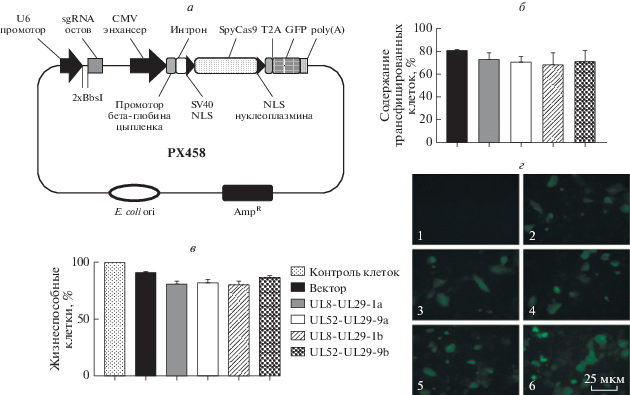
Таблица 1.
Олигонуклеотиды, использованные в работе
| Наименование | Последовательность 5' → 3' |
|---|---|
| HSV1-UL8-ch-F | ACACCAGGGCGTGGAAGTA |
| HSV1-UL8-ch-R | CCCCAACGCTGACATCATCC |
| HSV1-UL29-ch-F | GAGGGCGTCAGTTTCAGGGA |
| HSV1-UL29-ch-R | TACATATACCAACCGCATACC |
| HSV1-UL52-ch-F | GTCGGGTGTCGTCCAGGG |
| HSV1-UL52-ch-R | CCGTCCCCAATAAACAAAAGG |
| PX458-sgRNA-UL8-F | ATATAGAAGACCTCACCGAGACCGCCCGTGATCCTTAGGT TTTAGAGCTAGAAATAGCAAG |
| PX458-sgRNA-UL52-F | ATATAGAAGACCTCACCGTAGCGCGTTAACGACCGCCAGT TTTAGAGCTAGAAATAGCAAG |
| PX458-sgRNA-UL29-R | TAGAGGAAGACCCAAACGGGATACGTGTACGCTCGCGCG GTGTTTCGTCCTTTCCAC |
| PX458-sgRNA-UL29-ch-R | GGGATACGTGTACGCTCGCG |
| U6-ch-F | CGATACAAGGCTGTTAGAGAGA |
В экспериментах использовали четыре плазмиды CRISPR/Cas9 системы, нацеленной на гены ВПГ1: UL8-UL29-1a, UL8-UL29-1b, UL52-UL29-9a и UL52-UL29-9b, – где а и b обозначают плазмиды, несущие одинаковые спейсеры направляющей РНК, но выделенные из двух разных колоний E. coli. Все конструкции кодировали Cas9 из Streptococcus pyogenes, соединенный с зеленым флуоресцентным белком (GFP) через T2А-пептид вируса Thosea asigna. Пептид Т2А обеспечивает синтез Cas9 и GFP в соотношении 1 : 1, следовательно, сигнал флуоресценции GFP прямо пропорционален количеству Cas9. В качестве отрицательного контроля использовали плазмиду PX458. Плазмиды очищали из трансформированных клеток XL1Blue (“Евроген”, Россия) с помощью набора GeneJET Plasmid Purification Kit (“Thermo Fisher Scientific”, США) в соответствии с рекомендациями производителя. Препараты плазмид очищали от эндотоксина E. coli с помощью набора MiraCLEAN Endotoxin Removal Kit (“Mirus Bio LLC”, США).
Трансфекция. Клетки линии Vero трансфицировали с помощью Lipofectamine 3000 Reagent (“Thermo Fisher Scientific”, США) в соответствии с рекомендациями производителя. Клетки высаживали в 24-луночный планшет в концентрации 2 × 105 кл/мл в ростовой среде Игла МЕМ без антибиотиков. Через 24 ч в клетки вносили смесь исследуемых плазмид и Lipofectamine 3000 в соотношении 1 : 2. Эффективность трансфекции оценивали через 48 ч, используя инвертированный люминесцентный микроскоп Axio Scope A1 (“Zeiss”, Германия). Подсчитывали отношение числа флуоресцирующих клеток, экспрессирующих GFP, к общему числу клеток в популяции и выражали в процентах.
Цитотоксичность ДНК-конструкций оценивали по влиянию на жизнеспособность клеток и определяли методом исключения витального красителя трипанового синего. Клетки Vero трансфицировали, внося смесь исследуемых плазмид и Lipofectamine 3000 в соотношении 1 : 2 и инкубировали в течение 24 ч. После инкубации смесь удаляли и вносили ростовую среду. Через 72 ч в клетки добавляли 0.03%-ный раствор трипанового синего (ООО “БиолоТ”, Россия) и анализировали число жизнеспособных (неокрашенных) клеток при помощи инвертированного микроскопа Axio Vert A1 (“Zeiss”, Германия). В качестве контроля использовали интактные нетрансфицированные клетки Vero. Результаты представляли как процентное отношение числа жизнеспособных (неокрашенных) клеток к общему числу клеток в популяции.
Детекция ВПГ1. Клетки, продуцирующие ВПГ1, выявляли иммуноцитохимическим окрашиванием с помощью мышиных моноклональных антител против позднего структурного белка gB ВПГ1 (ab6506; “Abсam”, Великобритания). Клетки фиксировали охлажденным метанолом, обрабатывали моноклональными антителами и инкубировали 1 ч при 37°C. Антитела отмывали фосфатно-солевым буфером (PBS), наслаивали анти-мышиные антитела, коньюгированные с пероксидазой хрена (anti-mouse PX, PO260; “Dako”, Дания), и инкубировали 1 ч при 37°C. Препараты промывали PBS и наносили раствор 3,3'-диаминобензидина в концентрации 1 мг/мл в буфере 0.05 М Трис-HCl (pH 7.4) с добавлением 3% пероксида водорода. Реакцию останавливали через 10 мин внесением в лунки дистиллированной воды. Результаты оценивали с использованием инвертированного микроскопа Axio Vert A1. Подсчитывали число окрашенных клеток, содержавших белок gB ВПГ1, и представляли в процентах от общего числа клеток в популяции.
Чтобы оценить количество вновь образованных вирусных частиц в культурах клеток, трансфицированных плазмидами системы CRISPR/Cas9, отбирали культуральные жидкости и добавляли их к неинфицированным клеткам Vero, выращенным до монослоя. Через 24 и 48 ч детектировали наличие белка gB ВПГ1 иммуноцитохимически, как описано выше.
Статистическая обработка результатов. Статистическую обработку результатов проводили в программе GraphPadPrism 5.01. Статистическую значимость различий между средними значениями определяли с помощью двустороннего непарного t-test (критерий Стьюдента). Различия показателей считали статистически значимыми при p < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Эффективность трансфекции клеток Vero плазмидами CRISPR/Cas9-системы оценивали по количеству клеток, синтезирующих маркерный белок GFP. Через 48 ч после трансфекции флуоресценцию детектировали в более чем 50% клеток после введения всех полученных плазмид. Эффективность трансфекции плазмидами UL8-UL29-1a, UL8-UL29-1b, UL52-UL29-9a и UL52-UL29-9b составляла от 68.7 ± 10.5% до 73.3 ± 5.6% (рис. 1б) и значимо не отличалась от таковой для пустого вектора PX458 (p > 0.05). Следует отметить, что наиболее интенсивное свечение GFP наблюдалось в клетках, трансфицированных конструкцией UL52-UL29-9b (рис. 1г).
В результате проведенного анализа цитотоксичности ДНК-конструкций показано (рис. 1в), что через 72 ч после трансфекции содержание жизнеспособных клеток составляет 80.7–91.0% от всех клеток в популяции и статистически значимо не отличается от интактных нетрансфицированных клеток Vero (p > 0.05), из чего сделано заключение о низкой цитотоксичности всех изученных плазмид.
Противовирусную активность плазмид, экспрессирующих компоненты системы CRISPR/ Cas9, нацеленные на гены ВПГ1, оценивали в клеточной культуре Vero. Клетки, трансфицированные плазмидами, через 48 ч заражали ВПГ1 со множественностью инфекции (MOI) 0.1 БОЕ/кл и через 48 или 72 ч количество инфицированных клеток (бляшек) определяли иммуноцитохимически с использованием мышиных моноклональных антител против позднего структурного белка gB ВПГ1 (рис. 2). Подсчет инфицированных клеток проводили, как описано выше.
Рис. 2.
Подавление репликации ВПГ1 в клетках Vero, трансфицированных плазмидами системы CRISPR/Cas9. Морфология клеток Vero через 48 ч после заражения ВПГ1 при MOI 0.1 БОЕ/кл, где стрелками обозначены вирусспецифичные бляшки (а), и контрольных (неинфицированных) клеток (б). Окраска гематоксилином. Увеличение 100×. в – Анализ изменения содержания инфицированных ВПГ1 клеток в культуре, трансфицированной CRISPR/Cas9-плазмидами. Контроль – нетрансфицированные клетки Vero, зараженные ВПГ1 при MOI 0.1 БОЕ/кл; вектор – зараженные клетки Vero, трансфицированные пустым вектором PX458. Относительное содержание зараженных клеток (%) определяли по наличию в них белка gB ВПГ1, принимая за 100% число gB-позитивных клеток в контрольном образце. Результаты представлены как среднее ± SD на основании трех независимых экспериментов. *p < 0.05 (t-test); #p < 0.05 (t-test) при сравнении со всеми изученными вариантами.
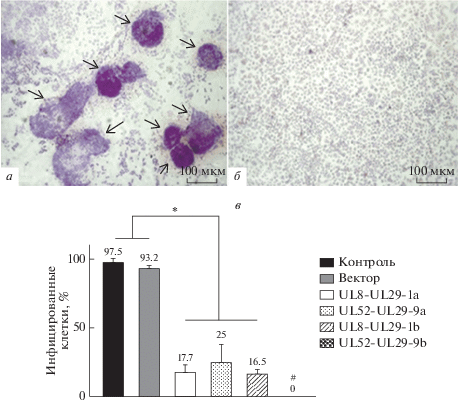
Установлено, что через 48 ч после инфекции содержание клеток, экспрессирующих белок gB ВПГ1, в культурах, трансфицированных плазмидами системы CRISPR/Cas9, снижалось в 4–6 раз (рис. 2в) по сравнению с отрицательными контролями: нетрансфицированными клетками и клетками, трансфицированными пустым вектором (р < 0.05). Следует отметить, что при использовании плазмиды UL52-UL29-9b белок gB ВПГ1 статистически значимо не детектировали. Учитывая, что синтез белка gB происходит после репликации ДНК ВПГ1, на поздних стадиях инфекции, можно думать, что под действием CRISPR/Cas9-системы, кодируемой этой плазмидой, происходит полное подавление инфекционного цикла вируса.
Представляло интерес выяснить, в какой мере система CRISPR/Cas9 подавляет продукцию инфекционного ВПГ1. С этой целью из трансфицированных культур, зараженных вирусом, отбирали культуральные жидкости и вносили их в свежие, незараженные, культуры. Через 24 и 48 ч клетки фиксировали, окрашивали мышиными моноклональными антителами к позднему структурному белку gB ВПГ1 и подсчитывали количество окрашиваемых клеток (рис. 3). В положительном контроле заражения через 24 ч после внесения культуральной жидкости от нетрансфицированных зараженных клеток 99.8 ± 0.8% клеток содержали вирусный белок gB, через 48 ч наблюдали 100%-ную гибель клеток. После внесения культуральной жидкости от клеток, трансфицированных пустым PX458 вектором, через 48 ч наблюдали образование вирусспецифических бляшек (рис. 3а), что свидетельствовало о прохождении всех стадий инфекционного цикла ВПГ1; при этом содержание инфицированных клеток снизилось менее чем на 10% (рис. 3б).
Рис. 3.
Ингибирование продуктивной ВПГ1-инфекции в клетках Vero, трансфицированных плазмидами системы CRISPR/Cas9. а – Влияние системы CRISPR/Cas9 на накопление белка gB в клетках Vero, зараженных ВПГ1. Клетки, инфицированные ВПГ1 (MOI 0.1 БОЕ/кл) и экспрессирующие белок gB: в нетрансфицированной культуре Vero (верхний ряд), трансфицированных вектором PX458 (средний ряд), трансфицированных плазмидой UL52-UL29-9b (нижний ряд). Иммунноцитохимическая детекция белка gB ВПГ1 проведена с использованием моноклональных антител, коньюгированных с пероксидазой хрена (коричневое окрашивание), и последующего доокрашивания клеток гематоксилином (синее окрашивание). Увеличение 400×. б – Содержание неинфицированных клеток в культурах Vero после внесения в них культуральных жидкостей от зараженных культур, трансфицированных указанными плазмидами. Результаты представлены как среднее значение ± стандартное отклонение (M ± SD). *Различия статистически значимы при p < 0.05 (t-test); #различия статистически значимы по сравнению со всеми изученными вариантами при p < 0.05 (t-test).
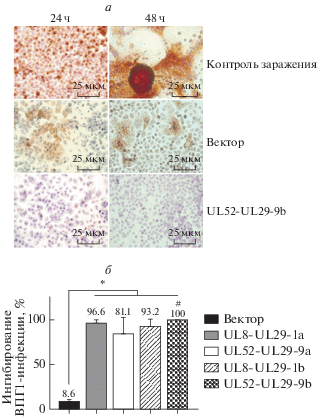
Наиболее важные результаты получены при анализе культуральных жидкостей от ВПГ1-зараженных клеток Vero, предварительно трансфицированных плазмидами UL8-UL29-1a, UL8-UL29-1b, UL52-UL29-9a и UL52-UL29-9b. Установлено, что первые три плазмиды снижали продукцию новых вирусных частиц более чем на 80%, тогда как плазмида UL52-UL29-9b полностью подавляла репродукцию вируса (рис. 3б).
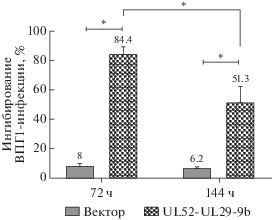
Чтобы оценить противовирусный эффект конструкции UL52-UL29-9b в течение более длительного времени, трансфицированные клетки заражали гораздо меньшей дозой ВПГ1 (MOI 0.001 БОЕ/кл) и анализировали репродукцию вируса через 3 и 6 суток после заражения. Полученные результаты представлены на рис. 4. Обнаружено, что через трое суток продукция ВПГ1 снижалась на 84.4%, а через шесть суток – на 51.3% по сравнению с контролем. Важно, что различия на обоих сроках инфекции были существенными и статистически значимыми по сравнению с контролем заражения (p < 0.001).
Рис. 4.
Динамика подавления ВПГ1-инфекции системой CRISPR/Cas9, направленной на гены UL52 и UL29 ВПГ1. Клетки Vero заражали ВПГ1 (MOI 0.001 БОЕ/кл) и трансфицировали пустым вектором PX458 (вектор) или плазмидой UL52-UL29-9b. Репликативную активность вируса оценивали по числу клеток, экспрессирующих gB ВПГ1. Результаты представлены как среднее значение ± стандартное отклонение (M ± SD). *p < 0.05.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Технология редактирования генома с помощью системы CRISPR/Cas9 открыла новые возможности в изучении важных генов вирусов, в том числе одного из самых распространенных альфагерпесвирусов – ВПГ1. Так, эффективность мутагенеза альфагерпесвирусов с использованием традиционных технологий составляет менее 1%, тогда как эффективность мутагенеза с помощью системы CRISPR/Cas9 – более 50% [25]. О необходимости получения новых данных свидетельствуют недостаточно полные знания о вирусных белках и их функциях, что объясняет как трудности, возникающие при лечении заболеваний герпесвирусной этиологии, так и отсутствие эффективных вакцин. Уже в первых работах [9–13] продемонстрирована перспективность использования CRISPR/Cas9-системы в поиске важных генов ВПГ1 и исследовании их структуры и функций. Получены новые сведения о белках тегумента – аморфного компонента, находящегося между оболочкой вируса и капсидом и содержащего более 20 белков, многие из которых пока не изучены. С помощью системы CRISPR/Cas9 впервые показано, что внесение мутации в ген белка тегумента UL7 приводит к 10-кратному снижению репликации ВПГ1 и аттенуации вирулентности. Отмечено, что ингибирующее влияние мутантный белок оказывал, воздействуя на транскрипцию сверхраннего гена α4 [12]. Недавно определена кристаллическая структура белка UL21 тегумента ВПГ [26], но это не позволило прояснить участие белка в жизненном цикле вируса и необходимость UL21 для репликации. С помощью системы CRISPR/Cas9 авторы получили панель мутантных штаммов ВПГ с делециями гена UL21 [27]. В результате сравнительного анализа мутантов выявлено, что белок UL21 участвует в распространении вируса, однако отсутствие белка UL21 в разной степени влияет на передачу ВПГ1 и ВПГ2 от клетки к клетке; типоспецифические различия выявлены также и при репликации мутантных вирусов.
Большой интерес представляет сверхранний белок ВПГ ICP0, который участвует в индукции ВПГ-инфекции, активируя экспрессию вирусных и клеточных генов. Roehm и соавт. [13] обнаружили, что внесение мутаций в экзон 2 гена ICP0 с помощью системы CRISPR/Cas9 в составе лентивирусного вектора значительно ингибировало развитие ВПГ-инфекции и защищало клетки от заражения. В то же время полного подавления репродукции ВПГ удалось достичь в результате воздействия CRISPR/Cas9-системы, направленной одновременно на три регуляторных гена ВПГ1: ICP0, ICP4 и ICP27. Это наводит на мысль, что для эффективного подавления инфекционного процесса необходимо воздействовать одновременно на два и более вирусных генов. К такому же заключению пришли Van Diemen и соавт. [11]. Они использовали CRISPR/Cas9-систему, нацеленную на 12 генов ВПГ1, с 4 направляющими РНК на ген, и показали, что эффективность подавления ВПГ-инфекции зависит от того, на какой ген нацелена система. Оказалось, что только система CRISPR/Cas9, нацеленная одновременно на 2 определенных гена, полностью ингибировала продукцию вирусных частиц в клетках человека. Следует отметить, что для доставки компонентов CRISPR/Cas9-системы van Diemen и др. использовали лентивирусный вектор [11].
Учитывая все вышеизложенное, в качестве мишеней CRISPR/Cas9-системы мы выбрали гены, участвующие в репликации ВПГ1 при литической инфекции (UL8, UL29 и UL52), потому что из всех исследованных к настоящему времени генов они считаются наиболее важными для жизненного цикла ВПГ1, и их “отключение” может привести к подавлению вирусной инфекции. Нами показано, что система CRISPR/Cas9, направленная одновременно на два гена репликативного комплекса ВПГ1 (UL8/UL29) и (UL52/UL29), в среднем на 90% подавляет репродукцию ВПГ1 в зараженных клетках Vero, а также более чем на 80% в среднем блокирует образование новых инфекционно активных вирусных частиц. Отметим, что одна из плазмидных конструкций (UL52-UL29-9b) полностью ингибировала продуктивную ВПГ-инфекцию. Интересно, что именно для этой конструкции получена максимальная интенсивность флуоресценции GFP в трансфицированных клетках. В плазмиде гены, кодирующие Cas9 и GFP соединены через ген T2А-пептида, который регулирует экспрессию целевых белков в соотношении 1 : 1. Следовательно, сигнал свечения GFP прямо пропорционален количеству белка Cas9. Ранее показано, что повышенный сигнал GFP в такой системе ассоциирован с повышенной активностью системы CRISPR/Cas9 [28].
Одним из ключевых факторов, определяющих эффективность и специфичность действия системы CRISPR/Cas9, служит последовательность направляющей РНК. В этой работе спейсеры направляющих РНК подобраны с помощью программы CRISPOR. Выбранные спейсеры имели от 1 до 10 внецелевых мишеней в геноме млекопитающих, отличающихся от целевых участков не менее чем на 4 нуклеотида. Несмотря на то, что Cas9 слабо связывается с мишенью ДНК, имеющей 4 и более неправильно спаренных основания со спейсером направляющей РНК [29], есть сообщения о том, что даже при наличии 6 неправильно спаренных оснований между спейсером направляющей РНК и ДНК-мишенью Cas9 может вносить двухцепочечный разрыв в ДНК [30]. Отсутствие высокой цитотоксичности у полученных плазмид (рис. 1в), возможно, связано с тем, что внецелевая активность Cas9 не влияет на функционирование жизненно важных для клетки генов, а способность плазмиды UL52-UL29-9b полностью подавлять репликацию ВПГ1 в течение двух суток (рис. 2в и 3б) свидетельствует о высокой активности системы. Недавно показано, что противовирусная активность системы CRISPR/Cas9 повышается, если направляющая РНК комплементарна транскрибируемой цепи ДНК [31]. Спейсер направляющей РНК против гена UL8 взаимодействовал с нетранскрибируемой цепью, а спейсеры против генов UL29 и UL52 – с транскрибируемой, что корреллирует с тем, что плазмида UL52-UL29-9b наиболее эффективна в подавлении репликации ВПГ1 (рис. 2в и 3б). Некоторая степень защиты от вирусной инфекции плазмидой системы CRISPR/Cas9, не имеющей спейсеров против вирусных генов (рис. 3б), может быть связана со способностью направляющей РНК активировать внутриклеточный противовирусный ответ [32, 33].
Одна из важных проблем генной терапии – доставка генетических конструкций. В рассмотренных выше опубликованных работах систему CRISPR/Cas9 вводили в клетки в составе лентивирусных векторов. Мы применили иной подход. Впервые система CRISPR/Cas9 введена в клетки в составе другого вектора – рекомбинантных ДНК (плазмид). Плазмидные векторы не встраиваются в геном клетки-мишени, не вызывают индукцию сильного неспецифического иммунного ответа в организме и используются для создания противовирусных ДНК-вакцин [34, 35]. Необходимо отметить, что сконструированная нами плазмида CRISPR/Cas9-системы эффективно доставляется в клетки и обеспечивает высокий уровень экспрессии гена Cas9 (70–90%).
Необходимое требование, предъявляемое к технологиям генной терапии, – безопасность получаемых лекарственных препаратов. Нами показано, что использование CRISPR/Cas9-плазмид не влияет на жизнеспособность трансфицированных клеток, и это позволяет рассматривать разработанные генетические конструкции как перспективные инструменты для дальнейших исследований in vivo и потенциальные таргетные препараты для противовирусной терапии.
Надежность терапевтических методов представляет особый интерес в связи с имеющимися данными о “прорыве” вирусной инфекции при длительной инкубации клеточных культур, защищенных системой CRISPR/Cas9. Так, van Diemen и др. [11] обнаружили, что CRISPR/Cas9-система эффективно подавляла репликацию ВПГ в течение 24 ч, но эффект полностью исчезал на третьи сутки. В проведенных нами экспериментах противовирусный эффект CRISPR/Cas9-конструкций сохранялся, по крайней мере, в течение 6 суток после заражения ВПГ1.
Таким образом, нами получена плазмида с системой CRISPR/Cas9, нацеленной против генов UL52 и UL29 ВПГ1, эффективно подавляющая репродукцию вируса. Впервые показано, что доставка системы CRISRP/Cas9 в виде плазмид не только подавляет вирусную инфекцию в зараженных клетках, но и способна полностью ингибировать продуктивную (литическую) инфекцию ВПГ1 в клеточной культуре в течение длительного времени. Это открывает перспективы для получения новых данных о функциях вирусных белков и создания новых противовирусных препаратов с более высокой специфичностью, безопасностью и эффективностью по сравнению с имеющимися в настоящее время.
Часть работы выполнена на оборудовании ЦКП “Геном” ИМБ РАН (http://www.eimb.ru/rus/ckp/ ccu_genome_c.php).
Работа выполнена при финансовой поддержке Программы фундаментальных исследований государственных академий наук на 2013–2020 годы (№ 01201363823) и гранта РФФИ № 18-29-07021.
Список литературы
Looker K.J., Magaret A.S., May M.T., Turner K.M., Vickerman P., Gottlieb S.L., Newman L.M. (2015) Global and regional estimates of prevalent and incident herpes simplex virus type 1 infections in 2012. PLoS One. 10, e0140765.
Otto W.R., Myers A.L., LaRussa B., Kimberlin D.W., Jackson M.A. (2017) Clinical markers and outcomes of neonates with herpes simplex virus deoxyribonucleic acid persistence in cerebrospinal fluid in disseminated and central nervous system infection. J. Pediatric Infect. Dis. Soc. 7, e30–e33.
Naumenko V., Tyulenev Y., Kurilo L., Shileiko L., Sorokina T., Evdokimov V., Yakovleva V., Kovalyk V., Malolina E., Kulibin A., Gomberg M., Kushch A. (2014) Detection and quantification of human herpes viruses types 4–6 in sperm samples of patients with fertility disorders and chronic inflammatory urogenital tract diseases. Andrology. 2, 687–694.
Schubert A., Gentner E., Bohn K., Schwarz M., Mertens T., Sauerbrei A. (2014) Single nucleotide polymorphisms of thymidine kinase and DNA polymerase genes in clinical herpes simplex virus type 1 isolates associated with different resistance phenotypes. Antiviral Res. 107, 16–22.
Piret J., Boivin G. (2011) Resistance of herpes simplex viruses to nucleoside analogues: mechanisms, prevalence, and management. Antimicrob. Agents Chemother. 55, 459–472.
Mali P., Yang L., Esvelt K.M., Aach J., Guell M., DiCarlo J.E., Norville J.E., Church G.M. (2013) RNA-guided human genome engineering via Cas9. Science. 339, 823–826.
Jinek M., East A., Cheng A., Lin S., Ma E., Doudna J. (2013) RNA-programmed genome editing in human cells. eLife. 2, e00471.
Cong L., Ran F.A., Cox D., Lin S., Barretto R., Habib N., Hsu P.D., Wu X., Jiang W., Marraffini L.A., Zhang F. (2013) Multiplex genome engineering using CRISPR/ Cas systems. Science. 339, 819–823.
Wang J., Quake S.R. (2014) RNA-guided endonuclease provides a therapeutic strategy to cure latent herpesviridae infection. Proc. Natl. Acad. Sci. USA. 111, 13157–13162.
Lin C., Li H., Hao M., Xiong D., Luo Y., Huang C., Yuan Q., Zhang J., Xia N. (2016) Increasing the efficiency of CRISPR/Cas9-mediated precise genome editing of HSV-1 virus in human cells. Sci. Rep. 6, 34531.
van Diemen F.R., Kruse E.M., Hooykaas M.J., Bruggeling C.E., Schurch A.C., van Ham P.M., Imhof S.M., Nijhuis M., Wiertz E.J., Lebbink R.J. (2016) CRISPR/ Cas9-mediated genome editing of herpesviruses limits productive and latent infections. PLoS Pathogens. 12, e1005701.
Xu X., Fan S., Zhou J., Zhang Y., Che Y., Cai H., Wang L., Guo L., Liu L., Li Q. (2016) The mutated tegument protein UL7 attenuates the virulence of herpes simplex virus 1 by reducing the modulation of alpha-4 gene transcription. Virol. J. 13, 152.
Roehm P.C., Shekarabi M., Wollebo H.S., Bellizzi A., He L., Salkind J., Khalili K. (2016) Inhibition of HSV-1 replication by gene editing strategy. Sci. Rep. 6, 23146.
von Kalle C., Deichmann A., Schmidt M. (2014) Vector integration and tumorigenesis. Human Gene Therapy. 25, 475–481.
Riviere I., Dunbar C.E., Sadelain M. (2012) Hematopoietic stem cell engineering at a crossroads. Blood. 119, 1107–1116.
Schroder A.R., Shinn P., Chen H., Berry C., Ecker J.R., Bushman F. (2002) HIV-1 integration in the human genome favors active genes and local hotspots. Cell. 110, 521–529.
Wu X., Li Y., Crise B., Burgess S.M. (2003) Transcription start regions in the human genome are favored targets for MLV integration. Science. 300, 1749–1751.
Ellis J. (2005) Silencing and variegation of gammaretrovirus and lentivirus vectors. Human Gene Therapy. 16, 1241–1246.
Izsvák Z., Chuah M.K., Vandendriessche T., Ivics Z. (2009) Efficient stable gene transfer into human cells by the Sleeping Beauty transposon vectors. Methods. 49 287–297.
Hardee C.L., Arevalo-Soliz L.M., Hornstein B.D., Zechiedrich L. (2017) Advances in non-viral DNA vectors for gene therapy. Genes. 8, 65.
Darwish A.S., Grady L.M., Bai P., Weller S.K. (2015) ICP8 filament formation is essential for replication compartment formation during herpes simplex virus infection. J. Virol. 90, 2561–2570.
Haeussler M., Schonig K., Eckert H., Eschstruth A., Mianne J., Renaud J.B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J., Joly J.S., Concordet J.P. (2016) Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17, 148.
Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. (2013) Genome engineering using the CRISPR-Cas9 system. Nat. Protoc. 8, 2281–2308.
Chiang T.W., le Sage C., Larrieu D., Demir M., Jackson S.P. (2016) CRISPR-Cas9(D10A) nickase-based genotypic and phenotypic screening to enhance genome editing. Sci. Rep. 6, 24356.
Chen Y.C., Sheng J., Trang P., Liu F. (2018) Potential application of the CRISPR/Cas9 system against herpesvirus infections. Viruses. 10, pii: E291.
Metrick C.M., Chadha P., Heldwein E.E. (2015) The unusual fold of herpes simplex virus 1 UL21, a multifunctional tegument protein. J. Virol. 89, 2979–2984.
Finnen R.L., Banfield B.W. (2018) CRISPR/Cas9 mutagenesis of UL21 in multiple strains of herpes simplex virus reveals differential requirements for pUL21 in viral replication. Viruses. 10, pii: E258.
Duda K., Lonowski L.A., Kofoed-Nielsen M., Ibarra A., Delay C.M., Kang Q., Yang Z., Pruett-Miller S.M., Bennett E.P., Wandall H.H., Davis G.D., Hansen S.H., Frodin M. (2014) High-efficiency genome editing via 2A-coupled co-expression of fluorescent proteins and zinc finger nucleases or CRISPR/Cas9 nickase pairs. Nucleic Acids Res. 42, e84.
Mali P., Aach J., Stranges P.B., Esvelt K.M., Moosburner M., Kosuri S., Yang L., Church G.M. (2013) CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nat. Biotechnol. 31, 833–838.
Fu Y., Foden J.A., Khayter C., Maeder M.L., Reyon D., Joung J.K., Sander J.D. (2013) High-frequency off-target mutagenesis induced by CRISPR-Cas nucleases in human cells. Nat. Biotechnol. 31, 822–826.
Clarke R., Heler R., MacDougall M.S., Yeo N.C., Cha vez A., Regan M., Hanakahi L., Church G.M., Marraffini L.A., Merrill B.J. (2018) Enhanced bacterial immunity and mammalian genome editing via RNA-polymerase-mediated dislodging of Cas9 from double-strand DNA breaks. Mol. Cell. 71, 42–55.
Kim S., Koo T., Jee H.G., Cho H.Y., Lee G., Lim D.G., Shin H.S., Kim J.S. (2018) CRISPR RNAs trigger innate immune responses in human cells. Genome Res. 28, 367–373.
Wienert B., Shin J., Zelin E., Pestal K., Corn J.E. (2018) In vitro-transcribed guide RNAs trigger an innate immune response via the RIG-I pathway. PLoS Biol. 16, e2005840.
Ferraro B., Morrow M.P., Hutnick N.A., Shin T.H., Lucke C.E., Weiner D.B. (2011) Clinical applications of DNA vaccines: current progress. Clin. Infect. Dis. 53, 296–302.
Borthwick N.J., Lane T., Moyo N., Crook A., Shim J.M., Baines I., Wee E.G., Hawkins P.N., Gillmore J.D., Hanke T., Pepys M.B. (2018) Randomized phase I trial HIV-CORE 003: depletion of serum amyloid P component and immunogenicity of DNA vaccination against HIV-1. PLoS One. 13, e0197299.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология