

Молекулярная биология, 2019, T. 53, № 1, стр. 84-90
Экспрессия генов контроля клеточного цикла, окислительного стресса и апоптоза (Chek1, Hmox1, Casp7) в печени крыс в ответ на тетрахлорметан
Г. Ф. Мухаммадиева a, *, Д. О. Каримов a, Т. Г. Кутлина a, Я. В. Валова a, Н. Ю. Хуснутдинова a, Э. Ф. Репина a, А. Б. Бакиров a
a Уфимский научно-исследовательский институт медицины труда и экологии человека
450106 Уфа, Россия
* E-mail: ufniimt@mail.ru
Поступила в редакцию 13.04.2018
После доработки 03.08.2018
Принята к публикации 03.08.2018
Аннотация
Тетрахлорметан относится к одному из наиболее хорошо изученных гепатотропных ядов. Отравление экспериментальных животных тетрахлометаном схоже с острыми поражениями печени различной этиологии у человека. Исследована экспрессия генов контроля клеточного цикла, апоптоза и окислительного стресса на модели индуцированного тетрахлорметаном токсического гепатита. В работе использованы белые беспородные крысы-самцы, которым однократно вводили 50%-ный масляный раствор тетрахлорметана в дозе 0.125–4.000 г/кг (экспериментальная группа) или оливковое масло (контрольная группа). Через 24 и 72 ч после введения тетрахлорметана животных декапитировали и, используя ПЦР в режиме реального времени, анализировали в печени экспрессию генов гемоксигеназы-1 (Hmox1), киназы-1 контрольной точки клеточного цикла (Chek1) и каспазы-7 (Casp7). Показано, что в печени животных из экспериментальной группы повышена экспрессия генов Hmox1 и Chek1, возможно, принимающих участие в патологических процессах, происходящих в печени при воздействии окислительного стресса. Можно предположить, что при формировании токсического поражения печени под действием тетрахлометана наибольший вклад в гибель клеток вносит некроз.
ВВЕДЕНИЕ
Токсическое поражение печени может привести к необратимым нарушениям функционирования организма человека [1, 2]. При попадании в печень токсических агентов происходят морфологические изменения ткани органа и связанные с ними обменные нарушения [3]. Один из главных механизмов повреждения клеток печени – образование свободных радикалов и активных метаболитов кислорода, которые стимулируют перекисное окисление липидов, тем самым повреждая мембраны клеток печени и эндоплазматической сети, что приводит к гибели гепатоцитов [4, 5]. Токсическое поражение печени часто сопровождается серьезным повреждением ДНК гепатоцитов, что приводит к активации путей репарации ДНК или к гибели клетки путем некроза или апоптоза.
Тетрахлорметан (CCl4) применяют для моделирования заболеваний печени. При воздействии CCl4 происходит развитие дистрофических и некротических изменений в ткани печени, а затем установление гистологической картины фиброза или цирроза [6].
Ведущую роль в поддержании функционального и структурного гомеостаза печени играет гемоксигеназная сигнальная система, способная активировать как защитно-адаптационные механизмы, так и деструктивно-патологические [7]. Изофермент гемоксигеназа-1 относится к белкам теплового шока, его экспрессия может быть индуцирована с помощью многих агентов, в том числе CCl4. Гемоксигеназа-1 принимает участие в адаптации клеток и тканей к действию окислительного стресса [8]. При гиперактивации гемоксигеназы-1 повышается резистентность клеток к апоптическим и некротическим изменениям, снижается воспалительная реакция, угнетается фиброгенная активность звездчатых клеток печени [9, 10]. Основные функциональные характеристики гемоксигеназы-1 обусловлены свойствами продуктов ее реакции: монооксида углерода (СО) и билирубина.
Протеолитическая система, включающая семейство каспаз, считается ключевым участником апоптоза. Эти белки принадлежат к классу цистеиновых протеаз [11]. Выделяют 14 видов каспаз, работающих по каскадному механизму. По выполняемой каспазами функции их можно разделить на 3 группы: активаторы цитокинов; каспазы-индукторы активации эффекторных каспаз; эффекторные каспазы-исполнители апоптотической программы [12, 13]. При активации эффекторных каспаз происходит деградация белков клетки, ответственных за различные жизненные функции, что в итоге приводит к образованию апоптотических телец. К эффекторным каспазам относится каспаза-7, которая принимает участие в финальной стадии процесса апоптоза [14].
Важную роль в поддержании геномной стабильности в клетке играет молекулярная система, вызывающая остановку клеточного цикла, экспрессию белков репарации или апоптотические сигнальные пути при невозможности удаления повреждения. Активируясь в ответ на разнообразные нарушения структуры ДНК, специфические протеинкиназы ATM (Ataxia-Telangiectasia Mutated) и ATR (ATM Related) фосфорилируют ряд своих мишеней, в том числе эффекторные киназы CHEK1 и CHEK2 (киназы 1 и 2 контрольных точек клеточного цикла); причем киназа ATM специфично фосфорилирует CHEK2, а ATR – CHEK1 [15–17]. Киназы CHEK1 и CHEK2 – это серин/треониновые киназы, не гомологичные по структуре, но сходные по функциям и субстратам фосфорилирования. Они служат связующим звеном между киназами АТМ/ATR и белковыми комплексами, непосредственно принимающими участие в процессах репарации ДНК. Эти киназы координируют процессы, происходящие по мере продвижения по клеточному циклу, играя роль своеобразного “переключателя” между репарацией и апоптозом. Активирующее фосфорилирование CHEK1 и CHEK2 приводит в итоге к остановке синтетических процессов и восстановлению стабильности генома [18].
Нами исследована экспрессия генов, которые кодируют ферменты, принимающие участие в контроле окислительного стресса, клеточного цикла и апоптоза, а именно гемоксигеназу-1 (Hmox1), киназу-1 контрольных точек (Chek1) и каспазу-7 (Casp7). Исследования проведены в условиях токсического поражения печени у крыс.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Животные. Работа проведена на 84 белых беспородных крысах-самцах массой 170–190 г. Животным контрольной группы (n = 12) подкожно вводили оливковое масло, а экспериментальной группы (n = 72) – 50%-ный масляный раствор CCl4 (однократно в дозе 0.125–4.000 г/кг массы тела животного). На каждую дозу CCl4 брали по 6 животных. Через 24 и 72 ч после начала опыта исследовали печень декапитированных крыс. Животных обеих групп одинаково кормили и содержали в одних и тех же условиях. При уходе за животными и проведении экспериментов руководствовались базисными нормативными документами: “Рекомендациями комитета по экспериментальной работе с использованием животных при Министерстве здравоохранения Российской Федерации”, рекомендациями ВОЗ, рекомендациями Европейской конвенции по защите позвоночных животных, используемых для экспериментальных и других целей [19].
Пробоподготовка и ПЦР-анализ. Экспрессию генов анализировали с помощью метода ПЦР с обратной транскрипцией в реальном времени. Для проведений исследований кусочки печени замораживали жидким азотом. Из полученных образцов выделяли тотальную РНК с помощью реагента ExtractRNA (“Евроген”, Россия) согласно протоколу производителя. Синтез кДНК проводили с матрицы выделенной тотальной РНК с использованием набора реактивов MMLV RT kit и праймеров олиго(dT)15 (“Евроген”, Россия). С полученными кДНК ставили ПЦР на амплификаторе Rotor-Gene Q (“Qiagen”, Германия) в присутствии SYBR Green. Олигонуклеотидные праймеры для ПЦР подбирали с помощью программы PrimerQuest (“Integrated DNA Technologies, Inc.”, США). Выбранные праймеры синтезированы фирмой “Евроген”.
Hmox1:
5'-CTCACAGACAGAGTTTCTTCGC-3' (прямой),
5'-GCTGATCTGGGATTTTCCTCGG-3' (обратный).
Chek1:
5'-ATACTGGTTGACTTCCGGCT-3' (прямой),
5'-CGCTGAGCTTCCCTTTAATCTT-3' (обратный).
Casp7:
5'-CCCGCTACAAGATCCCGGTG-3' (прямой),
5'-GCACAAACCAGGAGCCCTTTC-3' (обратный).
Каждую реакцию ПЦР, содержащую 2 мкл кДНК, проводили в объеме 25 мкл в следующих условиях: предварительный прогрев при 95°С в течение 3 мин, после чего следовало 45 основных циклов: 15 с при 95°С, 25 с при 59°С и 15 с при 72°С. В качестве гена сравнения использовали ген домашнего хозяйства Gapdh.
Статистическая обработка результатов. Статистическую обработку экспериментальных данных проводили с использованием t-критерия Стьюдента и однофакторного дисперсионного анализа (ANOVA), а также H-критерия Краскела–Уоллиса для попарного сравнения групп. Результаты считали достоверными при p < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Ген Hmox1. В результате проведенных исследований показано, что через 24 ч после введения крысам CCl4 в дозе 0.125 г/кг наблюдается незначительное снижение экспрессии гена Hmox1 по сравнению с контрольной группой животных (H = = 0.31, p = 0.76) (рис. 1). При более высоких дозах CCl4, 0.25 и 0.5 г/кг, уровень экспрессии Hmox1 увеличивался соответственно в 2.4 (H = 2.63, p = = 0.009) и в 1.9 раз (H = 2.46, p = 0.014). Транскрипционная активность этого гена достигала максимального значения (в 3.6 раз выше контрольного) при дозе CCl4 1 г/кг (H = 1.65, p = 0.163), но различия в этом случае не были статистически значимыми. При повышении дозы до 2 г/кг и 4 г/кг уровень экспрессии Hmox1 немного снижался, но оставался выше контрольных значений более чем в 2 раза (H = 3.48, p = 0.001 и H = 2.95, p = 0.003 соответственно).
Рис. 1.
Экспрессия гена Hmox1 в печени крыс через 24 (⚫) и 72 (◆) ч после обработки CCl4. Символом (*) обозначены значения, статистически значимо отличающиеся от контроля. Маркерами (┬) и (┴) отмечен 95%-ный доверительный интервал. Пунктирной линией отмечены уровни кратности экспрессии, равные 1 и –1.

Иной характер носила динамика экспрессии гена Hmox1 через 72 ч после введения CCl4 (рис. 1). При введении минимальной дозы CCl4 экспрессия Hmox1 практически на оличалась от контроля (H = 0.4, p = 0.65), с увеличением дозы от 0.25 г/кг до 4 г/кг наблюдалось незначительное снижение уровня мРНК гена Hmox1 (p > 0.05); при этом отличие от контрольной группы не превосходило 1.7 раза. Максимальное снижение экспрессии Hmox1 отмечено при дозе 1 г/кг (H = 0.83, p = 0.56).
Ген Chek1. Нами проанализирована экспрессия гена Chek1 через 24 и 72 ч после введения разных доз CCl4 (рис. 2). Через 24 ч отмечено снижение уровня экспрессии гена Chek1 в 1.6 раз при дозе CCl4 0.125 г/кг (H = 0.82, p = 0.415). Дальнейшее увеличение дозы CCl4 от 0.25 г/кг до 4 г/кг приводило к нарастанию уровня экспрессии. Максимально высокая транскрипционная активность (в 2.9 раз выше контрольной) зарегистрирована при дозе 4 г/кг (H = 1.74, p = 0.142). Таким образом, для гена Chek1 выявлена следующая тенденция: увеличение экспрессии гена Chek1 при повышении дозы токсиканта, – но без достижения уровня статистической значимости.
Рис. 2.
Экспрессии гена Chek1 в печени крыс через 24 (⚫) и 72 (◆) ч после обработки CCl4. Символом (*) обозначены значения, статистически значимо отличающиеся от контроля. Маркерами (┬) и (┴) отмечен 95%-ный доверительный интервал. Пунктирной линией отмечены уровни кратности экспрессии, равные 1 и –1.
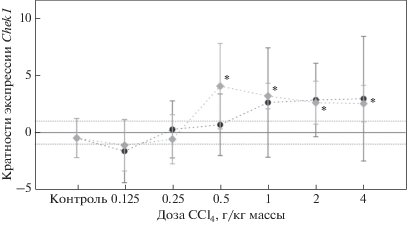
Через 72 ч после введения CCl4 в дозе 0.125 г/кг уровень транскриптов гена Chek1 снизился немного ниже контрольного – в 1.1 раз (H = 0.64, p = 0.52). При воздействии CCl4 в дозе 0.25 г/кг экспрессия гена Chek1 практически не отличалась от контрольного уровня (H = 0.19, p = 0.85). В группе крыс, обработанных CCl4 в дозе 0.5 г/кг, экспрессия гена Chek1 повышалась в 4.1 раз, достигая максимального значения (H = 3.00, p = 0.003); при увеличении дозы от 1 г/кг до 4 г/кг уровень экспрессии несколько снижался, но по-прежнему превышал исходный в 2–3 раза (p < 0.05).
Ген Casp7. Экспрессию гена Casp7 анализировали также через 24 и 72 ч после введения разных доз CCl4. В целом, уровень экспрессии этого гена оставался ниже исходных значений вне зависимости от дозы и времени воздействия токсиканта. Однако есть некоторые различия в динамике экспрессии гена Casp7 через 24 и 72 ч после введения CCl4 (рис. 3).
Рис. 3.
Экспрессии гена Casp7 в печени крыс через 24 (⚫) и 72 (◆) ч после обработки CCl4. Символом (*) обозначены значения, статистически значимо отличающиеся от контроля. Маркерами (┬) и (┴) отмечен 95%-ный доверительный интервал. Пунктирной линией отмечены уровни кратности экспрессии, равные 1 и –1.
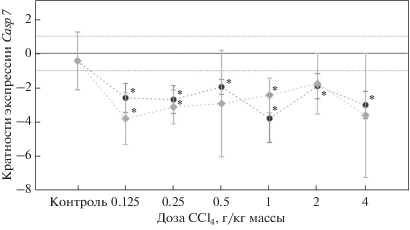
Тетрахлорметан при введении крысам в дозе 0.125 г/кг через 24 ч вызывал почти трехкратное снижение уровня экспрессии гена Casp7 относительно контрольного (H = 3.04, p = 0.002). Экспрессия сохранялась почти на том же уровне и при воздействии CCl4 в дозе 0.25 г/кг (H = 3.19, p = 0.001). Наиболее выраженное снижение экспрессии гена Casp7 происходило при дозе CCl4 1 г/кг и 4 г/кг – в 3.8 (H = 4.54, p = 0.001) и 3.0 (H = 3.94, p = 0.001) раза соответственно. При дозе 0.5 г/кг и 2 г/кг наблюдалось небольшое повышение уровня транскрипта, но при этом показатели сохранялись ниже исходного уровня (H = 2.46, p = = 0.013 и H = 2.20, p = 0.028 соответственно).
Экспрессия гена Casp7 через 72 ч после воздействия CCl4 в дозе 0.125 г/кг была более чем в 3 раза снижена в сравнении с контрольной группой (H = 3.87, p = 0.001). Выявлены определенные тенденции в изменении экспрессии этого гена при воздействии разных доз CCl4. При повышении дозы CCl4 от 0.25 г/кг (H = 3.32, p = 0.001) до 2 г/кг (H = 1.86, p = 0.063) уровень транскрипта гена Casp7 немного повышался, а воздействие CCl4 в дозе 4 г/кг сопровождалось снижением уровня экспрессии в 3.6 раза, хотя различия не были статистически значимыми (H = 1.62, p = 0.10).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Ген Hmox1 кодирует гемооксигеназу-1, которая принимает участие в окислении свободного гема и превращении его в антиоксидант билирубин. Индукция клетками гемоксигеназы-1 в ответ на внешние факторы способствует снижению интенсивности окислительных процессов, ингибирует синтез фактора некроза опухоли-α (TNF-α) и снижает активность каспазы-3, тем самым препятствует развитию деструктивных процессов в печени [20, 21]. Установлено, что подавление экспрессии гемоксигеназы-1 у животных приводит к некротическим воспалительным процессам в печени [22].
Нами показано, что через 24 ч после введения крысам CCl4 в печени животных экспрессия гена Hmox1 усиливалась. Повышение экспрессии Hmox1 в ответ на окислительный стресс, возможно, представляет собой один из механизмов сохранения структурно-функциональной целостности органа. Полученные результаты согласуются с данными других исследователей, согласно которым в небольших дозах CCl4 усиливает экспрессию гена Hmox1 [23]. Известно антиапоптотическое действие гемоксигеназы-1, обусловленное образованием продуктов реакции, главным образом CO. Антиапоптотический эффект CO может быть связан с угнетением активности каспаз [24].
Однако через 72 ч после введения крысам CCl4 происходило снижение экспрессии Hmox1, что может быть обусловлено преобладанием некротических процессов в тканях печени, которые сопровождаются изменениями в паттернах экспрессии генов.
Ген Chek1 кодирует синтез киназы 1 контрольной точки клеточного цикла, или чекпойнт-киназы-1. Белковый продукт гена Chek1 – “ключевой игрок” в механизме остановки клеточного цикла в S и G2/M фазах. Основной мишенью этой киназы служат белки семейства CDC25, которые удаляют гиперфосфорилирование регулируемых ими циклинзависимых киназ. В результате замедляется репликация ДНК и предотвращается вход клеток в митоз [25, 26].
При изучении экспрессии Chek1 в образцах ткани печени крыс, полученной через 24 ч после воздействия CCl4, заметна тенденция к повышению уровня транскриптов этого гена в зависимости от дозы токсического вещества. Возможно, это связано с тем, что в ответ на нарушения структуры ДНК происходит активация специфических киназ (ATM, ATR и их мишеней – CHEK1 и CHEK2), играющих ключевую роль в интеграции сигналов от поврежденной ДНК и их дальнейшей передаче разнообразным эффекторам [27].
В общих чертах в экспрессии гена Chek1 через 72 ч после воздействия CCl4 просматривается та же тенденция к повышению при увеличении дозы токсиканта. Максимальный уровень наблюдался при дозе 0.5 г/кг, при увеличении дозы вплоть до 4 г/кг выраженных изменений не зарегистрировано. По-видимому, активированная протеинкиназа ATR, накапливаясь в местах повреждений, фосфорилирует несколько эффекторов, включая CHEK1 [28], которые в свою очередь фосфорилируют и инактивируют белки семейства CDC25, приводя к опосредованному ингибированию комплекса CDK–циклин и, тем самым, вызывая задержку или остановку клеточного цикла [29, 30].
Ген Casp7 кодирует каспазу-7, которая относится к эффекторным каспазам. Каспаза-7 играет роль ключевого регулятора регенерации ткани. Активную форму каспазы-7 можно обнаружить в митохондриальной и микросомальной фракции гепатоцитов [31, 32].
При использованных дозах CCl4 в обоих временных промежутках в исследуемых группах животных наблюдалось снижение уровня транскриптов гена Casp7 в печени. Это значит, что в условиях, неблагоприятных для развития каспазного каскада, запускаемого каспазой-7, будет снижена апоптотическая активность клеток. Возможно, в случае инактивации каспазы-7 происходит запуск некроптоза. Ранее показано [33], что гепатотоксичность, которую оценивали с помощью метода ДНК-комет, обусловлена повреждением генома гепатоцитов. В группе животных, которым вводили CCl4 в дозе 0.4 мл/кг/сутки, в печени повышалась доля некрозо-апоптических клеток (поврежденные клетки с признаками некроза и апоптоза) по сравнению с контролем.
Таким образом, у крыс, подвергнутых однократному токсическому воздействию CCl4 в разных дозах и при разном времени воздействия, обнаружено повышение транскрипционной активности как гена, участвующего в регуляции окислительного стресса (Hmox1), так и гена, контролирующего клеточный цикл (Chek1), а также снижение экспрессии гена, вовлеченного в регуляцию апоптоза (Casp7). На основании полученных результатов можно предположить, что при формировании токсического поражения печени под действием CCl4 наибольший вклад в гибель клеток вносит некроз. Полученные данные расширяют наши представления о молекулярных механизмах повреждения печени при воздействии CCl4.
Список литературы
Валеева Э.Т. (2011) Профессиональные токсические поражения печени у работников производства жидкого топлива. Здравоохранение Российской Федерации. 4, 50–51.
Косарев В.В., Бабанов С.А. (2009) Токсические (токсико-аллергические) гепатиты. Гастроэнтерология. Приложение к журналу Consilium Medicum. 2, 29–30.
Владимиров Ю.А. (2000) Свободные радикалы в биологических системах. Соросовский образовательный журнал. 12, 13–19.
Голиков С.Н., Саноцкий И.В., Тиунов Л.А. (1986) Общие механизмы токсического действия. Ленинград: Медицина.
Кравченко Л.В, Трусов Н.В., Ускова М.А., Аксенов И.B., Авреньева Л.И., Гусева Г.В., Васильева М.А., Селифанов А.В., Тутельян В.А. (2009) Характеристика острого токсического действия четыреххлористого углерода как модели окислительного стресса. Токсикологический вестник. 1, 12–18.
Weber L.W., Boll M., Stampfl A. (2003) Hepatotoxity and mechanism of action of haloalcanes: carbon tetrachloride as a toxicological model. Crit. Ref. Toxicol. 33, 105–136.
Li Volti G., Sacerdoti D., Di Giacomo C., Barcellona M.L., Scacco A., Murabito P., Biondi A., Basile F., Gazzolo D., Abella R., Frigiola A., Galvano F. (2008) Natural heme oxygenase-1 inducers in hepatobiliary function. World J. Gastroenterol. 14, 6122–6132.
Wen T., Guan L., Zhang Y.L., Zhao J.Y. (2006) Dynamic changes of heme oxygenase-1 and carbon monoxide production in acute liver injury induced by carbon tetrachloride in rats. Toxicology. 228, 51–57.
Abraham N.G., Kappas A. (2008) Pharmacological and clinical aspects of heme oxygenase. Pharmacol. Rev. 60, 79–127.
Soares M.P., Bach F.H. (2009) Heme oxygenase-1: from biology to therapeutic potential. Trends Mol. Med. 15, 50–58.
Chinnaiyan A.M., Dixit V.M. (1996) The cell-death machine. Curr. Biol. 6, 555–562.
Владимирская Е.Б. (2002) Механизмы апоптотической смерти клеток. Гематология и трансфузиология. 47, 35–40.
Рыжов С.В., Новиков В.В. (2002) Молекулярные механизмы апоптотических процессов. Российский биотерапевтический журнал. 1, 27–33.
Lamkanfi M., Kanneganti T.D. (2010) Caspase-7: a protease involved in apoptosis and inflammation. Int. J. Biochem. Cell Biol. 42, 21–24.
Brown E.J., Baltimore D. (2003) Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 17, 615–628.
Liu Q., Guntuku S., Cui X.S., Matsuoka S., Cortez D., Tamai K., Luo G., Carattini-Rivera S., DeMayo F., Bradley A., Donehower L.A., and Elledge S.J. (2000) Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev. 14, 1448–1459.
Zhou B.B., Elledge S.J. (2000) The DNA damage response: putting checkpoints in perspective. Nature. 408, 433–439.
Hiom K. (2005) DNA repair: how to PIKK a partner. Curr. Biol. 15, 473–475.
Белоусова Ю.Б. (2005) Этическая экспертиза биомедицинских исследований: практические рекомендации. Москва: Оки.
Jais A., Einwallner E., Sharif O., Gossens K., Lu T.T., Soyal S.M., Medgyesi D., Neureiter D., Paier-Pourani J., Dalgaard K., Duvigneau J.C., Lindroos-Christensen J., Zapf T.C., Amann S., Saluzzo S., Jantscher F., Stiedl P., Todoric J., Martins R., Oberkofler H., Müller S., Hauser-Kronberger C., Kenner L., Casanova E., Sutterlüty-Fall H., Bilban M., Miller K., Kozlov A.V., Krempler F., Knapp S., Lumeng C.N., Patsch W., Wagner O., Pospisilik J.A., Esterbauer H. (2014) Heme oxygenase-1 drives metaflammation and insulin resistance in mouse and man. Cell. 158, 25–40.
Jou J., Choi S., Diehl A. (2008) Mechanisms of disease progression in nonalcoholic fatty liver disease. Semin. Liver Dis. 28, 370–379.
Yao P., Li K., Song F., Zhou S., Sun X., Zhang X., Nussler A.K., Liu L. (2007) Heme oxygenase-1 upregulated by Ginkgo biloba extract: potential protection against ethanol-induced oxidative liver damage. Food Chem. Toxicol. 45, 1333–1342.
Randle L.E., Goldring C.E., Benson C.A., Metcalfe P.N., Kitteringham N.R., Park B.K., Williams D.P. (2008) Investigation of the effect of a panel of model hepatotoxins on the Nrf2-Keap1 defence response pathway in CD-1 mice. Toxicology. 243, 249–260.
Conde de la Rosa L., Vrenken T.E., Hannivoort R.A., Buist-Homan M., Havinga R., Slebos D.J., Kauffman H.F., Faber K.N., Jansen P.L., Moshage H. (2008) Carbon monoxide blocks oxidative stress-induced hepatocyte apoptosis via inhibition of the p54 JNK isoform. Free Radic. Biol. Med. 44, 1323–1333.
Beck H., Nähse V., Larsen M.S., Groth P., Clancy T., Lees M., Jørgensen M., Helleday T., Syljuåsen R.G., Sørensen C.S. (2010) Regulators of cyclin dependent kinases are crucial for maintaining genome integrity in S phase. J. Cell Biol. 188, 629–638.
Cerqueira A., Santamaría D., Martínez-Pastor B., Cuadrado M., Fernández-Capetillo O., Barbacid M. (2009) Overall Cdk activity modulates the DNA damage response in mammalian cells. J. Cell Biol. 187, 773–780.
Заридзе Д.Г. (2004) Канцерогенез. Москва: Медицина.
Brown E.J., Baltimore D. (2003) Essential and dispensable roles of ATR in cell cycle arrest and genome maintenance. Genes Dev. 17, 615–628.
Morgan D. (2007) The Cell Cycle: Principles of Control. London: New Science Press.
Reinhardt H.C., Yaffe M.B. (2009) Kinases that control the cell cycle in response to DNA damage: Chk1, Chk2, and MK2. Curr. Opin. Cell Biol. 21, 245–255.
Chandler J.M., Cohen G.M., MacFarlane M. (1998) Different subcellular distribution of caspase-3 and caspase-7 following Fas-induced apoptosis in mouse liver. J. Biol. Chem. 273, 10815–10818.
Li F., Huang Q., Chen J., Peng, Y., Roop D.R., Bedford J.S., Li C.Y. (2010) Apoptotic cells activate the “phoenix rising” pathway to promote wound healing and tissue regeneration. Sci. Signal. 3, ra13.
Кропотов А.В., Челомин В.П., Слободскова В.В., Солодова Е.Е., Михайлов А.О. (2013) Оценка генотоксичности тетрахлорметана и защитного действия силибинина и хаурантина с помощью метода ДНК-комет в печени крыс. Тихоокеанский медицинский журнал. 2, 63–66.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология