

Молекулярная биология, 2019, T. 53, № 1, стр. 101-108
Влияние донора сероводорода GYY4137 на активацию нейтрофилов человека липополисахаридами E. сoli
И. Ю. Петрушанко a, Е. В. Мельникова a, М. М. Юринская a, b, М. Г. Винокуров b, А. В. Сусликов c, В. А. Митькевич a, А. А. Макаров a, *
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
b Институт биофизики клетки Российской академии наук
142290 Московская обл., Пущино, Россия
c Больница Пущинского научного центра Российской академии наук
142290 Московская обл., Пущино, Россия
* E-mail: aamakarov@eimb.ru
Поступила в редакцию 09.09.2018
После доработки 09.09.2018
Принята к публикации 14.09.2018
Аннотация
Липополисахариды, компоненты клеточной стенки грамотрицательных бактерий, активируют нейтрофилы, провоцируя многие патологические процессы, в том числе грамотрицательный сепсис. Липополисахариды ингибируют спонтанный апоптоз нейтрофилов, что индуцирует развитие воспаления. В работе охарактеризовано действие донора H2S (GYY4137) на активацию нейтрофилов человека липополисахаридами Escherichia сoli. Оценено изменение редокс-статуса нейтрофилов (уровня активных форм кислорода, внутриклеточного глутатиона, NO), апоптоза и митохондриального потенциала клеток под действием липополисахаридов в присутствии и в отсутствие GYY4137. GYY4137 снижает уровень активных форм кислорода в нейтрофилах, незначительно снижает уровень внутриклеточного глутатиона и не влияет на уровень NO, не оказывая при этом апоптогенного действия. Липополисахариды индуцируют повышение содержания активных форм кислорода и ингибируют спонтанный апоптоз нейтрофилов. Установлено, что GYY4137 предотвращает рост уровня активных форм кислорода, вызванный липополисахаридами, и приводит к снижению индуцированного липополисахаридами ингибирования апоптоза нейтрофилов. Таким образом, механизм защитного действия GYY4137 при воспалении, вызванном бактериальной инфекцией, связан с нейтрализацией эффекта липополисахаридов на нейтрофилы.
Клетки системы врожденного иммунитета – нейтрофилы, или полиморфноядерные лейкоциты, являются, наряду с макрофагами и дендритными клетками, первичными фагоцитами, которые привлекаются в очаг поражения для защиты от внешнего патогена. Нейтрофилы активируются в ответ на патоген, что сопровождается выделением активных форм кислорода (АФК) во внешнюю среду и способствует гибели чужеродных клеток. Однако чрезмерная активация нейтрофилов может вызывать развитие воспаления и гибель собственных клеток организма. Нейтрофилы несут на своей поверхности Толл-подобный рецептор 4 (TLR4), они способны активироваться под действием липополисахаридов (LPS) – основных компонентов клеточной стенки грамотрицательных бактерий. LPS высвобождаются из клеточной стенки и образуют комплекс с LBP (LPS-binding protein), который передает LPS на мембранную форму CD14, а затем на молекулу MD2. Далее в результате димеризации двух комплексов LPS-MD2-TLR4 формируется гетеродимерный рецепторный комплекс [1]. Этот комплекс активируется после присоединения большой группы адаптерных белков, что приводит к передаче сигнала на внутриклеточные сигнальные пути (с участием ERK1/2, p38MAPK, SAPK/JNK, PI3K). На следующем этапе эти сигналы достигают факторов транскрипции, таких как ядерный фактор (NF-κB) и белок AP-1, что приводит к секреции провоспалительных медиаторов, включая NO, АФК, TNFα [2]. Активация нейтрофилов под действием LPS вызывает увеличение продукции АФК, повышение экспрессии факторов адгезии в клеточной мембране и секреции ряда провоспалительных цитокинов и медиаторов воспаления. LPS ингибирует апоптоз нейтрофилов, что увеличивает время жизни активированных клеток. Активация нейтрофилов и других клеток врожденного иммунитета под действием LPS считается ключевым событием патогенеза грамотрицательного сепсиса и других заболеваний, связанных с LPS [3].
Работы последних лет показали, что газотрансмиттер сероводород (H2S) вовлечен в регуляцию многих биологических систем. Доноры сероводорода все чаще рассматривают в качестве возможных терапевтических средств, обладающих противовирусными и противовоспалительными свойствами. Среди них особое внимание уделяется GYY4137 (morpholin-4-ium 4-methoxyphenyl(morpholino) phosphinodithioate) – первой органической малой молекуле, охарактеризованной как медленный донор H2S, активно изучаемой на различных биологических моделях in vitro и in vivo [4]. Показано, что GYY4137 сдерживает развитие индуцированного LPS воспаления дыхательных путей у мышей, снижая, в частности, количество нейтрофилов в бронхоальвеолярной жидкости [5]. GYY4137 защищает также от LPS-индуцированного повреждения легких, снижая уровень окислительного стресса и воспаления [6]. Показано также, что GYY4137 защищает крыс от эндотоксического шока, индуцированного LPS, ингибируя продукцию провоспалительных цитокинов (TNF-α, IL-1 β, IL-6), снижая активацию NF-kB, миелопероксидазы и других провоспалительных факторов [7]. Механизмы защитного действия GYY4137 и снижения количества нейтрофилов в зоне воспаления до сих пор не установлены. Нами охарактеризовано действие донора сероводорода GYY4137 на нейтрофилы человека, подвергнутые воздействию LPS из Escherichia сoli. Оценено изменение редокс-статуса нейтрофилов, их апоптоза, жизнеспособности и способности к активации при действии LPS.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реагенты. В работе использованы LPS из E. coli О55:В5, культуральная среда RPMI-1640, йодид пропидия (РI), HEPES, раствор Хенкса (HBSS), L-глутамин, фиколл 400, фосфатно-солевой буфер (PBS), форбол-12-миристат-13-ацетат (РМА), хлорид дифенилениодония (diphenyleneiodonium chloride (DPI)), GYY4137 morpholin-4-ium 4-methoxyphenyl(morpholino) phosphinodithioate dichloromethane complex (“Sigma-Aldrich”, США). Эмбриональная телячья сыворотка, тестированная на присутствие эндотоксина (0.01 ед./мл) (“HyClone”, США).
Выделение нейтрофилов. Нейтрофилы выделяли из периферической крови здоровых доноров-добровольцев с помощью метода дифференциального центрифугирования на двухслойном градиенте фиколла–верографина (1.119 и 1.077 г/мл). Чистоту выделенных нейтрофилов контролировали по их морфологии. Содержание нейтрофилов в образце составляло ~98% от общего количества клеток. Жизнеспособность нейтрофилов во всех экспериментах контролировали на флуоресцентном микроскопе с использованием PI (30 мкМ) в качестве флуоресцентного зонда, измеряя долю клеток, непроницаемых для PI, в общем количестве клеток [8]. Жизнеспособность клеток после выделения составляла 98–99%.
Проточная цитометрия. Популяции нейтрофилов анализировали на проточном цитофлуориметре BD LSRFortessa™ (“Becton Dickinson”, США). Перед окрашиванием клетки центрифугировали, удаляли супернатант и ресуспендировали в 100 мкл PBS. Уровень АФК оценивали с помощью дигидрородамина 123 (DHR 123) (“Invitrogen”, США, Ex/Em = 507/525), который позволяет регистрировать АФК не только в цитозоле, но и в митохондриях. Клетки окрашивали, добавляя к ним DHR 123 до конечной концентрации 10 мкМ и инкубировали в течение 30 мин при 37°С в темноте. Уровень глутатиона оценивали с помощью ThiolTracker (Ex/Em = 405/525 нм), а уровень NO c помощью DAF-FM (Ex/Em = = 495/515 нм) (“Invitrogen”). К клеткам для окрашивания добавляли ThiolTracker до конечной концентрации 7.5 мкМ или DAF-FM до концентрации 10 мкМ и инкубировали в течение 30 мин при 37°С в темноте [9, 10]. Апоптотические клетки регистрировали с помощью аннексина V (“Invitrogen”), меченного флуоресцеином (FITC, Ex/Em = 494/518 нм). Апоптотическими клетками считали клетки, которые окрашивались аннексином V, но не окрашивались PI. Для определения апоптоза суспензию клеток центрифугировали (1000 об/мин, 10 мин), отбирали супернатант и ресуспендировали в буфере для связывания аннексина (10 мM HEPES, 140 мM NaCl, 2.5 мM CaCl2, pH 7.4). Затем к 100 мкл суспензии клеток (106 клеток/мл) добавляли 5 мкл раствора конъюгата аннексина. Пробы инкубировали в течение 15 мин в темноте при комнатной температуре, после чего добавляли 400 мкл буфера для связывания аннексина и ставили на лед [10]. Изменение величины митохондриального потенциала (Ψ) оценивали с помощью красителя MitoProbe DilC1(5) (Ex/Em = 638/658 нм) (“Invitrogen”) в клетках с неповрежденной мембраной [10]. При снижении митохондриального потенциала интенсивность флуоресценции DilC1(5) снижается. Для определения клеток с поврежденной мембраной за 1 мин до измерения добавляли PI до концентрации 10 мкг/мл (“Sigma”, США, Ex/Em = 535/617 нм). Уровень глутатиона, АФК, NO и величину митохондриального потенциала определяли по средней интенсивности флуоресценции в клетках, не окрашиваемых PI.
Статистический анализ. Приведены средние значения ± стандартные отклонения (SD).
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Влияние донора H2S GYY4137 и LPS из E. coli на редокс-статус нейтрофилов
Влияние донора сероводорода GYY4137 на редокс-статус нейтрофилов оценивали по его влиянию на уровень АФК, восстановленного глутатиона и оксида азота как в контроле, так и в присутствии LPS из E. coli.
Донор H2S снижает продукцию АФК в нейтрофилах и предотвращает повышение продукции АФК под действием LPS
Инкубация нейтрофилов с донором H2S GYY4137 (200 мкМ) в течение 17–18 ч приводит к снижению уровня АФК в нейтрофилах более чем на 35% (рис. 1). LPS (1 мкг/мл) индуцирует повышение уровня АФК в нейтрофилах более чем на 40%. Присутствие в среде инкубации GYY4137 (добавлен за 30 мин до LPS) предотвращает повышение продукции АФК под действием LPS, сохраняя его значение на уровне контроля.
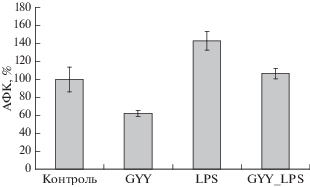
LPS и донор H2S снижают уровень восстановленного глутатиона в нейтрофилах
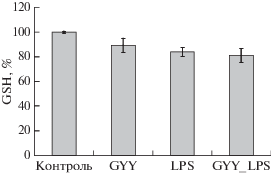
LPS, индуцирующие повышение продукции АФК в нейтрофилах, приводят к снижению уровня восстановленного глутатиона (GSH), что связано с развитием окислительного стресса (рис. 2). Инкубация нейтрофилов с донором H2S незначительно снижает уровень GSH. Совместная инкубация LPS с GYY4137 также вызывает снижение уровня GSH.
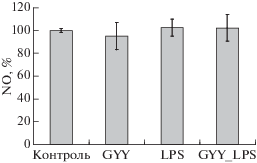
LPS и донор H2S не влияют на уровень NO в нейтрофилах
Инкубация нейтрофилов с LPS не приводит к изменению уровня NO. В присутствии донора сероводорода уровень NO также не меняется (рис. 3).
Влияние LPS и GYY4137 на жизнеспособность нейтрофилов
Жизнеспособность контрольных нейтрофилов постепенно снижается (рис. 4–7) вследствие спонтанного апоптоза. После инкубации в течение 17–18 ч существенно возрастает количество апоптотических клеток (более чем до 50%), на поверхности которых повышено количество фосфатидилсерина и снижен митохондриальный потенциал (рис. 4, 5). При этом количество мертвых клеток, т.е. клеток с поврежденной мембраной, составляет около 10% (рис. 6). Содержание мертвых и гибнущих клеток в контроле превышает 60% (рис. 7).
Рис. 4.
Изменение содержания (%) апоптотических клеток после инкубации нейтрофилов с донором H2S (GYY4137, 200 мкМ) и LPS (1 мкг/мл) в течение 17–18 ч.
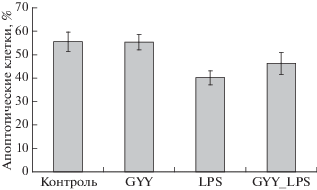
Рис. 5.
Изменение содержания (%) нейтрофилов со сниженным митохондриальным потенциалом после инкубации в течение 24 ч с донором H2S (GYY4137, 200 мкМ) и LPS (1 мкг/мл).
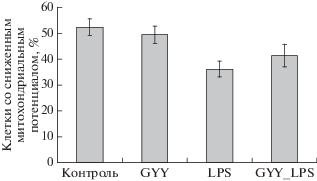
Рис. 6.
Изменение содержания (%) мертвых клеток, т.е. клеток с поврежденной мембраной (клетки, окрашиваемые PI) после инкубации в течение 24 ч с донором H2S (GYY4137, 200 мкМ) и LPS (1 мкг/мл).
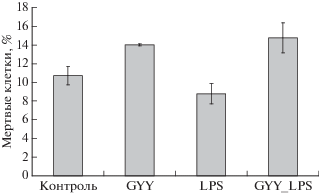
Рис. 7.
Изменение содержания (%) мертвых и гибнущих клеток (все клетки, окрашиваемые PI и/или Аннексином) после инкубации в течение 24 ч с донором H2S (GYY4137, 200 мкМ) и LPS (1 мкг/мл).
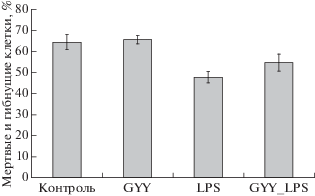
Инкубация нейтрофилов с LPS приводит к существенному повышению их жизнеспособности, снижая почти на 20% долю апоптотических клеток, клеток со сниженным митохондриальным потенциалом и общее число мертвых и гибнущих клеток (рис. 4–7). Донор сероводорода GYY4137 не влияет существенно на жизнеспособность нейтрофилов, не индуцирует апоптоз. Однако в его присутствии наблюдается тенденция к отмене ингибирования апоптоза, вызванного LPS (рис. 4, 5). Суммарная доля гибнущих и мертвых клеток в присутствии GYY4137 и LPS также выше, чем в присутствии одного лишь LPS (рис. 7).
Донор сероводорода влияет на активацию нейтрофилов
Охарактеризовано влияние GYY4137 и LPS на активацию нейтрофилов форбол-12-миристат-13-ацетатом (PMA). Предынкубация нейтрофилов с GYY4137 перед добавлением PMA приводит к стимуляции продукции АФК, тогда как LPS практически не влияет на активацию нейтрофилов PMA. Одновременное присутствие LPS и GYY4137 также повышает продукцию АФК под действием PMA (рис. 8).
Рис. 8.
Изменение уровня АФК в нейтрофилах под действием PMA в присутствии GYY4137 (GYY4137, 200 мкМ), LPS (1 мкг/мл) и GYY4137 c LPS. Клетки инкубировали в течение 30 мин с GYY4137, LPS или GYY4137 c LPS, после чего добавляли 100 нМ PMA и инкубировали в течение еще 30 мин. В половину проб за 15 мин до добавления GYY4137 и LPS вносили ингибитор NADPH-оксидазы DPI (5 мкМ). Уровень АФК в контроле в каждом случае (контроль, GYY, LPS, GYY_LPS) принят за 100%.
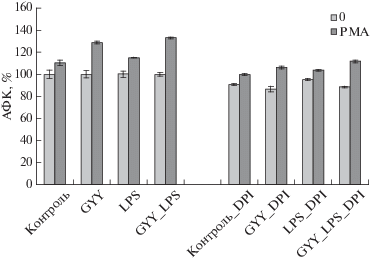
Ингибирование NADPH-оксидазы в присутствии DPI приводит к снижению уровня АФК во всех случаях, более выраженному в присутствии GYY4137. Однако эффект повышения ответа на PMA в среде с GYY4137 сохраняется (рис. 8).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Нейтрофилы – первичные эффекторные клетки острого воспаления – одна из самых многочисленных популяций циркулирующих лейкоцитов. Главной функцией нейтрофилов является защита организма от микроорганизмов путем фагоцитоза. Захваченные нейтрофилами клетки бактерий подвергаются фагоцитозу и погибают, при этом ускоряется апоптоз нейтрофилов [11]. Затем эти апоптотические нейтрофилы удаляются макрофагами без усиления воспаления. При взаимодействии LPS с нейтрофилами наблюдается их активация и ингибирование апоптоза, что проявляется, в первую очередь, в сборке и активации NADPH-оксидазы, которая может непосредственно взаимодействовать с TLR4 (после связывания LPS) [12].
За сутки в кровоток человека поступает около 1011 клеток и столько же гибнет путем апоптоза. Нейтрофилы отличаются коротким временем жизни. В норме нейтрофилы циркулируют в крови менее 24 ч, а затем гибнут путем спонтанного апоптоза [13]. Этим объясняется высокое содержание апоптотических клеток в нашей популяции нейтрофилов через 17 ч инкубации (рис. 4). Сопоставление числа апоптотических (рис. 4) и мертвых (PI-положительных) клеток с поврежденной мембраной (рис. 6) позволяет заключить, что основным путем гибели исследуемых клеток является апоптоз. Необходимо отметить, что содержание апоптотических клеток в популяции совпадает с числом клеток со сниженным митохондриальным потенциалом (рис. 4, 5). Кроме того, для всех апоптотических клеток характерно снижение митохондриального потенциала. Таким образом, развитие апоптоза нейтрофилов в данных условиях протекает по митохондриальному (внутреннему пути). Это согласуется с опубликованными данными, согласно которым внутренний путь апоптоза критичен как для спонтанного (конститутивного), так и для стимулированного апоптоза нейтрофилов [13]. Сигнал, индуцирующий гибель стареющих нейтрофилов, еще не установлен, однако показано, что одним из ключевых ранних событий апоптоза является пермеабилизация внешней митохондриальной мембраны [13].
При инфицировании тканей или их повреждении под действием хемоаттрактантов, к которым относятся и LPS, нейтрофилы мигрируют в ткани, где могут функционировать еще несколько дней. Регуляция апоптоза нейтрофилов очень важна для нормализации воспалительной реакции. При инфицировании и АФК, и протеазы могут высвобождаться из клеток, вызывая локальное повреждение тканей в месте заражения. При воспалительных заболеваниях нейтрофилы, активированные цитокинами и хемокинами, также могут вызывать повреждение тканей [14]. В здоровом организме работают антиоксидантные системы, предотвращающие или снижающие повреждение тканей. В частности, изменяется уровень экспрессии таких ферментов, как глутатионпероксидаза и супероксиддисмутаза, в ряде случаев наблюдается даже выход супероксиддисмутазы из клеток [15]. Важную роль играют также такие антиоксиданты, как глутатион, витамины А, E, С и коэнзим Q, которые восстанавливают пероксид водорода и предотвращают дальнейшее образование АФК. Однако при многих заболеваниях активность антиоксидантных систем снижется, что приводит к существенному поражению собственных тканей вследствие активации нейтрофилов.
Направленная регуляция времени жизни нейтрофилов необходима для разрешения воспаления, так как это предотвращает высвобождение внутриклеточных компонентов, которые могут повреждать здоровую ткань. Некоторые патогены удлиняют жизнь нейтрофилов, поскольку это способствует поддержанию необходимых для них условий, а другие снижают жизнеспособность нейтрофилов или вызывают их лизис, чтобы избежать атаки с их стороны [13]. Одним из факторов, индуцирующих увеличение времени жизни нейтрофилов, являются LPS. Согласно нашим данным, обработка нейтрофилов человека LPS в течение 17 ч повышает продукцию АФК (рис. 1) и вызывает существенное уменьшение числа апоптотических клеток и клеток со сниженным митохондриальным потенциалом (рис. 4–7), т.е. снижает уровень конститутивного (спонтанного) апоптоза. Это согласуется с опубликованными данными [13, 16]. Ингибирование апоптоза нейтрофилов наблюдается как при бактериальной инфекции, так и при сердечно-сосудистых и иных заболеваниях, сопровождающихся развитием воспаления.
Во временной задержке апоптоза нейтрофилов, мигрирующих из кровотока в инфицированные и воспаленные ткани, участвуют несколько частично перекрывающихся сигнальных путей с активацией PI3K и Akt, ERK и NF-kB, которые действуют после активации рецепторов к факторам роста, рецепторов адгезии и TLR4. Решающую роль в выживаемости нейтрофилов под действием LPS играет сигнальный путь PI3K/Akt [17]. В клетках, обработанных LPS, активируется MAP-киназа [18] и увеличивается стабильность белка Mcl-1 [19], ингибирующего Bax, который индуцирует митохондриальный апоптоз, и XIAP [20], ингибирующего каспазу 9, что приводит к увеличению времени жизни нейтрофилов. TLR4-зависимая задержка апоптоза связана с ростом уровня Mcl-1 и A1, антиапоптотических членов семейства Bcl-2. TLR4-зависимое подавление апоптоза нейтрофилов ингибиторами PI3K и NF-kB имеет обратимый характер [21]. Под действием LPS наблюдается не только активация NF-kB и PI3K, но также фосфорилирование ERK и подавление каспазы 3. В присутствии антител против CD11b/CD18 и TLR4 полностью блокируется LPS-индуцированная задержка апоптоза нейтрофилов [22]. Значительную роль в регуляции апоптоза нейтрофилов играют MAP-киназы, в том числе р38МАРК [23], фосфорилирование которой увеличивается при спонтанном и Fas-индуцированном апоптозе, а также при праймировании нейтрофилов LPS. Установлено взаимодействие между ERK и р38МАРК-сигнальными путями, которое определяет регуляцию апоптоза нейтрофилов при грамотрицательном сепсисе [24]. Кроме того, p38MAPK может непосредственно фосфорилировать каспазы 8 и 3 и ингибировать их активность, препятствуя таким образом апоптозу нейтрофилов [25]. Под действием LPS в поверхностной мембране нейтрофилов возрастает экспрессия β2-интегринов (CD11b/CD18). При кластеризации этих рецепторов сигнал активации передается по цепочке MEK–ERK–XIAP, последний член которой подавляет активность каспазы 3 [20].
Задача направленной регуляции времени жизни нейтрофилов очень актуальна, поскольку ее решение позволило бы сократить или замедлить развитие воспаления. В настоящее время идет поиск соединений, индуцирующих апоптоз нейтрофилов. Так, инкубация нейтрофилов с препаратами иммуноглобулинов, выделенных из крови (так называемых внутривенных иммуноглобулинов) индуцирует апоптоз LPS-индуцированных нейтрофилов, приводя к повышению содержания АФК и снижению уровня GSH. При этом ингибитор NADPH-оксидазы (DPI) и антиоксиданты предотвращали этот процесс [26]. Таким образом, в данном случае индукция апоптоза в LPS-стимулированных нейтрофилах связана с образованием АФК NADPH-оксидазой. Ранее было показано, что экзогенные антиоксиданты ингибируют апоптоз нейтрофилов [27, 28], в то время как окислители его ускоряют [3, 29].
Однако мы обнаружили, что обработка нейтрофилов сильным восстановителем – донором сероводорода в концентрации 200 мкМ, стабильно снижает уровень АФК (рис. 1), не влияя при этом на спонтанный апоптоз. В присутствии донора сероводорода LPS не вызывают повышения уровня АФК (рис. 1). При этом наблюдается тенденция к отмене ингибирования апоптоза нейтрофилов, индуцированного LPS. Это может свидетельствовать о положительной роли сероводорода в снижении воспаления, индуцированного нейтрофилами, активированными LPS. Понять механизм, лежащий в основе действия сероводорода, мы попытались, оценив влияние сероводорода на редокс-статус нейтрофилов в присутствии и в отсутствие LPS. Под действием сероводорода уровень АФК в клетках понижается, уровень GSH снижается не очень значительно, а уровень NO не меняется. LPS приводят к росту продукции АФК и снижают содержание GSH, не влияя на уровень NO. Донор сероводорода подавляет индуцированный LPS рост АФК, практически не влияя при этом на уровень глутатиона (рис. 1–3).
Таким образом, снижение уровня АФК и GSH также может приводить к предотвращению LPS-индуцированного ингибирования апоптоза. Роль редокс-статуса в регуляции активации нейтрофилов и продолжительности их жизни до конца не установлена. Данные о роли АФК в регуляции продолжительности жизни нейтрофилов несколько противоречивы. В ряде работ показано, что АФК способны участвовать в активации спонтанного апоптоза нейтрофилов – антиоксиданты ингибируют апоптоз, снижение уровня глутатиона приводит к росту АФК и снижению продолжительности жизни нейтрофилов [30]. Продолжительность жизни нейтрофилов у больных хроническим гранулематозом с нарушенным функционированием NADPH-оксидазы больше, чем у здоровых доноров [31]. Однако в другой работе было показано, что нейтрофилы со сниженным уровнем АФК неустойчивы к апоптозу. Это свидетельствует о том, что АФК, образуемые NADPH, не являются основными медиаторами апоптоза нейтрофилов [32].
Исходя из данных об увеличении доли апоптотических нейтрофилов в присутствии LPS и H2S по сравнению с LPS, можно предположить, что в предотвращении LPS-индуцированного ингибирования апоптоза под действием H2S важную роль играют редокс-чувствительный путь NF-kB, редокс-чувствительные факторы транскрипции и белки, в том числе каспазы. В частности, ингибирование апоптоза LPS сопровождается активацией NF-kB [3] при повышении уровня АФК, в то время как H2S предотвращает рост АФК под действием LPS и, следовательно, предположительно будет предотвращать активацию NF-kB. Также в задержке апоптоза нейтрофилов важную роль играет AФК-зависимая деградация проапоптотического фактора Bad [33]. Поэтому можно полагать, что снижение АФК под действием сероводорода приводит к отмене ингибирования апоптоза, активируя Bad.
Даже кратковременная инкубация нейтрофилов с ингибитором NADPH-оксидазы DPI приводит к снижению общего уровня АФК, что подтверждает участие этого фермента в повышении продукции АФК под действием различных стимулов. Нами установлено, что кратковременная инкубация нейтрофилов с донором сероводорода увеличивает их способность активироваться под действием PMA. По-видимому, это происходит вследствие того, что GYY4137, снижая уровень АФК, позволяет клетке сохранить больший пул восстановительных эквивалентов, в частности NADPH, увеличивая тем самым ресурсы клеток и обеспечивая большую активацию. Ингибитор NADPH DPI не влияет на различия в уровне АФК в активированных PMA нейтрофилах в присутствии GYY4137, что свидетельствует о связи этих различий с функционированием какой-то другой системы. Можно предположить, что GYY4137 влияет на уровень митохондриальных АФК.
Таким образом, полученные нами данные позволяют предположить, что защитное действие донора сероводорода GYY4137 при воспалении может быть связано со снижением продукции АФК, вызванным LPS, и LPS-индуцированного ингибирования апоптоза нейтрофилов.
Работа выполнена при финансовой поддержке Российского научного фонда (проект № 17-74-30030).
Список литературы
Kim S.J., Kim H.M. (2017) Dynamic lipopolysaccharide transfer cascade to TLR4/MD2 complex via LBP and CD14. BMB Rep. 50(2), 55–57.
Liu J., Tang J., Zuo Y., Yu Y., Luo P., Yao X., Dong Y., Wang P., Liu L., Zhou H. (2016) Stauntoside B inhibits macrophage activation by inhibiting NF-κB and ERK MAPK signalling. Pharmacol. Res. 111, 303–315.
Винокуров М.Г., Юринская М.М. (2010) Регуляция апоптоза нейтрофилов при действии липополисахаридов. Биол. мембраны. 27, 18–27.
Rose P., Dymock B.W., Moore P.K. (2015) GYY4137, a novel water-soluble, H2S-releasing molecule. Methods Enzymol. 554, 143–167
Kaya-Yasar Y., Karaman Y., Bozkurt T.E., Onder S.C., Sahin-Erdemli I. (2017) Effects of intranasal treatment with slow (GYY4137) and rapid (NaHS) donors of hydrogen sulfide in lipopolysaccharide-induced airway inflammation in mice. Pulm. Pharmacol. Ther. 45, 170–180.
Zhang H.X., Liu S.J., Tang X.L., Duan G.L., Ni X., Zhu X.Y., Liu Y.J., Wang C.N. (2016) H2S Attenuates LPS-induced acute lung injury by reducing oxidative/nitrative stress and inflammation. Cell. Physiol. Biochem. 40(6), 1603–1612.
Li L., Salto-Tellez M., Tan C.H., Whiteman M., Moore P.K. (2009) GYY4137, a novel hydrogen sulfide-releasing molecule, protects against endotoxic shock in the rat. Free Radic. Biol. Med. 47(1), 103–113.
Vinokurov M., Ostrov V., Yurinskaya M., Garbuz D., Murashev A., Antonova O., Evgen’ev M. (2012) Recombinant human Hsp70 protects against lipoteichoic acid-induced inflammation manifestations at the cellular and organismal levels. Cell Stress Chaperones. 7, 89–101.
Лакунина В.А., Бурнышева К.М., Митькевич В.А., Макаров А.А., Петрушанко И.Ю. (2017) Изменение рецепторной функции Na,K-АТРзы в условиях гипоксии и ишемии. Молекуляр. биология. 51(1), 172–179.
Mitkevich V.A., Petrushanko I.Y., Yegorov Y.E., Simonenko O.V., Vishnyakova K.S., Kulikova A.A., Tsvetkov P.O., Makarov A.A., Kozin S.A. (2013) Isomerization of Asp7 leads to increased toxic effect of amyloid-β42 on human neuronal cells. Cell Death Dis. 4, e939.
Watson G., Redmond P.H., Wang J.H., Condron C., Bouchier-Hayes D. (1996) Neutrophils undergo apoptosis following ingestion of Escherichia coli. J. Immunol. 156(10), 3986–3992.
Park H.S., Jung H.Y., Park E.Y., Kim J., Lee W.J., Bae Y.S. (2004) Direct interaction of TLR4 with NAD(P)H oxidase 4 isozyme is essential for lipopolysaccharide-induced production of reactive oxygen species and activation of NF-kappa B. J. Immunol. 173 (6), 3589–3593.
McCracken J.M., Allen L.A. (2014) Regulation of human neutrophil apoptosis and lifespan in health and disease. J. Cell. Death. 7, 15–23.
Glennon-Alty L., Hackett A.P., Chapman E.A., Wright H.L. (2018) Neutrophils and redox stress in the pathogenesis of autoimmune disease. Free Radic. Biol. Med. 125, 25–35.
Iversen M.B., Gottfredsen R.H., Larsen U.G., Enghild J.J., Praetorius J., Borregaard N., Petersen S.V. (2016) Extracellular superoxide dismutase is present in secretory vesicles of human neutrophils and released upon stimulation. Free Radic. Biol. Med. 97, 478–488.
van Raam B.J., Verhoeven A.J., Kuijpers T.W. (2006) Mitochondria in neutrophil apoptosis. Int. J. Hematol. 84, 199–204.
Rane M.J., Klein J.B. (2009) Regulation of neutrophil apoptosis by modulation of PKB/ Akt activation. Front. Biosci. 14, 2400–2412.
Sweeney J.F., Nguyen P.K., Omann G., Hinshaw D.B. (1999) Granulocyte-macrophage colony-stimulating factor rescues human polymorphonuclear leukocytes from ultraviolet irradiation-accelerated apoptosis. J. Surg. Res. 81(1), 108–112.
Derouet M., Thomas L., Cross A., Moots R.J., Edwards S.W. (2004) Granulocyte macrophage colony-stimulating factor signaling and proteasome inhibition delay neutrophil apoptosis by increasing the stability of Mcl-1. J. Biol. Chem. 279(26), 26915–26921.
Gardai S.J., Whitlock B.B., Qui Xiao Y.Q., Bratton D.B., Henson P.M. (2004) Oxidants inhibit Erk/MAPK and prevent its ability to delay neutrophil apoptosis downstream of mitochondrial changes and at the level of XIAP. J. Biol. Chem. 279, 44695–44703.
Francois S., El Benna J., Dang P.M., Pedruzzi E., Gougerot-Pocidalo M.A., Elbim C. (2005) Inhibition of neutrophil apoptosis by TLR agonists in whole blood: involvement of the phosphoinositide 3-kinase/ Akt and NF-kappa B signaling pathways, leading to increased levels of Mcl-1, A1, and phosphorylated Bad. J. Immunol. 174, 3633–3642.
Nagaoka I., Yomogida S., Tamura H., Hirata M. (2004) Antibacterial cathelicidin peptide CAP11 inhibits the lipopolysaccharide (LPS)-induced suppression of neutrophil apoptosis by blocking the binding of LPS to target cells. Inflamm. Res. 53, 609–622.
Aoshiba K., Nakajima Y., Yasui S., Tamaoki J., Nagai A. (1999) Red blood cells inhibit apoptosis of human neutrophils. Blood. 93, 4006–4010.
Sheth K., Friel J., Nolan B., Bankey P. (2001) Inhibition of p38 mitogen activated protein kinase increases lipopolysaccharide induced inhibition of apoptosis in neutrophils by activating extracellular signal-regulated kinase. Surgery. 130, 242–248.
Alvarado-Kristensson M., Melander F., Leandersson K., Ronnstrand L., Wernstedt C., Andersson T. (2004) p38-MAPK signals survival by phosphorylation of caspase-8 and caspase-3 in human neutrophils . J. Exp. Med. 199, 449–458.
Takeshita S., Tsujimoto H., Nakatani K. (2005) Intravenous immunoglobulin preparations promote apoptosis in lipopolysaccharide-stimulated neutrophils via an oxygen-dependent pathway in vitro. APMIS. 113, 269–277.
Oishi K., Machida K. (1997) Inhibition of neutrophil apoptosis by antioxidants in culture medium. Scand. J. Immunol. 45, 21–27.
Aoshiba K., Yasui S., Hayashi M., Tamaoki J., Nagai A. (1999) Role of p38-mitogen-activated protein kinase in spontaneous apoptosis of human neutrophils. J. Immunol. 162, 1692–1700.
Rollet-Labelle E., Grange M.J., Elbim C., Marquetty C., Gougerot-Pocidalo M.A., Pasquier C. (1998) Hydroxyl radical as a potential intracellular mediator of polymorphonuclear neutrophil apoptosis. Free Radic. Biol. Med. 24, 563–572.
Scheel-Toellner D., Wang K., Craddock R., Webb P.R., McGettrick H.M., Assi L.K., Parkes N., Clough L.E., Gulbins E., Salmon M., Lord J.M. (2004) Reactive oxygen species limit neutrophil life span by activating death receptor signaling. Blood. 104, 2557–2564.
Kasahara Y., Iwai K., Yachie A., Ohta K., Konno A., Seki H., Miyawaki T., Taniguchi N. (1997) Involvement of reactive oxygen intermediates in spontaneous and CD95 (Fas/APO-1)-mediated apoptosis of neutrophils. Blood. 89, 1748–1753.
Fadeel B., Ahlin A., Henter J.I., Orrenius S., Hampton M.B. (1998) Involvement of caspases in neutrophil apoptosis: regulation by reactive oxygen species. Blood. 92, 4808–4818.
Barcellos-de-Souza P., Canetti C., Barja-Fidalgo C., Arruda M.A. (2012) Leukotriene B(4) inhibits neutrophil apoptosis via NADPH oxidase activity: redox control of NF-kappaB pathway and mitochondrial stability. Biochim. Biophys. Acta. 1823(10), 1990–1997.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология