

Молекулярная биология, 2019, T. 53, № 3, стр. 393-401
Сравнение эффективности связывания факторов транскрипции с аллельными вариантами регуляторных участков генов человека методами иммунопреципитации и ПЦР в реальном времени
Н. А. Митькин a, К. В. Корнеев a, А. М. Горбачева a, b, Д. В. Купраш a, b, *
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
b Биологический факультет Московского государственного университета им. М.В. Ломоносова
119234 Москва, Россия
* E-mail: kuprash@gmail.com
Поступила в редакцию 30.10.2018
После доработки 30.11.2018
Принята к публикации 04.12.2018
Аннотация
Изменение эффективности взаимодействия специфических факторов транскрипции с ДНК, обусловленное присутствием однонуклеотидных полиморфизмов, – один из важных механизмов регуляции экспрессии генов человека, влияющий на индивидуальную предрасположенность к различным заболеваниям. Накоплено огромное количество генетических и эпигенетических данных, позволяющих предсказывать функциональные полиморфные варианты, а также факторы транскрипции, на связывание которых они влияют. Тем не менее, в большинстве случаев необходима экспериментальная верификация подобных предсказаний. В представленной статье рассмотрен отработанный в нашей лаборатории метод иммунопреципитации ДНК-белковых комплексов in vitro с последующей оценкой количества связавшейся ДНК при помощи ПЦР в реальном времени. Метод не требует химической модификации ДНК-пробы и воспроизводимо работает на полных ядерных экстрактах из культивируемых клеток человека.
ВВЕДЕНИЕ
Однонуклеотидные полиморфизмы (SNP от “single nucleotide polymorphisms”) – основной элемент генетического разнообразия человеческой популяции. Постоянно появляющиеся данные об эпигенетических механизмах регуляции генома указывают на то, что индивидуальные особенности регуляции экспрессии генов зависят не только от вариаций нуклеотидных последовательностей [1, 2], однако, нет сомнений в крайней важности этого фактора [3, 4]. Большие объемы накопленных клинических данных, а также совершенствование технологических подходов к их анализу позволяют разрабатывать новые алгоритмы предсказания функциональных эффектов, в частности, механизмов прогрессии заболеваний с ярко выраженной генетической составляющей [5–7]. Полногеномный поиск SNP, ассоциированных с этими эффектами, в первую очередь с предрасположенностью к прогрессии определенных заболеваний (GWAS от “genome-wide association studies”), в большинстве случаев приводит к обнаружению SNP, имеющих лишь прогностическую значимость [8]. Для формулировки гипотезы о механистической причине ассоциации, а, значит, о потенциальных терапевтических мишенях, на которые она указывает, требуется функциональная аннотация [3, 9]. Подобное исследование можно проводить с использованием широкого спектра методов молекулярной и клеточной биологии [10–12]. Технологии геномного редактирования и полногеномных скринингов на основе геномных нуклеаз позволяют проводить прямое тестирование функциональных гипотез в релевантных клетках и обнаруживать дальнодействующие эффекты затронутых вариациями энхансеров [13, 14]. Необходимой стадией любого подобного проекта является прямое измерение эффекта нуклеотидной вариации на связывание релевантного фактора транскрипции с ДНК.
“Золотым стандартом” анализа ДНК-белковых взаимодействий считается метод гель-ретардации (electrophoretic mobility shift assay, EMSA), основанный на способности белков специфично связываться с меченым фрагментом дцДНК, что приводит к уменьшению подвижности такого фрагмента при электрофорезе в полиакриламидном геле [15]. EMSA подразумевает возможность флуоресцентной, иммуногистохимической или хемилюминесцентной детекции. В случае использования радиоактивно меченного зонда метод обладает высокой чувствительностью, что в принципе делает возможной гибридизацию с минимальными количествами белка и ДНК (100 пМ и менее). Кроме того, метод позволяет работать с фрагментами нуклеиновых кислот размером от десятков до сотен, а в некоторых случаях до нескольких тысяч нуклеотидов, и использовать как клеточные экстракты, так и очищенные белки [16]. Однако метод EMSA технически достаточно сложен и капризен. При использовании клеточных экстрактов, а не очищенных белков он позволяет получить отчетливые полосы на геле и проанализировать связывание только некоторых семейств факторов транскрипции. Метод не дает точной информации о нуклеотидной последовательности сайта связывания белка, а электрофоретическая подвижность комплекса в общем случае нелинейно зависит от размера белка, поэтому для идентификации белковых компонентов комплексов необходим так называемый супершифт-анализ с использованием специфических антител [17, 18]. Возможности подхода также часто ограничены низкой стабильностью ДНК-белковых комплексов в условиях гель-электрофореза [19]. Подобная проблема отсутствует в ряде альтернативных подходов, не требующих проведения электрофореза, в частности, основанных на иммунопреципитации.
Футпринтинг белковых комплексов на ДНК позволяет определить сайт связывания белка путем анализа меченых фрагментов ДНК после их обработки ферментом или химическим агентом, вносящим разрывы, имеющие случайное положение [20]. Как и EMSA, футпринтинг ДНК не требует предварительных знаний о связывающемся белке, дает возможность радиоактивной или флуоресцентной детекции и позволяет работать с клеточными экстрактами. К преимуществам метода относится также возможность определения in vitro и in vivo сайтов связывания сразу нескольких факторов транскрипции с одним фрагментом ДНК, формирования ДНК-белковых комплексов в нативных условиях [21] и получение количественных характеристик ДНК-белковых взаимодействий [22]. С другой стороны, метод футпринтинга, не менее трудоемкий и капризный, чем EMSA, можно использовать для всех семейств факторов транскрипции.
Классический метод фиксации ДНК-белковых комплексов на твердофазном носителе (например, на нитроцеллюлозной мембране) является относительно простым, чувствительным и быстрым способом изучения кинетики диссоциации комплексов [23]. Этот метод позволяет работать с фрагментами ДНК большой длины [24], однако анализ ДНК-белковых взаимодействий в случае двух и более белков, связывающихся с фрагментом нуклеиновой кислоты, становится затруднительным [23]. Кроме того, одноцепочечные фрагменты нуклеиновых кислот при определенных условиях могут задерживаться на фильтре без связывания с белком и повышать общий фоновый уровень сигнала, что усложняет идентификацию целевых сигналов связывания [25]. Поскольку образцы не находятся в химическом равновесии, количественные оценки связывания белка с нуклеиновыми кислотами требуют определения удерживающей способности твердого носителя [26].
Развитие высокопроизводительных технологий не обошло стороной и ДНК-белковые взаимодействия, где находят активное применение микрочипы [27–30], и системы, использующие микрофлюидику [31] в комбинации с глубоким секвенированием [32]. Подобные подходы позволяют эффективно тестировать связывание изучаемых белков сразу с большим количеством разных фрагментов ДНК, но требуют специального оборудования и в целом являются слишком дорогостоящими и медленными для определения связывания белков с несколькими вариантами конкретного фрагмента ДНК.
Важнейшее значение имеет комплекс методов, основанных на иммунопреципитации хроматина (ChIP от “Chromatin ImmunoPrecipitation”), т.е. на обработке живых клеток химическими агентами, ковалентно сшивающими ДНК и белки, с последующей фрагментацией ДНК и обогащением комплексами, содержащими целевой белок, с использованием специфических антител [33, 34]. ДНК, оказавшуюся в составе комплексов вследствие ДНК-белковых контактов, можно анализировать при помощи количественной ПЦР, гибридизации с микрочипами (ChIP-chip) или глубокого секвенирования (ChIP-seq) [35, 36]. ChIP позволяет определять образование ДНК-белковых комплексов in vivo в зависимости от фазы клеточного цикла или типа клеточной активации, а также минимизировать детекцию артефактных нерелевантных взаимодействий [37]. ChIP не требует работы с радиоактивной меткой, этот метод позволяет относительно просто и дешево охарактеризовать при необходимости единичный локус. Важное ограничение любых методов на основе ChIP состоит в применении специфических антител высокого качества (“ChIP grade”), которые часто не распознают различные изоформы одного белка [36, 38]. Применение классического варианта ChIP ограничивает необходимость использования нескольких миллионов клеток, что может вызывать затруднения при работе с редкими клеточными популяциями или с небольшими модельными организмами. Однако в таком случае можно использовать модифицированный метод nano-ChIP-seq [39]. В силу особенностей дизайна ChIP подходит для идентификации геномных областей, взаимодействующих с факторами транскрипции, но не индивидуальных сайтов связывания, однако это ограничение можно существенным образом ослабить благодаря увеличению статистической мощности экспериментов ChIP-seq [40, 41]. Еще один недостаток метода – невозможность выделить конкретный белковый комплекс из совокупности всех комплексов, имеющих общую субъединицу; на эффективность метода также сильно влияет состояние хроматина в областях связывания [42].
Интересные возможности предоставляет классический вариант метода осаждения ДНК (“DNA pull-down”), основанный на использовании биотинилированной ДНК-пробы, что позволяет провести обогащение ДНК-белковыми комплексами с использованием стрептавидина и твердофазного носителя – магнитных частиц или агарозы [43]. К преимуществам этого метода относится возможность работы с интактными ДНК-белковыми комплексами в отсутствие артефактов, вызванных химическими сшивками, а также возможность определения белка как вестерн-блотингом, так и при помощи масс-спектрометрии. К ограничениям метода следует отнести возможность его использования только in vitro и быстрый рост неспецифического связывания с увеличением длины изучаемого фрагмента ДНК, а также при отсутствии нуклеаз. Кроме того, потенциально наиболее интересная детекция белка с помощью масс-спектрометрии, не требующая предварительной гипотезы и антител для ее проверки, чувствительна к концентрации и степени очистки белка и поэтому не всегда применима для изучения сложных белковых смесей [18, 44].
Разработанный нами метод напоминает DNA pull-down по постановке реакции связывания и классический ChIP по дальнейшему обогащению специфическими ДНК-белковыми комплексами и их детекции. При этом не используются ни ковалентные сшивки, ни биотинилирование ДНК-пробы. В данной статье детально рассмотрен наш метод, его сильные стороны и ограничения, которые он унаследовал от этих двух подходов, а также наш опыт по выбору SNP и факторов транскрипции для анализа.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Описанный ниже протокол имеет достаточно универсальный характер, он применим к любым факторам транскрипции, к которым имеются специфические антитела, и полиморфным генетическим элементам, не подверженным эпигенетическим модификациям. Протокол будет изложен на конкретном примере SNP rs630923 в промоторе гена хемокинового рецептора CXCR5, ассоциированного с предрасположенностью к прогрессии рассеянного склероза и затрагивающего сайт связывания фактора транскрипции MEF2C [45].
Используемые в эксперименте контроли. В качестве контролей используют (рис. 1):
Рис. 1.
Типы контролей, используемых при анализе ДНК-белковых взаимодействий методом иммунопреципитации.
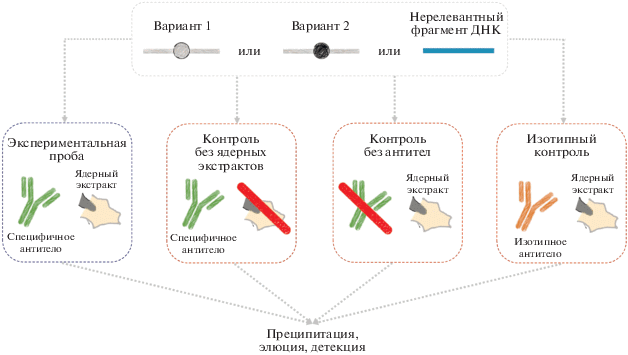
– Нерелевантную последовательность ДНК. Отражает неспецифическое связывание интересующего фактора транскрипции с ДНК.
– Контроль без ядерных экстрактов. Позволяет детектировать неспецифическое связывание используемых антител с ДНК.
– Контроль без антител. Позволяет контролировать неспецифическое связывание ДНК-белковых комплексов с частицами белок-А-сефарозы.
– Изотипный контроль. Позволяет контролировать неспецифическое связывание антител с ДНК-белковыми комплексами.
Технически успешным необходимо считать эксперимент, в котором значения для этих контролей идентичны во всех пробах и не зависят от характера используемого фрагмента ДНК. В таком случае эти значения можно принять за фоновые и при расчетах вычитать их из экспериментальных значений.
Изготовление ДНК-проб. Амплифицируют фрагменты промотора CXCR5 длиной 155 п.н. (рекомендуемая длина фрагментов от 150 до 250 п.н.; также можно использовать фрагменты длиной до нескольких тысяч пар нуклеотидов, это ухудшит соотношение сигнал/шум, но позволит лучше учесть хромосомный контекст) в следующих вариантах:
– Оригинальный участок промотора с “С” вариантом полиморфизма.
– Участок промотора с “A” вариантом полиморфизма.
– Участок промотора с “A” вариантом полиморфизма и мутациями в сайте связывания MEF2C.
Амплифицируют контрольный нерелевантный участок ДНК длиной 155 п.н., не содержащий сайтов связывания MEF2C (проверка осуществлена с использованием сервиса JASPAR (см. далее) и моделей сайтов связывания факторов транскрипции, содержащихся в данной базе). В дальнейшем данный контроль подвергают всем тем же манипуляциям, что и остальные ДНК-пробы (рис. 1). Хорошим вариантом контрольного фрагмента, особенно при использовании ДНК-пробы большей длины, является идентичный фрагмент ДНК, содержащий делецию предполагаемого участка связывания изучаемого фактора транскрипции.
Выделение ядерных экстрактов (источники факторов транскрипции). Клетки (30 млн.) линии Raji или Daudi осаждают центрифугированием (4°C, 1200 g, 5 мин), однократно промывают охлажденным фосфатно-солевым буфером (PBS). Осадок ресуспендируют в буфере А (3 мл буфера на 30 млн. клеток), встряхивают в течение 10 с и инкубируют на льду в течение 5 мин, лизат центрифугируют (13 000 g, 4°C, 15 с), супернатант удаляют. Гелеобразный осадок ресуспендируют в низкоионном буфере B (1 : 1 по объему), к суспензии добавляют высокоионный буфер С (2 : 1 по объему) и инкубируют в течение 15 мин на льду, после чего центрифугируют (13 000 g, 4°C, 20 мин).
Работы проводят на льду. Супернатант аккуратно переносят в криопробирки (100 мкл на пробирку). В дальнейшем аликвоты хранят в жидком азоте.
Буфер A: 10 мМ Трис-HCl pH 8.0, 0.32 M сахароза, 2 мМ ацетат магния, 0.1 мМ EDTA, 3 мМ CaCl2, 0.5% NP-40, 1 мМ DTT, 0.5 мМ PMSF, коктейль ингибиторов протеаз (SigmaFast). Буфер B: 20 мM HEPES pH 7.9, 1.5 мМ MgCl2, 0.02 M KCl, 0.2 мМ EDTA, 25%-ный глицерин, 0.5 мМ DTT, 0.5 мМ PMSF, коктейль ингибиторов протеаз (SigmaFast). Буфер C: 20 мМ HEPES pH 7.9, 1.5 мM MgCl2, 0.8 M KCl, 0.2 мМ EDTA, 25%-ный глицерин, 1% NP-40, 0.5 мМ DTT, 0.5 мМ PMSF, коктейль ингибиторов протеаз (SigmaFast).
Смешивание ДНК-фрагментов, ядерных экстрактов и специфичных антител. В 100 мкл буфера для инкубации смешивают 100 нг ДНК-фрагмента и 10 мкл ядерного экстракта (около 50 мкг суммарного белка). К раствору также добавляют 5 мкг оцДНК-конкурента (ДНК из спермы лосося), 1 мМ DТТ и коктейль ингибиторов протеаз (SigmaFast). Раствор инкубируют на льду в течение 1 ч. На данном этапе целесообразно включить контроль без ядерных экстрактов (рис. 1). Пробы этого контроля подвергаются тем же манипуляциям, что и остальные.
К раствору добавляют 2 мкг антител к MEF2C (ab79436, Abcam) или 2 мкг изотипных антител для контроля (неспецифические IgG антитела кролика). Раствор инкубировали на льду в течение 1 ч. На этом этапе целесообразно поставить контроль без антител (рис. 1).
Буфер для инкубации: 12 мМ HEPES pH 8.0, 60 мМ KCl, 4 мМ Tрис-HCl pH 8.0, 5%-ный глицерин, 0.5 мМ EDТА.
Подготовка частиц сефарозы с белком А. Частицы сефарозы с белком А промывают 3 раза буфером RIPA, после последней промывки буфер удаляют. Осуществляют забивку частиц: добавляют оцДНК из спермы лосося из расчета 75 нг на 1 мкл частиц, бычий сывороточный альбумин из расчета 0.1 мкг на 1 мкл частиц и RIPA-буфер (1 : 1 по объему). Инкубируют при 25°С в течение 30 мин при постоянном перемешивании. Частицы промывают 1 раз буфером RIPA.
Буфер RIPA: 50 мМ Трис-HCl pH 8.0, 2 мМ EDTA pH 8.0, 150 мМ NaCl, 1% NP-40, 0.1% SDS, 0.5% дезоксихолата натрия, коктейль ингибиторов протеаз (SigmaFast).
Иммунопреципитация ДНК-белковых комплексов на подготовленных частицах сефарозы с белком А. К растворам с ДНК, ядерными экстрактами и антителами добавляют по 60 мкл подготовленных частиц сефарозы с белком А. Суспензию инкубируют на ротаторе в течение ночи при 4°С, после чего образцы центрифугируют (2000 g, 1 мин), а супернатант удаляют. Проводят серию однократных отмывок (частицы центрифугируют после каждой отмывки при 2000 g в течение 1 мин, супернатант удаляют): в низкосолевом буфере, в высокосолевом буфере, в промывочном LiCl буфере.
Низкосолевой буфер: 20 мМ Трис-HCl pH 8.0, 150 мМ NaCl, 0.1% SDS, 2 мМ EDTA, 1% Triton X-100. Высокосолевой буфер: 20 мМ Трис-HCl pH 8.0, 500 мМ NaCl, 0.1% SDS, 2 мМ EDTA, 1% Triton X-100. Промывочный LiCl буфер: 10 мМ Трис-HCl pH 8.0, 1% NP-40, 1% дезоксихолата натрия, 0.25 М LiCl, 1 мМ EDТА.
Элюция ДНК с частиц сефарозы. К частицам добавляют 120 мкл буфера для элюции, инкубируют при 30°С в течение 15 мин при интенсивном перемешивании, центрифугируют (2000 g, 1 мин), супернатант переносят в чистую пробирку. Буфер для элюции: 1% SDS, 100 мМ NaHCO3, pH 8.0.
Количество целевой ДНК в супернатанте оценивают методом ПЦР в реальном времени с помощью праймеров, использованных для амплификации исходных фрагментов ДНК.
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Общий план эксперимента по анализу дифференциального связывания факторов транскрипции с аллельными вариантами SNP генов человека можно разделить на несколько этапов (рис. 2).
Рис. 2.
Общая схема эксперимента по идентификации факторов транскрипции, различия в связывании которых с разными аллельными вариантами полиморфизмов в регуляторных элементах генов могут влиять на активность этих элементов.
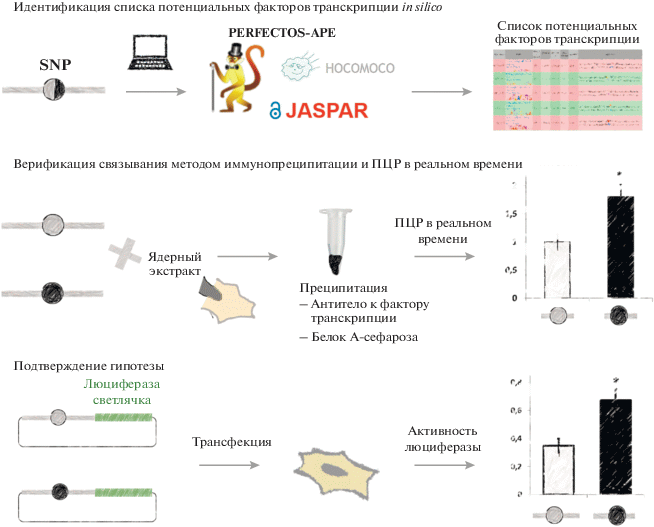
Выбор факторов транскрипции для исследования
Список факторов транскрипции составляли in silico с использованием биоинформатических подходов. Решить данную задачу позволяет онлайн-сервис Perfectos-Ape [46], который использует курируемые модели сайтов связывания факторов транскрипции из ряда баз данных: HOCOMOCO [47], JASPAR [48], HT-SELEX [49], SwissRegulon [50], HOMER (http://homer.ucsd.edu/homer/). По нашему опыту предсказания, наилучшим образом подтверждаемые в ходе дальнейших исследований, получают из результатов, общих для моделей из HOCOMOCO и JASPAR. Дополнительно из списка исключаются факторы транскрипции с низкой экспрессией в используемой клеточной модельной системе. Для оценки уровня экспрессии мы использовали данные, представленные в базе Cancer Cell Line Encyclopedia [51]. Наконец, на основе анализа опубликованных данных исключают факторы транскрипции, не имеющие функционального отношения к исследуемому биологическому явлению. Полученный список по-прежнему носит сугубо предсказательный характер и требует дальнейшей функциональной верификации, но подобная оптимизация позволяет значительно сократить затраты времени и материальных ресурсов.
Экспериментальная проверка биоинформатических предсказаний
Фрагменты ДНК, содержащие разные варианты исследуемого полиморфизма, инкубировали с ядерными экстрактами, полученными из релевантной для конкретного эксперимента клеточной линии. Далее с использованием антител к идентифицированному фактору транскрипции проводили иммунопреципитацию целевых ДНК-белковых комплексов. Количество целевой ДНК в препаратах после преципитации оценивали методом ПЦР в реальном времени. Поскольку концентрация фактора транскрипции в ядерном экстракте не известна, эта процедура не позволяет оценить сродство целевого белка к соответствующим нуклеотидным последовательностям. Однако сравнение сигналов, полученных с ДНК-пробами, содержащими разные аллельные варианты SNP, показывает, зависит ли эффективность связывания от исследуемого полиморфизма.
Отдельно необходимо отметить, что рекомендуется использовать не только фрагменты ДНК, содержащие природные аллели SNP, но и фрагменты, несущие точечные мутации в других позициях предсказанного сайта связывания, которые могли бы привести к его полному разрушению. При планировании подобных мутаций необходимо ориентироваться на лого-диаграммы и позиционно-весовые матрицы для консенсусных сайтов связывания, представленные в базах данных HOCOMOCO и JASPAR, и проводить замены в наиболее консервативных позициях. Это важный контроль, позволяющий наилучшим образом оценить уровень фонового сигнала.
Функциональное изучение корреляции между связыванием фактора транскрипции и уровнем активности исследуемого регуляторного элемента
Эту задачу мы решали с помощью люциферазных репортерных конструкций (в частности, на базе вектора pGL3 basic, “Promega”, США), содержащих регуляторные элементы гена с разными вариантами изучаемого полиморфизма, и тестирования их активности в релевантной клеточной модели. Под релевантной клеточной моделью следует понимать ту линию клеток человека, для которой физиологична как экспрессия исследуемого гена, так и активность идентифицированного фактора транскрипции. Например, для изучения роли фактора транскрипции MEF2C [52] в rs630923-зависимой регуляции промотора гена хемокинового рецептора CXCR5 [53] использовали B-лимфо-бластоидные клеточные линии Raji и Daudi [45], а для выявления роли rs928413 и CREB1 [54] в модуляции активности промотора гена IL33 [55] – линию клеток рака легкого NCIH-196 [56, 57].
Хорошей стратегией представляется также оценка активности данных конструкций в условиях либо подавления экспрессии, либо активации исследуемого фактора транскрипции. В области подавления экспрессии хорошо зарекомендовали себя малые интерферирующие РНК (siRNA), подобрать которые можно с использованием как базы данных NCBI probe [58], так и опубликованных данных (например, [59]).
Активация фактора транскрипции необходима в том случае, если базовой активности фактора недостаточно для получения достоверных эффектов. Например, фактор CREB1 в клетках рака легкого активировали с использованием провоспалительного цитокина фактора некроза опухолей [56], фактор ASCL2 в B-лимфобластоидных и моноцитоподобной линиях – посредством индукции Wnt-сигнального каскада [60] с использованием LiCl [61], а фактора MEF2C в B-лимфобластоидных клетках – через запуск B-клеточного ответа с использованием липополисахарида и иономицина [45].
В представленной работе приведено подробное описание отработанного в нашей лаборатории метода иммунопреципитации ДНК-белковых комплексов in vitro с последующей оценкой количества связавшейся ДНК при помощи ПЦР в реальном времени. Метод не требует химической модификации ДНК-пробы и воспроизводимо работает на полных ядерных экстрактах из культивируемых клеток человека. Полученные результаты позволяют говорить о валидности подхода в целом и о перспективности его использования для функциональной аннотации однонуклеотидных полиморфизмов.
Работа выполнена при поддержке Российского научного фонда (№ 14-14-01140) и Программы фундаментальных исследований государственных академий наук на 2013-2020 годы (тема № 01201363817).
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Slattery M., Zhou T., Yang L., Dantas Machado A.C., Gordân R., Rohs R. (2014) Absence of a simple code: How transcription factors read the genome. Trends Biochem. Sci. 39, 381–399.
Deplancke B., Alpern D., Gardeux V. (2016) The genetics of transcription factor DNA binding variation. Cell. 166, 538–554.
Albert F.W., Kruglyak L. (2015) The role of regulatory variation in complex traits and disease. Nat. Rev. Genet. 16, 197–212.
Schmiedel B.J., Singh D., Madrigal A., Valdovino-Gonzalez A.G., White B.M., Zapardiel-Gonzalo J., Ha B., Altay G., Greenbaum J.A., McVicker G., Seumois G., Rao A., Kronenberg M., Peters B., Vijaya-nand P. (2018) Impact of genetic polymorphisms on human immune cell gene expression. Cell. 175, 1701–1715.
Goris A., Pauwels I., Dubois B. (2012) Progress in multiple sclerosis genetics. Curr. Genomics. 13, 646–663.
Ward L.D., Kellis M. (2016) HaploReg v4: systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucl. Acids Res. 44, D877–D881.
McLaren W., Gil L., Hunt S.E., Riat H.S., Ritchie G.R.S., Thormann A., Flicek P., Cunningham F. (2016) The ensembl variant effect predictor. Genome Biol. 17, 122.
Visscher P.M., Wray N.R., Zhang Q., Sklar P., McCarthy M.I., Brown M.A., Yang J. (2017) 10 Years of GWAS discovery: biology, function, and translation. Am. J. Hum. Genet. 101, 5–22.
Carey M.F., Peterson C.L., Smale S.T. (2012) Confirming the functional importance of a protein-DNA interaction. Cold Spring Harb. Protoc. 2012, 733–757.
De Gobbi M., Viprakasit V., Hughes J.R., Fisher C., Buckle V.J., Ayyub H., Gibbons R.J., Vernimmen D., Yoshinaga Y., De Jong P., Cheng J.F., Rubin E.M., Wood W.G., Bowden D., Higgs D.R. (2006) A regulatory SNP causes a human genetic disease by creating a new transcriptional promoter. Science. 312, 1215–1217.
Musunuru K., Strong A., Frank-Kamenetsky M., Lee N.E., Ahfeldt T., Sachs K. V., Li X., Li H., Kuperwasser N., Ruda V.M., Pirruccello J.P., Muchmore B., Prokunina-Olsson L., Hall J.L., Schadt E.E., Morales C.R., Lund-Katz S., Phillips M.C., Wong J., Cantley W., Racie T., Ejebe K.G., Orho-Melander M., Melander O., Koteliansky V., Fitzgerald K., Krauss R.M., Cowan C.A., Kathiresan S., Rader D.J. (2010) From noncoding variant to phenotype via SORT1 at the 1p13 cholesterol locus. Nature. 466, 714–719.
Awah C.U., Tamm S., Hedtfeld S., Steinemann D., Tümmler B., Tsiavaliaris G., Stanke F. (2016) Mechanism of allele specific assembly and disruption of master regulator transcription factor complexes of NF-KBp50, NF-KBp65 and HIF1a on a non-coding FAS SNP. Biochim. Biophys. Acta – Gene Regul. Mech. 1859, 1411–1428.
Soldner F., Stelzer Y., Shivalila C.S., Abraham B.J., Latourelle J.C., Barrasa M.I., Goldmann J., Myers R.H., Young R.A., Jaenisch R. (2016) Parkinson-associated risk variant in distal enhancer of α-synuclein modulates target gene expression. Nature. 39, 381–399.
Gupta R.M., Hadaya J., Trehan A., Zekavat S.M., Roselli C., Klarin D., Emdin C.A., Hilvering C.R.E., Bianchi V., Mueller C., Khera A. V., Ryan R.J.H., Engreitz J.M., Issner R., Shoresh N., Epstein C.B., de Laat W., Brown J.D., Schnabel R.B., Bernstein B.E., Kathiresan S. (2017) A genetic variant associated with five vascular diseases is a distal regulator of endothelin-1 gene expression. Cell. 170, 522–533.
Fried M., Crothers D.M. (1981) Equilibria and kinetics of lac repressor-operator interactions by polyacrylamide gel electrophoresis. Nucl. Acids Res. 9, 6505–6525.
Hellman L.M., Fried M.G. (2007) Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2, 1849.
Adams C., Fried M.G. (2007) Protein Interactions: Biophysical Approaches for the Study of Multicomponent Systems. New York: Springer Science & Business Media.
Singh B., Nath S.K. (2019) Identification of proteins interacting with single nucleotide polymorphisms (SNPs) by DNA pull-down assay. In: Electrophoretic Separation of Proteins. New York: Humana Press, 355–362.
Hellman L.M., Fried M.G. (2007) Electrophoretic mobility shift assay (EMSA) for detecting protein-nucleic acid interactions. Nat. Protoc. 2, 1849–1861.
Galas D.J., Schmitz A. (1978) DNAase footprinting a simple method for the detection of protein-DNA binding specificity. Nucl. Acids Res. 5, 3157–3170.
Hesselberth J.R., Chen X., Zhang Z., Sabo P.J., Sandstrom R., Reynolds A.P., Thurman R.E., Neph S., Kuehn M.S., Noble W.S., Fields S., Stamatoyannopoulos J.A. (2009) Global mapping of protein-DNA interactions in vivo by digital genomic footprinting. Nat. Methods. 6, 283.
Shcherbakova I., Mitra S., Beer R.H., Brenowitz M. (2006) Fast Fenton footprinting: a laboratory-based method for the time-resolved analysis of DNA, RNA and proteins. Nucl. Acids Res. 34, e48–e48.
Woodbury C.P., von Hippel P.H. (1983) On the determination of deoxyribonucleic acid-protein interaction parameters using the nitrocellulose filter-binding assay. Biochemistry. 22, 4730–4737.
Sanger F., Coulson A.R., Hong G.F., Hill D.F., Petersen G.B. (1982) Nucleotide sequence of bacteriophage λ DNA. J. Mol. Biol. 162, 729–773.
Radding C.M., Beattie K.L., Holloman W.K., Wiegand R.C. (1977) Uptake of homologous single-stranded fragments by superhelical DNA. IV. Branch migration. J. Mol. Biol. 116, 783–803.
Oehler S., Alex R., Barker A. (1999) Is nitrocellulose filter binding really a universal assay for protein-DNA interactions? Anal. Biochem. 268, 330–336.
Mukherjee S., Berger M.F., Jona G., Wang X.S., Muz-zey D., Snyder M., Young R.A., Bulyk M.L. (2004) Rapid analysis of the DNA-binding specificities of transcription factors with DNA microarrays. Nat. Genet. 36, 1331–1339.
Berger M.F., Philippakis A.A., Qureshi A.M., He F.S., Estep P.W., Bulyk M.L. (2006) Compact, universal DNA microarrays to comprehensively determine transcription-factor binding site specificities. Nat. Biotechnol. 24, 1429.
Siggers T., Chang A.B., Teixeira A., Wong D., Williams K.J., Ahmed B., Ragoussis J., Udalova I.A., Smale S.T., Bulyk M.L. (2011) Principles of dimer-specific gene regulation revealed by a comprehensive characterization of NF-κB family DNA binding. Nat. Immunol. 13, 95.
Zasedateleva O.A., Vasiliskov V.A., Surzhikov S.A., Sazykin A.Y., Putlyaeva L. V., Schwarz A.M., Kuprash D.V., Rubina A.Y., Barsky V.E., Zasedatelev A.S. (2014) UV fluorescence of tryptophan residues effectively measures protein binding to nucleic acid fragments immobilized in gel elements of microarrays. Biotechnol. J. 9, 1074–1080.
Fordyce P.M., Gerber D., Tran D., Zheng J., Li H., Derisi J.L., Quake S.R. (2010) De novo identification and biophysical characterization of transcription-factor binding sites with microfluidic affinity analysis. Nat. Biotechnol. 28, 970–975.
Le D.D., Shimko T.C., Aditham A.K., Keys A.M., Longwell S.A., Orenstein Y., Fordyce P.M. (2018) Comprehensive, high-resolution binding energy landscapes reveal context dependencies of transcription factor binding. Proc. Natl. Acad. Sci. USA. 115, E3702–E3711.
Gilmour D.S., Lis J.T. (1984) Detecting protein-DNA interactions in vivo: distribution of RNA polymerase on specific bacterial genes. Proc. Natl. Acad. Sci. USA. 81, 4275–4279.
Solomon M.J., Larsen P.L., Varshavsky A. (1988) Mapping proteinDNA interactions in vivo with formaldehyde: evidence that histone H4 is retained on a highly transcribed gene. Cell. 53, 937–947.
Mardis E.R. (2007) ChIP-seq: welcome to the new frontier. Nat. Methods. 4, 613–614.
Park P.J. (2009) ChIP-seq: Advantages and challenges of a maturing technology. Nat. Rev. Genet. 10, 669.
Collas P. (2010) The current state of chromatin immunoprecipitation. Mol. Biotechnol. 45, 87–100.
Buck M.J., Lieb J.D. (2004) ChIP-chip: considerations for the design, analysis, and application of genome-wide chromatin immunoprecipitation experiments. Genomics. 83, 349–360.
Adli M., Bernstein B.E. (2011) Whole-genome chromatin profiling from limited numbers of cells using nano-ChIP-seq. Nat. Protoc. 6, 1656.
Tompa M., Li N., Bailey T.L., Church G.M., De Moor B., Eskin E., Favorov A.V., Frith M.C., Fu Y., Kent W.J., Makeev V.J., Mironov A.A., Noble W.S., Pavesi G., Pesole G., Régnier M., Simonis N., Sinha S., Thijs G., Van Helden J., Vandenbogaert M., Weng Z., Workman C., Ye C., Zhu Z. (2005) Assessing computational tools for the discovery of transcription factor binding sites. Nat. Biotechnol. 23, 137–144.
Jothi R., Cuddapah S., Barski A., Cui K., Zhao K. (2008) Genome-wide identification of in vivo protein-DNA binding sites from ChIP-Seq data. Nucl. Acids Res. 36, 5221–5231.
Wong D., Teixeira A., Oikonomopoulos S., Humburg P., Lone I.N., Saliba D., Siggers T., Bulyk M., Angelov D., Dimitrov S., Udalova I.A., Ragoussis J. (2011) Extensive characterization of NF-κB binding uncovers non-canonical motifs and advances the interpretation of genetic functional traits. Genome Biol. 12, R70.
Tone Y., Furuuchi K., Kojima Y., Tykocinski M.L., Greene M.I., Tone M. (2008) Smad3 and NFAT cooperate to induce Foxp3 expression through its enhancer. Nat. Immunol. 9, 194.
Wu K.K. (2006) Analysis of protein-DNA binding by streptavidin-agarose pulldown. In: Gene Mapping, Discovery, and Expression. New York: Humana Press, 281–290.
Mitkin N.A., Muratova A.M., Schwartz A.M., Kuprash D.V. (2016) The A allele of the single-nucleotide polymorphism rs630923 creates a binding site for MEF2C resulting in reduced cxcr5 promoter activity in B-cell lymphoblastic cell lines. Front. Immunol. 7, 515.
Vorontsov I.E., Kulakovskiy I. V., Khimulya G., Nikolaeva D.D., Makeev V.J. (2015) PERFECTOS-APE: Predicting regulatory functional effect of SNPs by approximate P-value estimation. Proc. Bioinforma. 2015 – 6th Int. Conf. Bioinforma. Model. Methods Algorithms. 1, 102–108.
Kulakovskiy I.V., Vorontsov I.E., Yevshin I.S., Sharipov R.N., Fedorova A.D., Rumynskiy E.I., Medvedeva Y.A., Magana-Mora A., Bajic V.B., Papatsenko D.A., Kolpakov F.A., Makeev V.J. (2018) HOCOMOCO: Towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucl. Acids Res. 46, D252–D259.
Khan A., Fornes O., Stigliani A., Gheorghe M., Castro-Mondragon J.A., Van Der Lee R., Bessy A., Chèneby J., Kulkarni S.R., Tan G., Baranasic D., Arenillas D.J., Sandelin A., Vandepoele K., Lenhard B., Ballester B., Wasserman W.W., Parcy F., Mathelier A. (2018) JASPAR 2018: Update of the open-access database of transcription factor binding profiles and its web framework. Nucl. Acids Res. 46, D260–D266.
Jagannathan V., Roulet E., Delorenzi M., Bucher P. (2006) HTPSELEX – a database of high-throughput SELEX libraries for transcription factor binding sites. Nucl. Acids Res. 34, D90–D94.
Carninci P., Kasukawa T., Katayama S., et al. (2005) The transcriptional landscape of the mammalian genome. Science. 309, 1559–1563.
Barretina J., Caponigro G., Stransky N., Venkatesan K., Margolin A.A., Kim S., Wilson C.J., Lehár J., Kryukov G. V., Sonkin D., Reddy A., Liu M., Murray L., Berger M.F., Monahan J.E., Morais P., Meltzer J., Korejwa A., Jané-Valbuena J., Mapa F.A., Thibault J., Bric-Furlong E., Raman P., Shipway A., Engels I.H., Cheng J., Yu G.K., Yu J., Aspesi P., De Silva M., Jagtap K., Jones M.D., Wang L., Hatton C., Palescandolo E., Gupta S., Mahan S., Sougnez C., Onofrio R.C., Liefeld T., MacConaill L., Winckler W., Reich M., Li N., Mesirov J.P., Gabriel S.B., Getz G., Ardlie K., Chan V., Myer V.E., Weber B.L., Porter J., Warmuth M., Finan P., Harris J.L., Meyerson M., Golub T.R., Morrissey M.P., Sellers W.R., Schlegel R., Garraway L.A. (2012) The cancer cell line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature. 483, 603.
Swanson B.J., Jäck H.M., Lyons G.E. (1998) Characterization of myocyte enhancer factor 2 (MEF2) expression in B and T cells: MEF2C is a B cell-restricted transcription factor in lymphocytes. Mol. Immunol. 35, 445–458.
Legler D.F., Loetscher M., Roos R.S., Clark-Lewis I., Baggiolini M., Moser B. (1998) B cell-attracting chemokine 1, a human CXC chemokine expressed in lymphoid tissues, selectively attracts B lymphocytes via BLR1/CXCR5. J. Exp. Med. 187, 655–660.
Bartel S., Schulz N., Alessandrini F., Schamberger A.C., Pagel P., Theis F.J., Milger K., Noessner E., Stick S.M., Kicic A., Eickelberg O., Freishtat R.J., Krauss-Etschmann S. (2017) Pulmonary microRNA profiles identify involvement of Creb1 and Sec14l3 in bronchial epithelial changes in allergic asthma. Sci. Rep. 7, 46026.
Lloyd C.M. (2010) IL-33 family members and asthma – bridging innate and adaptive immune responses. Curr. Opin. Immunol. 22, 800–806.
Gorbacheva A., Korneev K., Kuprash D., Mitkin N. (2018) The Risk G allele of the single-nucleotide polymorphism rs928413 creates a CREB1-binding site that activates IL33 Promoter in lung epithelial cells. Int. J. Mol. Sci. 19, 2911.
Gorbacheva A.M., Kuprash D.V., Mitkin N.A. (2018) Glucocorticoid receptor binding inhibits an intronic IL33 enhancer and is disrupted by rs4742170 (T) allele associated with specific wheezing phenotype in early childhood. Int. J. Mol. Sci. 19, 3956.
Coordinators (2013) Database resources of the national center for biotechnology information. Nucl. Acids Res. 41, D8.
Hasson S.A., Kane L.A., Yamano K., Huang C.H., Sliter D.A., Buehler E., Wang C., Heman-Ackah S.M., Hessa T., Guha R., Martin S.E., Youle R.J. (2013) High-content genome-wide RNAi screens identify regulators of parkin upstream of mitophagy. Nature. 504, 291.
Jubb A.M., Chalasani S., Frantz G.D., Smits R., Grabsch H.I., Kavi V., Maughan N.J., Hillan K.J., Quirke P., Koeppen H. (2006) Achaete-scute like 2 (ascl2) is a target of Wnt signalling and is upregulated in intestinal neoplasia. Oncogene. 25, 3445.
Mitkin N.A., Muratova A.M., Korneev K.V., Pavshintsev V.V., Rumyantsev K.A., Vagida M.S., Uvarova A.N., Afanasyeva M.A., Schwartz A.M., Kuprash D.V. (2018) Protective C allele of the single-nucleotide polymorphism rs1335532 is associated with strong binding of Ascl2 transcription factor and elevated CD58 expression in B-cells. Biochim. Biophys. Acta – Mol. Basis Dis. 1864, 3211–3220.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология