

Молекулярная биология, 2019, T. 53, № 5, стр. 741-754
Провоспалительные цитокины и заживление кожных ран у мышей
М. А. Носенко a, b, С. Г. Амбарян a, b, М. С. Друцкая a, *
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
b Кафедра иммунологии биологического факультета Московского государственного университета
им. М.В. Ломоносова
119234 Москва, Россия
* E-mail: marinadru@gmail.com
Поступила в редакцию 14.03.2019
После доработки 14.03.2019
Принята к публикации 22.03.2019
Аннотация
Cложно регулируемый процесс заживления кожных ран, в который вовлечено большое количество клеточных популяций и молекулярных медиаторов, считается одним из ключевых механизмов обеспечения барьерных функций кожи и поддержания гомеостаза организма. Эффективность этого процесса во многом определяется балансом воспалительных и прорегенеративных сигналов, в значительной степени контролируемых цитокинами. В обзоре суммированы последние данные о роли провоспалительных цитокинов, преимущественно фактора некроза опухоли, интерлейкинов-6 и -1, интерферонов типа 1 и 2 в заживлении кожных ран, полученные на животных моделях с использованием технологий редактирования генома. Рассмотрена роль этих медиаторов на различных этапах регенерации кожи как в обычном состоянии, так и на фоне системных патологий, в частности при сахарном диабете, обсуждаются также перспективные направления терапии незаживающих ран.
ВВЕДЕНИЕ
Независимо от типа раны и сопутствующей патологии заживление кожи можно разделить на несколько ключевых этапов: первичный гемостаз, воспалительный ответ и формирование грануляционной ткани, пролиферация кератиноцитов и фибробластов, приводящая к закрытию повреждения, и последующая реорганизация внеклеточного матрикса [1–3]. При небольшом повреждении кожи и отсутствии отягчающих условий происходит полноценное восстановление кожного покрова на месте ранения, что включает в себя, в первую очередь, восстановление эпидермального слоя и слоя дермы с сопутствующими придатками, такими как волосяные фолликулы и железы секреции [4]. Однако такая последовательность событий наблюдается не во всех случаях, что приводит либо к незаживающей ране, либо к неполноценному восстановлению кожного покрова. Второе чаще всего связано с образованием фиброзной соединительной ткани, неспособной полноценно выполнять функции нормальной кожи, и не содержащей характерных кожных придатков [5, 6]. Борьба с незаживающими ранами и предотвращение фиброза и по сей день остаются ключевыми нерешенными задачами терапии кожных ран [7]. Идет активный поиск мишеней, определяющих ход регенеративного процесса в коже. Ключевыми регуляторами этого процесса являются провоспалительные цитокины [8].
Провоспалительные цитокины продуцируются одними из первых в ответ на повреждение кожи и регулируют функции иммунных клеток в ходе регенерации, а также влияют на ключевые стромальные клеточные популяции – кератиноциты и фибробласты [3, 9, 10] (рис. 1). Хронически повышенная экспрессия провоспалительных цитокинов характерна для незаживающих ран кожи на фоне системных аутоиммунных и метаболических заболеваний [11]. В то же время, пониженная продукция этих факторов способствует инфицированию кожных ран и препятствует полноценному заживлению [12]. Интересно, что индукция умеренного воспаления с помощью биоматериалов способствует заживлению ран кожи, что говорит о важности баланса провоспалительных сигналов в процессе регенерации [13, 14]. Ключевыми провоспалительными цитокинами, участвующими в регенерации кожи, являются фактор некроза опухоли (TNF), интерлейкин-6 (IL-6) и интерлейкин-1 (IL-1) [15]. Поскольку все эти факторы выполняют многочисленные функции, их детальный вклад в процесс регенерации кожи до сих пор не полностью установлен. Физиологические функции цитокинов активно изучают на мышах, в том числе с полной или тканеспецифической инактивацией этих цитокинов или их рецепторов, с привлечением методов обратной генетики. Ключевой вопрос, ответ на который позволяет получить эта технология, ‒ определение клеточного источника того или иного цитокина, вовлеченного в заживление кожи. Известно, что различные популяции миелоидных клеток и, в первую очередь, макрофаги вносят ключевой вклад в регенерацию кожи при повреждении [16], в то время как Т-клетки не играют существенной роли в этом процессе [17]. Однако детальные молекулярные механизмы участия этих клеток в регенерации кожи пока не установлены.
Рис. 1.
Схема заживления ран кожи с указанием роли провоспалительных цитокинов. После повреждения кожного покрова (Этап 1) происходит индукция воспаления за счет попадания в рану патогенных бактерий и микрофлоры кожи, а также за счет сигналов опасности, высвобождаемых поврежденными кератиноцитами и фибробластами (например, дцДНК). Это приводит к быстрой экспрессии провоспалительных цитокинов (TNF, IL-6, IL-1 и IFN), которые запускают иммунный ответ против возможных патогенных микроорганизмов. В дальнейшем (Этап 2) формируется грануляционная ткань, защищающая повреждения от инфекции, происходит запуск пролиферации фибробластов и кератиноцитов. На этом этапе важную роль играют IL-6 и TNF. В зависимости от баланса сигналов на этом этапе регенерация может завершиться функциональным восстановлением участка кожи (Этап 3а), либо образованием фиброза (Этап 3б). И TNF, и IL-6 необходимы для регенерации волосяных фолликулов, однако они также могут стимулировать пролиферацию фибробластов и экспансию соединительной ткани. Эти различия, по-видимому, определяются несколькими клеточными источниками цитокинов, а также их плейотропным влиянием на многие типы клеток. Синими стрелками показаны установленные клеточные источники цитокинов, зеленые стрелки указывают на положительную роль цитокинов, красные – на их негативное участие в заживлении кожи. Рисунок сделан с использованием материалов с сайта https://smart.servier.com/
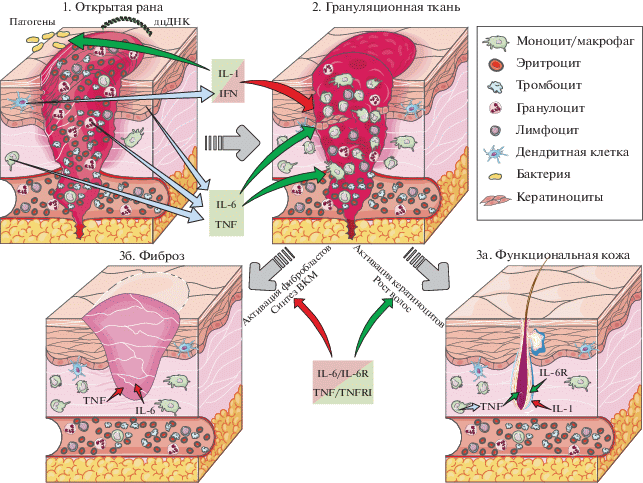
Ситуация осложняется тем, что использование мышиных моделей в изучении механизмов заживления ран кожи у человека сильно ограничивают принципиальные различия в регенерации повреждений кожи у мышей и людей. Так, у мышей есть специальный слой подкожной мускулатуры, поэтому заживление ран у них происходит преимущественно с помощью быстрой контракции с последующей реэпителизацией, тогда как для человека характерен более длительный процесс заживления, связанный с образованием грануляционной ткани [18]. Имеющиеся данные о роли цитокинов в заживлении кожи получены без учета этого факта. Тем не менее, разработана мышиная модель заживления повреждений кожи, в которой края раны фиксируют силиконовыми кольцами, препятствующими быстрому затягиванию повреждения [19]. Эта модель считается самой релевантной для изучения механизмов заживления кожных ран у человека. В связи с этим данные о роли провоспалительных цитокинов в заживлении кожи, полученные на более простой модели, должны быть в дальнейшем подтверждены в опытах с предотвращением контракции кожных ран. Далее будут отдельно рассмотрены основные провоспалительные цитокины, участвующие в заживлении ран кожи, и суммированы основные результаты, полученные на трансгенных животных и животных с нокаутом генов (рис. 1).
TNF
TNF – многофункциональный цитокин, роль которого, с одной стороны, заключается в индукции и контроле воспаления, защите от инфекций, а с другой, – в иммунорегуляции и поддержании структуры вторичных лимфоидных органов. Поскольку TNF – ключевой индуктор многих воспалительных процессов, довольно давно стало понятно, что он входит в число основных участников патогенеза ряда аутовоспалительных и аутоиммунных заболеваний, таких как ревматоидный артрит, рассеянный склероз, псориаз и болезнь Крона [20]. При этих заболеваниях широко применяют системные блокаторы TNF. Однако системная терапия зачастую сопряжена с рядом побочных и нежелательных эффектов, а при рассеянном склерозе она оказалась неэффективной. Интересно, что одним из наиболее частых побочных эффектов некоторых блокаторов, в частности адалимумаба, является замедленная регенерация кожи при повреждении (по данным EМА, 2018, https://www.ema.europa.eu/en/medicines/human/EPAR/humira#product-information-section). При этом опубликованные данные о роли TNF в заживлении кожи неоднозначны и даже противоречивы. Мало изучен и вклад цитокина из конкретных клеточных источников.
TNF экспрессируется не только клетками иммунной системы, но и стромальными, эндотелиальными клетками, адипоцитами, кардиомиоцитами и нейронами. Интересно, что в коже TNF экспрессируется конститутивно, а его первоначальная индукция происходит постнатально при заселении кожи микробиотой вскоре после рождения [21]. Изначально TNF экспрессируется на поверхности клетки в виде гомотримера в мембраносвязанной форме (tmTNF), которая в дальнейшем подвергается протеолизу ферментом TACE (TNF-α converting enzyme) и превращается в растворимую форму (sTNF). Внутриклеточная передача сигнала в клетках, на которые действует TNF, происходит после его связывания с двумя рецепторами: TNFR1 (p55) и TNFR2 (p75). При взаимодействии TNF с обоими рецепторами происходит привлечение внутриклеточных адаптерных белков и запуск сигнальных каскадов, приводящий к активации факторов транскрипциии NF-κB и AP-1, которые опосредуют выживаемость и пролиферацию клеток, иммунорегуляцию и экспрессию провоспалительных цитокинов, в том числе и самого TNF [22, 23]. При этом TNFR1, в отличие от TNFR2, содержит так называемый “домен смерти” (death domain) в своей цитоплазматической части, поэтому его связывание с TNF, помимо активации NF-κB, в некоторых случаях может приводить к индукции клеточной гибели [24]. Экспрессия антиапоптотических факторов cFLIP, Bcl-xL, индуцированная активацией NF-κB-сигналинга, наоборот, обеспечивает выживание клетки [25, 26]. NF-κB является фактором транскрипции для большого числа генов, кодирующих провоспалительные цитокины, хемокины и их рецепторы [27]. Более того, NF-κB может индуцировать транскрипцию самого гена Tnf, амплифицируя активность сигнальных путей TNF/TNFR [28, 29].
Считается, что sTNF связывается преимущественно с TNFR1, а tmTNF может взаимодействовать с обоими рецепторами. При этом TNFR1 представлен почти на всех типах клеток, тогда как экспрессия TNFR2 характерна для клеток гемопоэтического происхождения, а также нейронов и олигодендроцитов. Интересно, что индукция системного воспаления, в том числе при развитии аутоиммунных патологий, осуществляется через сигнальный путь TNF–TNFR1 [30, 31]. Этот же сигнальный каскад вовлечен в защиту от внутриклеточных патогенов [32, 33] и формирование герминальных центров [34]. Долгое время считалось, что TNFR2 выполняет второстепенную роль в гомеостазе и TNF-зависимых патологиях. Однако недавно показали, что регуляторные эффекты TNF связаны с передачей сигнала через TNFR2, в частности, для поддержания функций миелоидных супрессоров [25, 35] и Т-регуляторных клеток [36]. Наконец, есть данные, свидетельствующие о том, что при связывании с рецептором tmTNF не только передает сигнал через рецептор в клетку, но также способен активировать сигнальный каскад внутри той клетки, на которой представлен сам лиганд, так называемая “обратная сигнализация” [37]. Создание мышей с тканеспецифической интактивацией TNF позволило выявить некоторые невырожденные функции этого цитокина из различных клеточных источников как в контексте иммунного ответа [34, 38–40], так и в других системах, в том числе в процессах, связанных с регенерацией кожи [41–44] (табл. 1).
Таблица 1.
Заживление кожи у мышей с генетическим нарушением сигнального пути TNF
| Мыши с нокаутом (KO) или трансгенные (Tg) | Модель повреждения кожи | Основной фенотип и вывод исследования | Ссылка |
|---|---|---|---|
| TNF KO | Полнослойная рана | Заживление полнослойных ран кожи сопряжено с пониженной реэпителизацией на фоне разрастания грануляционной ткани, что свидетельствует о неэффективном заживлении кожи в отсутствие TNF | [46] |
| TNF KO; TNFR1 KO;
TNFR2 KO; TNF Tg;
TNF-Luc-eGFP Tg Тканеспецифическая инактивация TNF в миелоидных клетках (LysMCre/wtTNFfl/fl) |
» | TNF-продуцирующие Ly6C+ макрофаги, но не CX3CR1+ резидентные макрофаги кожи необходимы для WIH-A и WIHN. Сверхэкспрессия TNF сопряжена с увеличением WIH-A и, наоборот, с существенным снижением WIHN. Экспрессия TNFR1, но не TNFR2 на Lgr5+ стволовых клетках волосяных фолликулов, необходима для эффективного восстановления роста волос. | [43] |
| TNFR1 KO | » | Мыши с нокаутом TNFR1 характеризуются ускоренной динамикой заживления кожи. Передача сигнала через TNFR1 необходима для привлечения лейкоцитов в область ранения и негативно влияет на ангиогенез и накопление коллагена. | [45] |
| TNF KO | Повреждение, индуцированное выщипыванием волос | TNF-продуцирующие макрофаги регулируют синхронную активацию стволовых клеток волосяных фолликулов. Инактивация TNF или удаление макрофагов препятствуют восстановлению волосяного покрова. | [47] |
| TNFR1 KO; TNFR2 KO; TNFR1/TNFR2 KO; TNF/LTa KO | Некроз кожи, индуцированный TNF или LPS | Мыши TNFR2 KO чувствительны к TNF- или LPS-индуцированному некрозу кожи. Мыши TNFR1 KO, TNFR1/TNFR2 KO, TNF/LTa KO характеризуются полной или частичной устойчивостью к развитию патологии. | [48] |
| TNFR1 KO; TNFR2 KO | Контактный дерматит | Миграция дендритных клеток кожи нарушена у мышей TNFR2 KO, но не у TNFR1 KO, что свидетельствует о ключевой роли сигнала через TNFR2 в запуске иммунного ответа в коже. | [49] |
| Тканеспецифическая инактивация TNF в миелоидных клетках (LysMCre/wtTNFfl/fl) | Контактный дерматит | TNF из тучных клеток регулирует созревание и миграцию CD8+ дендритных клеток кожи в лимфоузлы и последующую активацию иммунного ответа. | [41] |
Так, у мышей с генетической инактивацией TNFR1 наблюдается ускоренная динамика заживления ран, что может свидетельствовать о патогенной роли TNF в регенерации кожи [45]. Однако у мышей с дефицитом TNFR1 качество заживления полнослойных ран кожи было хуже и характеризовалось образованием фиброзной ткани. Инфильтрация лейкоцитов, а также экспрессия генов, кодирующих молекулы адгезии и провоспалительные цитокины, были значительно снижены у мышей с генетической инактивацией TNFR1. Эти наблюдения показывают, что опосредуемые TNFR1 сигналы способствуют привлечению лейкоцитов в область ранения, что важно для эффективной регенерации кожи.
Позднее эти результаты подтвердили и на мышах с полным нокаутом TNF. Так, в аналогичной модели полнослойных ран кожи у мышей с нокаутом снижена реэпителизация на фоне разрастания грануляционной ткани, что еще раз свидетельствует о неэффективном заживлении кожи при полном отсутствии TNF [46]. Детальный механизм действия TNF при заживлении кожи на сегодняшний день недостаточно изучен. При повреждении кожи, как показано, активируется Notch1, что приводит к экспрессии TNF и хемокинов CCL20 и CXCL13, привлекающих лимфоциты врожденного иммунитета класса 3 (ILC3) в кожу [50]. ILC3, в свою очередь, стимулируют инфильтрацию кожи миелоидными клетками, необходимыми для полноценной регенерации [16]. Локальное применение рекомбинантного TNF ускоряет, а его блокировка, наоборот, замедляет заживление кожи, что указывает на ключевую роль этого цитокина в заживлении кожи в отсутствие системной патологии. Кроме того, доказана ключевая роль TNF из макрофагов в процессе полноценного восстановления придатков кожи при повреждении, а именно, в активации роста волосяных фолликулов [43, 47]. Этот процесс опосредован передачей сигнала через TNFR1, так как удаление p55 (TNFR1), но не p75 (TNFR2), нарушало рост волосяных фолликулов при регенерации. Еще более интересная ситуация наблюдалась в случае de novo регенерации волосяных фолликулов (WIHN), существенно нарушенной как у мышей с генетической инактивацией TNF, так и у трансгенных мышей, сверхэкспрессирующих TNF [43], что указывает на необходимость соблюдения оптимального уровня медиаторов воспаления при заживлении ран кожи.
В подтверждение этого установлено, что хронически повышенная экспрессия TNF негативно влияет на регенерацию повреждений кожи. Так, показана терапевтическая роль локального блокирования TNF у мышей с хронически незаживающими кожными ранами [51], а также в регенерации ран у мышей, больных диабетом [52]. В обоих случаях избыточное системное воспаление, опосредованное TNF, приводило к замедлению заживления ран, что объясняет эффективность подавления этого цитокина. Это подтверждается опытами с некрозом кожи, индуцированным липополисахаридом (LPS), ключевую роль в развитии которого играет сигнальный путь TNF–TNFR1 [48]. Клеточный источник патогенного TNF при заживлении ран в условиях хронического воспаления пока неизвестен. Важным продуцентом TNF в коже, необходимым для запуска полноценного иммунного ответа, оказались тучные клетки ‒ еще один тип миелоидных клеток. Первоначально роль сигнального каскада TNF–TNFR2 в активации миграции дендритных клеток кожи была показана в модели контактного дерматита [49]. Позднее установили, что TNF, продуцируемый именно тучными клетками, необходим для созревания и миграции дендритных клеток кожи [41]. Причем при LPS-индуцированном воспалении кожи сначала происходит дегрануляция тучных клеток с последующим захватом TNF-содержащих гранул дендритными клетками и их миграцией в дренирующий лимфоузел, что приводит к активации адаптивного иммунного ответа [44]. По всей видимости, в здоровом организме умеренная продукция TNF при появлении кожных ран благоприятно действует на регенерацию, способствуя привлечению иммунных клеток и активируя переход процесса заживления в фазу пролиферации, приводящую к восстановлению функциональной архитектуры кожи (рис. 1). Напротив, в условиях системного воспаления избыточная продукция TNF негативно воздействует на регенерацию кожи, удлиняя воспалительную фазу и препятствуя, таким образом, пролиферации кератиноцитов и фибробластов. Тем не менее, сбалансированная продукция TNF при воспалении, по-видимому, важна для защиты организма в случае инфицирования кожной раны.
IL-6
IL-6 – это еще один многофункциональный цитокин, первоначально описанный как медиатор воспаления и дифференцировки лимфоцитов. IL-6 функционирует через связывание с рецептором, состоящим из двух субъединиц: gp130 (который также участвует в передаче сигнала от некоторых других цитокинов семейства IL-6) и IL-6R (CD126). В то время как gp130 экспрессируется на многих типах клеток, экспрессия IL-6R ограничена гепатоцитами и клетками иммунной системы [53]. Связывание IL-6 с рецептором индуцирует его гомодимеризацию (т.е. образование димера из димеров gp130/IL-6R) и активацию киназ семейства Janus: JAK1, JAK2 и TYK2. JAK фосфорилируют STAT3, вызывая его димеризацию, транслокацию в ядро и индукцию транскрипции IL-6-зависимых генов [54]. Активация STAT3 препятствует клеточной гибели, приводит к пролиферации и дифференцировке клеток и индукции воспаления. Передача сигнала от IL-6 может происходить также через растворимую форму рецептора IL-6R – sIL-6R, которая связывает IL-6 и впоследствии может индуцировать димеризацию gp130 на клетках, не экспрессирующих IL-6R. Этот процесс известен как транссигнализация и, парадоксально, делает клетки, которые не экспрессируют IL-6R, чувствительными к IL-6 [55]. Считается, что транссигнализация посредством IL-6/sIL6-R обеспечивает запуск провоспалительных каскадов, тогда как классическая передача сигнала IL-6 через мембраносвязанный IL-6R необходима преимущественно для регенеративной или противовоспалительной активности цитокина. Ингибирование растворимой формы IL-6R in vivo обеспечивает блокирование многих известных функций IL-6, связанных, в первую очередь, с модуляцией воспалительного ответа [56]. Кроме того, для сигналинга IL-6 характерна так называемая транспрезентация, при которой секретируемые IL-6 и IL-6R димеризуются во внутриклеточных эндосомах дендритных клеток, а затем представляются Т-лимфоцитам, что и приводит к дифференцировке Т-клеток в направлении Th17 при развитии аутоиммунных заболеваний [57].
Первые работы по регенерации кожи у мышей с дефицитом IL-6 выявили у них более медленное заживление ран по сравнению с мышами дикого типа [58, 59] (табл. 2). Это сопряжено со слабой пролиферацией кератиноцитов, уменьшением воспаления и снижением количества нейтрофилов и макрофагов в ране. В поврежденной ткани мышей с нокаутом Il6 отмечено снижение активации фактора AP-1 через 16 ч после ранения по сравнению с мышами дикого типа. Методом гибридизации in situ поврежденной ткани мышей дикого типа экспрессия мРНК IL-6 выявлена преимущественно в кератиноцитах на переднем крае раны, а также в макрофагах и фибробластах дермального слоя. При этом инъекция рекомбинантного IL-6 мышам с дефицитом IL-6 приводила к нормализации динамики заживления, что подтверждает участие IL-6 непосредственно в процессе регенерации кожи. Эти результаты коррелировали с данными тканеспецифического нокаута фактора транскрипции STAT3 в кератиноцитах, который также вызывал замедление заживления ран кожи, что указывает на ключевую роль сигнального пути IL-6/STAT3 в кожной регенерации [60]. Интересно, что роль IL-6 в заживлении кожи с возрастом усиливается. Так, в кератиноцитах старых мышей, локализованных на краю раны, снижен уровень активации STAT3, что, в свою очередь, приводит к снижению экспрессии факторов семейства Skint, ответственных за привлечение и активацию гамма-дельта Т-клеток кожи (DETC) [19]. При этом пониженная активация STAT3 в кератиноцитах старых мышей коррелировала со сниженной продукцией IL-6 в кожных ранах. Эти результаты подчеркивают, что эпителиально-иммунное взаимодействие, в целом, и Skint, в частности, являются важнейшими регуляторами, которые приводят к нарушениям регенерации в коже старых мышей.
Таблица 2.
Заживление кожи у мышей с генетическим нарушением сигнального пути IL-6
| Мыши с нокаутом (KO) или трансгенные мыши (Tg) | Модель повреждения кожи | Основной фенотип и вывод исследования | Ссылка |
|---|---|---|---|
| IL-6 KO | Полнослойная рана | Замедление заживления ран кожи, сопряженное со сниженной инфильтрацией иммунными клетками и сниженной пролиферацией кератиноцитов | [58, 59] |
| IL-6 KO IL-6R KO IL-6/IL-6R KO |
То же | Замедление контракции раны наблюдали у мышей IL-6 KO, но не IL-6Rα KO или c двойным нокаутом. Во всех трех нокаутах наблюдали нарушение реэпителизации раны | [61] |
| TLR3 KO IL-6R KO |
» | Нарушение de novo регенерации волосяных фолликулов (WIHN) у мышей с нокаутом | [62] |
| IL-6 KO IL-6R KO |
» | Удаление IL-6Rα блокировало, а удаление IL-6 ускоряло WIHN после повреждения. Удаление IL-6 приводило к повышенной экспрессии OsM и IL-11, а также к активации STAT3 в коже | [63] |
Влияние IL-6 на заживление ран в значительной степени связано с активацией фибробластов кожи, что способствует активной миграции клеток в ранах и экспрессии прорегенеративных факторов. Так, установлено, что IL-6 участвует в индукции экспрессии TGFβ, количество которого снижено в коже мышей с дефицитом IL-6 [64]. При этом с использованием специфических ингибиторов внутриклеточных сигналов показано, что этот эффект IL-6 опосредуется активацией MAP-каскада и киназы ERK1/2 в фибробластах. Кроме того, фибробласты кожи мышей IL-6 KO увеличивали экспрессию металлопротеиназы MMP-2 и имели сниженную способность к миграции, что, по-видимому, объясняет нарушения в образовании грануляционной ткани при заживлении ран [65]. Для более детального понимания механизма действия IL-6 изучено заживление кожи у мышей с генетическим дефицитом IL-6 или IL-6R [61]. Поскольку функции, опосредованные IL-6, требуют связывания со специфическим рецептором IL-6R, ожидалось, что мыши, лишенные рецептора, будут иметь такой же фенотип, как и мыши с дефицитом IL-6. Однако, хотя у мышей с дефицитом IL-6R выявлены те же нарушения воспалительного ответа, как и у мышей с дефицитом IL-6, у них отсутствовала задержка процесса регенерации кожных ран. Более того, мыши с комбинированным дефицитом IL-6 и IL-6R также не отличались от мышей дикого типа по скорости закрытия раны. Тем не менее, у всех мышей с нарушенной передачей сигнала IL-6 наблюдалась замедленная реэпителизация. По-видимому, это указывает, с одной стороны, на участие IL-6 в функциональном заживлении ран кожи, а с другой, на возможное участие других членов семейства IL-6, выполняющих компенсаторную функцию на фоне дефицита IL-6/IL-6R, в регуляции контракции раны. Вероятно также, что различные клеточные источники IL-6 могут вносить разный вклад в заживление кожи, что должно быть выяснено в дальнейшем с использованием тканеспецифических нокаутов IL-6.
Помимо собственно заживления раны, IL-6 также играет ключевую роль в процессе WIHN. Показано, что при повреждении кожи происходит высвобождение фрагментов дцДНК, которая активирует рецептор врожденного иммунитета TLR3. В результате наблюдается экспрессия IL-6 и активация STAT3, обеспечивающая WIHN [62]. В подтверждение этого у мышей с генетическим нокаутом TLR3 или IL-6R отсутствует WIHN. Удивительно, но удаление IL-6 ускоряло WIHN в ходе регенерации кожи [63]. Это парадоксальное увеличение WIHN у мышей IL-6 KO побудило рассмотреть существование возможных компенсаторных механизмов. IL-6 и другие цитокины семейства IL-6, такие как онкостатин M (OsM) и IL-11, связывают gp130, что приводит к активации STAT3 [66]. Интересно, что после повреждения кожи экспрессия OsM возрастала у мышей как IL-6 KO, так и дикого типа, однако у мышей IL-6 KO была примерно в 3 раза выше. Кроме того, через 6 ч после ранения экспрессия IL-11 у мышей IL-6 KO была значительно выше, чем у мышей дикого типа, хотя общий уровень IL-11 оставался относительно низким. Эти данные свидетельствуют о том, что в отсутствие IL-6 повышается экспрессия других цитокинов семейства IL-6, которые могут обеспечивать активацию STAT3 на фоне дефицита IL-6. Действительно, в неповрежденной коже мышей IL-6 KO уровень p-STAT3 был в 10 раз выше, чем у мышей дикого типа, что согласуется с ускоренным WIHN у мышей IL-6 KO, так как STAT3 участвует в морфогенезе волосяных фолликулов, заживлении раны и WIHN [60, 62]. Вместе эти данные показывают, что, сигнальный путь STAT3 функционирует даже в отсутствие IL-6 и необходим для WIHN [63].
Интересно, что в некоторых случаях рекомбинантный IL-6 может способствовать ускорению регенерации кожи, например при системной иммуносупрессии. Так, отсроченное заживление кожных ран у мышей, обусловленное индуцированной глюкокортикоидами иммуносупрессией, может быть отменено с помощью рекомбинантного IL-6, о чем свидетельствуют эпителизация, образование грануляционной ткани и закрытие раны [67]. Следует отметить, что у контрольных мышей рекомбинантный IL-6 не усиливал заживление ран, а скорее задерживал процесс, указывая на необходимость оптимального баланса провоспалительных медиаторов. Иммунохимические исследования выявили повышение экспрессии ММР-10 у мышей, получавших дексаметазон, а обработка IL-6 снижала экспрессию ММР-10, показывая, что IL-6 может влиять на образование дермального матрикса и, в частности, на синтез коллагена. Таким образом, IL-6 может восстанавливать заживление раны, нарушенное при иммунодефиците.
IL-1
IL-1 – один из древнейших цитокинов животных, его сигнальный путь связан с активацией каскада, характерного для Toll-подобных рецепторов врожденного иммунитета [68, 69]. Известны два варианта этого цитокина – IL-1α и IL-1β, которые передают сигнал через рецепторный комплекс IL-1R1 и IL-1RAcP (IL-1R3), содержащий TIR-домен и активирующий классический NF-κB, а также MAP-киназы [70]. Помимо этого основного сигнального пути существуют также два регуляторных пути, препятствующих передаче сигнала. Один связан с рецепторным антагонистом IL-1Ra, который конкурирует с IL-1 за связывание с IL-1R1, но, в отличие от цитокина, связывание антагониста не приводит к запуску сигнального каскада [71]. Кроме того, существует другой рецептор IL-1 – IL-1R2, который не имеет TIR-домена, в результате чего связывание с ним также не приводит к активации сигнальных путей [72].
IL-1, наряду с другими провоспалительными цитокинами, активно экспрессируется в коже при повреждении [10]. Однако его вклад в процесс реэпителизации кожных повреждений не до конца изучен (табл. 3). Результаты, полученные ранее на модели ран кожи свиней, свидетельствовали о терапевтическом потенциале рекомбинантного IL-1α [73]. Тем не менее, с развитием технологий обратной генетики появилась возможность более детального изучения механизма действия этого цитокина. Так, удаление IL-1R1 приводило к замедлению заживления ран мягкого неба, но не кожи, что объяснялось большей микробной нагрузкой в ротовой полости, так как использование антибиотиков ускоряло заживление повреждений неба у мышей с нокаутом IL-1R1 [74]. Более того, в ряде работ показана патогенная роль воспалительного ответа, запускаемого IL-1, в регенерации повреждений кожи и роговицы как в обычном состоянии [75, 76], так и при сахарном диабете [77, 78]. Особая роль отводится рецепторному антагонисту IL-1Ra, так как его отсутствие приводит к замедлению регенерации кожи [75], а применение рекомбинантного белка способствует заживлению роговицы при диабете [78]. Обнаружено также негативное влияние IL-1 на рост волосяных фолликулов у мышей и человека [79–82]. По-видимому, продукция IL-1 исключительно важна для предотвращения инфицирования раны, что особенно характерно в случае ран ротовой полости, тогда как в условиях отсутствия патогенных микроорганизмов IL-1-опосредованное воспаление замедляет переход к реэпителизации повреждений кожи у SPF (specific pathogen free) животных. Это заключение подтверждается эффективностью терапии гнойных ран у человека с использованием рекомбинантного IL-1β [83]. В то же время, роль IL-1 в активации стволовых клеток кожи показана на модели гиперпролиферации кератиноцитов, индуцируемой генетическим удалением каспазы-8 в эпителиальных клетках, что указывает на участие этого сигнального пути не только в воспалительном ответе, но также в процессе реэпителизации раны [84, 85]. Более того, показана ключевая роль IL-1 в адаптации стволовых клеток кожи к воспалительному ответу, приводящая к ускорению последующего заживления ран. Так, локальный воспалительный ответ, вызванный лигандами рецепторов TLR или повреждением, посредством передачи сигнала от IL-1, приводит к эпигенетическим перестройкам в стволовых клетках кожи, что способствует ускоренному заживлению ран даже через полгода после воспалительного стимула [86].
Таблица 3.
Заживление кожи у мышей с генетическим нарушением сигнального пути IL-1
| Мыши с нокаутом (KO) или трансгенные (Tg) | Модель повреждения кожи | Основной фенотип и вывод исследования | Ссылка |
|---|---|---|---|
| IL-1R1 KO | Модель полнослойной раны кожи и раны мягкого неба | Замедление заживления раны мягкого неба, но не кожи спины, при удалении IL-1R1. Использование антибиотиков ускоряло регенерацию у животных с нокаутом. | [74] |
| IL-1R1 KO | Модель полнослойной раны кожи | Сниженная инфильтрация ран иммунными клетками и сниженное образование фиброзной ткани у мышей с нокаутом. | [76] |
| IL-1Ra KO | Модель полнослойной раны кожи | Замедленное заживление ран кожи, высокая инфильтрация иммунными клетками и повышенный уровень активации NF-κB по сравнению с мышами дикого типа. | [75] |
| Макрофаги мышей IL-1R1 KO. | In vitro стимуляция макрофагов раневым эксудатом из полнослойных ран кожи у мышей db/db | Удаление IL-1R1 на макрофагах блокирует экспрессию провоспалительных медиаторов и восстанавливает продукцию прорегенеративных факторов при активации клеток раневым эксудатом из полнослойных ран мышей с диабетом типа 2. | [77] |
| IL-1R1/IL-1α Tg, сверхэкспрессия в кератиноцитах | Спонтанное воспаление кожи | Спонтанное воспаление кожи, сопровождающееся утолщением эпителиального слоя, увеличением инфильтрации иммунных клеток у двойной трансгенной мыши. | [81] |
| IL-1R1 KO | Модель заживления кожи, индуцируемая эпидермальным нокаутом каспазы-8. | Сигнальный путь IL-1 необходим для запуска пролиферации стволовых клеток кожи и активации γδТ-клеток. | [85] |
| IL-1R1 KO | Модель полнослойной фиксированной раны кожи | Удаление IL-1R1 блокировало ускоренное заживление полнослойных ран кожи через месяц после индукции воспаления, что указывает на необходимость сигнального пути IL-1 для активации стволовых клеток кожи. | [86] |
ИНТЕРФЕРОНЫ
Семейство интерферонов (IFN) подразделяют на три типа: IFN типа I (IFNα, IFNβ и некоторые другие), единственный IFN типа II (IFNγ) и IFN типа III (IFNλ) [87]. Типичный рецептор IFN представляет собой комплекс из двух субъединиц: IFNRA1 и IFNRA2 ‒ для IFN типа I, IFNGR1 и IFNGR2 для IFNγ и IFNLR1, IL10R2 для IFN типа III. В результате передачи сигнала происходит активация JAK1/2 и TYK2, что приводит к фосфорилированию STAT1, STAT2 и IRF9 и экспрессии IFN-зависимых генов [87]. Кроме того, существует также альтернативный путь передачи сигнала, приводящий к активации MAP-киназ и фактора транскрипции STAT3 [87]. IFN типа I продуцируются преимущественно плазмацитоидными дендритными клетками, они являются основными противовирусными цитокинами [88], в то время как IFNγ – это цитокин Th1-ветви адаптивного иммунитета, играющий ключевую роль в активации провоспалительных макрофагов [89].
Неожиданностью стало обнаружение терапевтической активности IFN типа I при ряде заболеваний, таких как рассеянный склероз и некоторые злокачественные новообразования [90], хотя эта терапия в лучшем случае замедляет течение болезни. Несмотря на это, роль IFN в заживлении кожи оказалась во многих случаях отрицательной (табл. 4). Так, локальная инъекция IFN типа I замедляла заживление повреждений кожи за счет ингибирования ангиогенеза [91], а системное введение IFNγ блокировало дифференцировку миофибробластов, секрецию коллагена, что также приводило к замедлению регенерации кожи [92]. Эти результаты были подтверждены в опытах на мышах с генетической инактивацией IFNγ, у которых наблюдалось ускоренное заживление ран кожи, сниженная инфильтрация иммунных клеток в очаг повреждения и более активный ангиогенез [93], ассоциированные с повышенной продукцией TGF-β1, участвующего в дифференцировке миофибробластов и продукции коллагена, и VEGF, необходимого для роста кровеносных сосудов. Тем не менее, будучи важными провоспалительными молекулами, IFN типа I и II участвуют в индукции иммунного ответа в местах повреждения, что, как и в случае с IL-1, может играть важную роль в терапии инфицированных ран. Недавно обнаружена положительная роль IFN типа I, продуцируемых кератиноцитами и плазмацитоидными дендритными клетками в ответ на повреждение, в запуске иммунного ответа [94, 95], показано участие IFNγ в продукции TNF и NO макрофагами в кожных ранах [96].
Таблица 4.
Заживление кожи у мышей с генетическим нарушением сигнального пути IFN
| Мыши с нокаутом (KO) или трансгенные (Tg) | Модель повреждения кожи | Основной фенотип и вывод исследования | Ссылка |
|---|---|---|---|
| IFNγ KO | Полнослойная рана | Ускоренное заживление ран, более активный ангиогенез, повышенная продукция коллагена и прорегенеративного фактора TGFβ1 | [93] |
| IFNγ KO | Подкожная имплантация PVA-матриксов | Сниженная продукция TNF и NO в ранах мышей с нокаутом | [96] |
| IFNAR KO | Повреждение кожи с помощью адгезивной пленки | Блокировка сигнального пути IFN типа I препятствовала реэпителизации кожного повреждения, приводила к снижению продукции IL-6, IL-17A и IL-22 в поврежденной коже | [94] |
ЗАКЛЮЧЕНИЕ
Таким образом, провоспалительные цитокины играют ключевую роль как в регуляции кожного гомеостаза, так и в контроле регенеративного процесса. Динамика экспрессии цитокинов определяет последовательность защитных реакций кожи после повреждения. Так, в первые дни после ранения важнейшей задачей является предотвращение инфицирования, что обеспечивается быстрой (в течение часов после повреждения) продукцией провоспалительных цитокинов, преимущественно IL-1 и TNF. Это может приводить как к активации резидентных стромальных и иммунных клеток, так и к привлечению лейкоцитов, преимущественно нейтрофилов, из кровотока с последующей активацией фагоцитоза и секреции антимикробных пептидов. Интересно, что у мышей без микробиоты (germ-free, GF) в отсутствие комменсальных бактерий увеличивается скорость закрытия раны кожи [97]. Гистологический анализ выявил ускоренную эпителизацию ран у таких мышей по сравнению с контрольными. Отмечено значительное снижение инфильтрации нейтрофилами и повышение инфильтрации тучными клетками и макрофагами области ранения у GF-мышей. Интересно, что гены, кодирующие альтернативно активированные регенеративные факторы макрофагов, активно экспрессируются в раневой ткани GF-мышей. Более того, экспрессия противовоспалительного цитокина IL-10, ангиогенного фактора роста VEGF и уровень ангиогенеза в раневой ткани мышей GF были выше, а рубцевание и уровень TGF-β1 значительно ниже, чем у контрольных мышей. В целом, эти данные свидетельствуют о том, что при отсутствии контакта с микробиотой заживление кожных ран ускоряется и происходит без образования рубцов, частично из-за уменьшения накопления нейтрофилов, увеличения количества альтернативно активированных регенеративных макрофагов и более активного ангиогенеза в участках раны.
В дальнейшем, при нивелировании угрозы инфицирования и закрытия повреждения встает необходимость полноценного восстановления функционального покрова кожи. В этот момент избыточное воспаление тормозит регенеративный процесс, поэтому в норме экспрессия таких цитокинов, как IL-1 и IFN, которые обеспечивали до этого защиту от заражения, снижается, тогда как TNF и IL-6 продолжают экспрессироваться на высоком уровне. Их роль на этом этапе заключается в активации и стимуляции деления кератиноцитов и фибробластов, которые составляют основу нормальной кожи, через NF-κB и STAT3. Эти два цитокина также играют особую роль в восстановлении кожных придатков, в первую очередь, волосяных фолликулов, что указывает на качественную регенерацию кожи.
Тем не менее, несмотря на полученные за последнее время знания в области регенеративной медицины, ясно, что значительная часть повреждений кожи завершается фиброзом, т.е. механическим закрытием повреждения без восстановления функциональной кожной архитектуры. Механизм выбора между этими двумя физиологическими исходами на сегодняшний день не установлен, однако известно, что важную роль в обоих процессах играют провоспалительные цитокины. Это указывает на необходимость строгого баланса в продукции цитокинов, детали которого не могут быть поняты только с использованием системной блокировки или полного генетического нокаута. Новые технологии, включая кондиционный нокаут и особенно кондиционный индуцируемый нокаут (именно эта модальность более полно соответствует фармакологической блокировке) конкретных цитокинов и их рецепторов, необходимы для более детального понимания механизмов регенерации кожи. Это, в свою очередь, откроет новые перспективы в терапии кожных ран различной этиологии, которая будет направлена на установление определенного баланса провоспалительных и других медиаторов с целью достижения полноценного функционального восстановления кожного покрова.
Работа поддержана Российским фондом фундаментальных исследований (№ 19-04-01094) и Российским научным фондом (№ 19-75-30032).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Eming S.A., Martin P., Tomic-Canic M. (2014) Wound repair and regeneration: mechanisms, signaling, and translation. Sci. Transl. Med. 6, 1–16.
Landén N.X., Li D., Ståhle M. (2016) Transition from inflammation to proliferation: a critical step during wound healing. Cell. Mol. Life Sci. 73, 3861–3885.
Larouche J., Sheoran S., Maruyama K., Martino M.M. (2018) Immune regulation of skin wound healing: Mechanisms and novel therapeutic targets. Adv. Wound Care. 7, 209–231.
Martin P. (1997) Wound healing – aiming for perfect skin regeneration. Science. 276, 75–81.
Wick G., Grundtman C., Mayerl C., Wimpissinger T.-F., Feichtinger J., Zelger B., Sgonc R., Wolfram D. (2013) The immunology of fibrosis. Annu. Rev. Immunol. 31, 107–135.
White E.S., Mantovani A.R. (2013) Inflammation, wound repair, and fibrosis: Reassessing the spectrum of tissue injury and resolution. J. Pathol. 229, 141–144.
Walmsley G.G., Maan Z.N., Wong V.W., Duscher D., Hu M.S., Zielins E.R., Wearda T., Muhonen E., McArdle A., Tevlin R., Atashroo D.A., Senarath-Yapa K., Lorenz H.P., Gurtner G.C., Longaker M.T. (2015) Scarless wound healing: chasing the holy grail. Plast. Reconstr. Surg. 135, 907–917.
Behm B., Babilas P., Landthaler M., Schreml S. (2012) Cytokines, chemokines and growth factors in wound healing. J. Eur. Acad. Dermatology Venereol. 26, 812–820.
Kondo T., Ohshima T. (1996) The dynamics of inflammatory cytokines in the healing process of mouse skin wound: A preliminary study for possible wound age determination. Int. J. Legal Med. 108, 231–236.
Sato Y., Ohshima T. (2000) The expression of mRNA of proinflammatory cytokines during skin wound healing in mice: A preliminary study for forensic wound age estimation (II). Int. J. Legal Med. 113, 140–145.
Zhao R., Liang H., Clarke E., Jackson C., Xue M. (2016) Inflammation in chronic wounds. Int. J. Mol. Sci. 17, 1–14.
Wu G., Zhu B., Hong X., Luo P., Xia Z. (2017) Role of cytokines in host defense against Staphylococcus aureus skin infection. Histol. Histopathol. 32, 761–766.
Архипова А.Ю., Носенко М.А., Малюченко Н.В., Зварцев Р.В., Мойсенович А.М., Жданова А.С., Васильева Т.В., Горшкова Е.А., Агапов И.И., Друцкая М.С., Недоспасов С.А., Мойсенович М.М. (2016) Влияние фиброиновых микроносителей на воспаление и регенерацию полнослойных ран кожи у мышей. Биохимия. 81, 1251–1260.
Nosenko M.A., Moysenovich A.M., Zvartsev R. V, Arkhipova A.Y., Zhdanova A.S., Agapov I.I., Vasilieva T.V., Bogush V.G., Debabov V.G., Nedospasov S., Moisenovich M.M., Drutskaya M.S. (2018) Novel biodegradable polymeric microparticles facilitate scarless wound healing by promoting re-epithelialization and inhibiting fibrosis. Front. Immunol. 9, 2851.
Turabelidze A., Dipietro L.A. (2012) Inflammation and wound healing. Endod. Top. 24, 26–38.
Goren I., Allmann N., Yogev N., Schu C., Linke A., Holdener M., Waisman A., Pfeilschifter J., Frank S. (2009) A transgenic mouse model of inducible macrophage depletion. Am. J. Pathol. 175, 132–147.
Chen L., Mehta N.D., Zhao Y., DiPietro L.A. (2014) Absence of CD4 or CD8 lymphocytes changes infiltration of inflammatory cells and profiles of cytokine expression in skin wounds, but does not impair healing. Exp. Dermatol. 23, 189–194.
Ansell D.M., Holden K.A., Hardman M.J. (2012) Animal models of wound repair: Are they cutting it? Exp. Dermatol. 21, 581–585.
Keyes B.E., Liu S., Asare A., Naik S., Levorse J., Polak L., Lu C.P., Nikolova M., Pasolli H.A., Fuchs E. (2016) Impaired epidermal to dendritic T cell signaling slows wound repair in aged skin. Cell. 167, 1323–1338.
Feldmann M. (2002) Development of anti-TNF therapy for rheumatoid arthritis. Nat. Rev. Immunol. 2, 364–371.
Южакова Д.В., Ширманова М.В., Бочаров А.А., Астраханцева И.В., Василенко Е.А., Горшкова Е.Н., Друцкая М.С., Загайнова Е.В., Недоспасов С.А., Круглов А.А. (2016) Микрофлора индуцирует экспрессию фактора некроза опухолей в коже в постнатальный период у мышей. Биохимия. 81, 1553–1558.
Karin M., Ben-Neriah Y. (2000) Phosphorylation meets ubiquitination: the control of NF-kB activity. Annu. Rev. Immunol. 18, 621–663.
Kankaanranta H., Ilmarinen P., Zhang X., Adcock I.M., Lahti A., Barnes P.J., Giembycz M.A., Lindsay M.A., Moilanen E. (2014) Tumour necrosis factor-alpha regulates human eosinophil apoptosis via ligation of TNF-receptor 1 and balance between NF-kappaB and AP-1. PLoS One. 9, e90298.
Park Y.-H., Jeong M.S., Jang S.B. (2014) Death domain complex of the TNFR-1, TRADD, and RIP1 proteins for death-inducing signaling. Biochem. Biophys. Res. Commun. 443, 1155–1161.
Zhao X., Rong L., Zhao X., Li X., Liu X., Deng J., Wu H., Xu X., Erben U., Wu P., Syrbe U., Sieper J., Qin Z. (2012) TNF signaling drives myeloid-derived suppressor cell accumulation. J. Clin. Invest. 122, 4094–4104.
Kumar A., Gordy L.E., Bezbradica J.S., Stanic A.K., Hill T.M., Boothby M.R., Van Kaer L., Joyce S. (2017) NF-kappaB protects NKT cells from tumor necrosis factor receptor 1-induced death. Sci. Rep. 7, 15594.
Tak P.P., Firestein G.S. (2001) NF-kappaB: a key role in inflammatory diseases. J. Clin. Invest. 107, 7–11.
Sica A., Mantovani A. (2012) Macrophage plasticity and polarization: in vivo veritas. J. Clin. Invest. 122, 787–795.
Wang N., Liang H., Zen K. (2014) Molecular mechanisms that influence the macrophage m1-m2 polarization balance. Front. Immunol. 5, 614.
Zalevsky J., Secher T., Ezhevsky S.A., Janot L., Steed P.M., O’Brien C., Eivazi A., Kung J., Nguyen D.-H.T., Doberstein S.K., Erard F., Ryffel B., Szymkowski D.E. (2007) Dominant-negative inhibitors of soluble TNF attenuate experimental arthritis without suppressing innate immunity to infection. J. Immunol. 179, 1872–1883.
Kalliolias G.D., Ivashkiv L.B. (2016) TNF biology, pathogenic mechanisms and emerging therapeutic strategies. Nat. Rev. Rheumatol. 12, 49–62.
Allie N., Grivennikov S.I., Keeton R., Hsu N.-J., Bourigault M.-L., Court N., Fremond C., Yeremeev V., Shebzukhov Y., Ryffel B., Nedospasov S.A., Quesniaux V.F.J., Jacobs M. (2013) Prominent role for T cell-derived tumour necrosis factor for sustained control of Mycobacterium tuberculosis infection. Sci. Rep. 3, 1809.
Olleros M.L., Chavez-Galan L., Segueni N., Bouri-gault M.L., Vesin D., Kruglov A.A., Drutskaya M.S., Bisig R., Ehlers S., Aly S., Walter K., Kuprash D.V., Chouchkova M., Kozlov S.V., Erard F., Ryffel B., Quesniaux V.F.J., Nedospasov S.A., Garcia I. (2015) Control of mycobacterial infections in mice expressing human tumor necrosis factor (TNF) but not mouse TNF. Infect. Immun. 83, 3612–3623.
Tumanov A.V., Grivennikov S.I., Kruglov A.A., Shebzukhov Y.V., Koroleva E.P., Piao Y., Cui C., Kuprash D.V., Nedospasov S.A. (2010) Cellular source and molecular form of TNF specify its distinct functions in organization of secondary lymphoid organs. Blood. 116, 3456–3464.
Atretkhany K.S.N., Nosenko M.A., Gogoleva V.S., Zvartsev R.V., Qin Z., Nedospasov S.A., Drutskaya M.S. (2016) TNF neutralization results in the delay of transplantable tumor growth and reduced MDSC accumulation. Front. Immunol. 7, 147.
Atretkhany K.N., Mufazalov I.A., Dunst J., Kuchmiy A.A., Gogoleva V.S., Andruszewski D., Drutskaya M.S., Faustman D.L., Schwabenland M., Prinz M., Kruglov A.A., Waisman A., Nedospasov S.A. (2018) Intrinsic TNFR2 signaling in T regulatory cells provides protection in CNS autoimmunity. Proc. Natl. Acad. Sci. USA. 115, 13051–13056.
Eissner G., Kolch W., Scheurich P. (2004) Ligands working as receptors: Reverse signaling by members of the TNF superfamily enhance the plasticity of the immune system. Cytokine Growth Factor Rev. 15, 353–366.
Grivennikov S.I., Tumanov A. V., Liepinsh D.J., Kruglov A.A., Marakusha B.I., Shakhov A.N., Murakami T., Drutskaya L.N., Förster I., Clausen B.E., Tessarollo L., Ryffel B., Kuprash D.V., Nedospasov S.A. (2005) Distinct and nonredundant in vivo functions of TNF produced by T cells and macrophages/neutrophils: Protective and deleterious effects. Immunity. 22, 93–104.
Benezech C., Luu N.-T., Walker J.A., Kruglov A.A., Loo Y., Nakamura K., Zhang Y., Nayar S., Jones L.H., Flores-Langarica A., McIntosh A., Marshall J., Barone F., Besra G., Miles K., Allen J.E., Gray M., Kollias G., Cunningham A.F., Withers D.R., Toellner K.M., Jones N.D., Veldhoen M., Nedospasov S.A., McKenzie A.N.J., Caamano J.H., Bénézech C., Luu N.-T., Walker J.A., Kruglov A.A., Loo Y., Nakamura K., Zhang Y., Nayar S., Jones L.H., Flores-Langarica A., McIntosh A., Marshall J., Barone F., Besra G., Miles K., Allen J.E., Gray M., Kollias G., Cunningham A.F., Withers D.R., Toellner K.M., Jones N.D., Veldhoen M., Nedospasov S.A., McKenzie A.N.J., Caamaño J.H. (2015) Inflammation-induced formation of fat-associated lymphoid clusters. Nat. Immunol. 16, 819–828.
Kruglov A.A., Lampropoulou V., Fillatreau S., Nedospasov S.A. (2011) Pathogenic and protective functions of TNF in neuroinflammation are defined by its expression in T lymphocytes and myeloid cells. J. Immunol. 187, 5660–5670.
Dudeck J., Ghouse S.M., Lehmann C.H.K., Hoppe A., Schubert N., Nedospasov S.A., Dudziak D., Dudeck A. (2015) Mast-cell-derived TNF amplifies CD8+ dendritic cell functionality and CD8+ T-cell priming. Cell Rep. 13, 399–411.
Awad A.S., You H., Gao T., Cooper T.K., Nedospasov S.A., Vacher J., Wilkinson P.F., Farrell F.X., Brian Reeves W. (2015) Macrophage-derived tumor necrosis factor-α mediates diabetic renal injury. Kidney Int. 88, 722–733.
Wang X., Chen H., Tian R., Zhang Y., Drutskaya M.S., Wang C., Ge J., Fan Z., Kong D., Wang X., Cai T., Zhou Y., Wang J., Wang J., Wang S., Qin Z., Jia H., Wu Y., Liu J., Nedospasov S.A., Tredget E.E., Lin M., Liu J., Jiang Y., Wu Y. (2017) Macrophages induce AKT/beta-catenin-dependent Lgr5+ stem cell activation and hair follicle regeneration through TNF. Nat. Commun. 8, 14091.
Dudeck J., Froebel J., Kotrba J., Lehmann C.H.K., Dudziak D., Speier S., Nedospasov S.A., Schraven B., Dudeck A. (2018) Engulfment of mast cell secretory granules on skin inflammation boosts dendritic cell migration and priming efficiency. J. Allergy Clin. Immunol. 143(5), 1849–1864. e4. https://doi.org/10.1016/j.jaci.2018.08.052
Mori R., Kondo T., Ohshima T., Ishida Y., Mukaida N. (2002) Accelerated wound healing in tumor necrosis factor receptor p55-deficient mice with reduced leukocyte infiltration. FASEB J. 16, 963–974.
Shinozaki M., Okada Y., Kitano A., Ikeda K., Saika S., Shinozaki M. (2009) Impaired cutaneous wound healing with excess granulation tissue formation in TNFα-null mice. Arch. Dermatol. Res. 301, 531–537.
Chen C.-C., Wang L., Plikus M. V, Jiang T.X., Murray P.J., Ramos R., Guerrero-Juarez C.F., Hughes M.W., Lee O.K., Shi S., Widelitz R.B., Lander A.D., Chuong C.M. (2015) Organ-level quorum sensing directs regeneration in hair stem cell populations. Cell. 161, 277–290.
Amar S., Van Dyke T.E., Eugster H.P., Schultze N., Koebel P., Bluethmann H. (1995) Tumor necrosis factor (TNF)-induced cutaneous necrosis is mediated by TNF receptor 1. J. Inflamm. 47, 180—189.
Wang B., Fujisawa H., Zhuang L., Kondo S., Shivji G.M., Kim C.S., Mak T.W., Sauder D.N. (1997) Depressed Langerhans cell migration and reduced contact hypersensitivity response in mice lacking TNF receptor p75. J. Immunol. 159, 6148–6155.
Li Z., Hodgkinson T., Gothard E.J., Boroumand S., Lamb R., Cummins I., Narang P., Sawtell A., Coles J., Leonov G., Reboldi A., Buckley C.D., Cupedo T., Siebel C., Bayat A., Coles M.C., Ambler C.A. (2016) Epidermal Notch1 recruits RORγ+ group 3 innate lymphoid cells to orchestrate normal skin repair. Nat. Commun. 7, 11394.
Ashcroft G.S., Jeong M.-J., Ashworth J.J., Hardman M., Jin W., Moutsopoulos N., Wild T., McCartney-Francis N., Sim D., McGrady G., Song X., Wahl S.M. (2012) Tumor necrosis factor-alpha (TNF-α) is a therapeutic target for impaired cutaneous wound healing. Wound Repair Regen. 20, 38–49.
Goren I., Müller E., Schiefelbein D., Christen U., Pfeilschifter J., Mühl H., Frank S. (2007) Systemic anti-TNFα treatment restores diabetes-impaired skin repair in ob/ob mice by inactivation of macrophages. J. Invest. Dermatol. 127, 2259–2267.
Taga T., Hibi M., Hirata Y., Yamasaki K., Yasukawa K., Matsuda T., Hirano T., Kishimoto T. (1989) Interleukin-6 triggers the association of its receptor with a possible signal transducer, gp130. Cell. 58, 573–581.
Wegenka U.M., Buschmann J., Lütticken C., Heinrich P.C., Horn F. (1993) Acute-phase response factor, a nuclear factor binding to acute-phase response elements, is rapidly activated by interleukin-6 at the posttranslational level. Mol. Cell. Biol. 13, 276–288.
Rose-John S. (2012) IL-6 trans-signaling via the soluble IL-6 receptor: Importance for the proinflammatory activities of IL-6. Int. J. Biol. Sci. 8, 1237–1247.
Rabe B., Chalaris A., May U., Waetzig G.H., Seegert D., Williams A.S., Jones S.A., Rose-John S., Scheller J. (2008) Transgenic blockade of interleukin 6 transsignaling abrogates inflammation. Blood. 111, 1021–1028.
Heink S., Yogev N., Garbers C., Herwerth M., Aly L., Gasperi C., Husterer V., Croxford A.L., Moller-Hackbarth K., Bartsch H.S., Sotlar K., Krebs S., Regen T., Blum H., Hemmer B., Misgeld T., Wunderlich T.F., Hidalgo J., Oukka M., Rose-John S., Schmidt-Supprian M., Waisman A., Korn T. (2017) Trans-presentation of IL-6 by dendritic cells is required for the priming of pathogenic TH17 cells. Nat. Immunol. 18, 74–85.
Gallucci R.M., Simeonova P.P., Matheson J.M., Kommineni C., Guriel J.L., Sugawara T., Luster M.I. (2000) Impaired cutaneous wound healing in interleukin-6-deficient and immunosuppressed mice. FASEB J. 14, 2525–2531.
Lin Z.-Q., Kondo T., Ishida Y., Takayasu T., Mukaida N. (2003) Essential involvement of IL-6 in the skin wound-healing process as evidenced by delayed wound healing in IL-6-deficient mice. J. Leukoc. Biol. 73, 713–721.
Sano S., Itami S., Takeda K., Tarutani M., Yamaguchi Y., Miura H., Yoshikawa K., Akira S., Takeda J. (1999) Keratinocyte-specific ablation of Stat3 exhibits impaired skin remodeling, but does not affect skin morphogenesis. EMBO J. 18, 4657–4668.
McFarland-Mancini M.M., Funk H.M., Paluch A.M., Zhou M., Giridhar P.V., Mercer C., Kozma S.C., Drew A.F. (2010) Differences in wound healing in mice with deficiency of IL-6 versus IL-6 receptor. J. Immunol. 184, 7219–7228.
Nelson A.M., Reddy S.K., Ratliff T.S., Hossain M.Z., Katseff A.S., Zhu A.S., Chang E., Resnik S.R., Page C., Kim D., Whittam A.J., Miller L.S., Garza L.A. (2015) dsRNA released by tissue damage activates TLR3 to drive skin regeneration. Cell Stem Cell. 17, 139–151.
Nelson A.M., Katseff A.S., Resnik S.R., Ratliff T.S., Zhu A.S., Garza L.A. (2016) Interleukin 6 null mice paradoxically display increased Stat3 activity and wound-induced hair neogenesis. J. Invest. Dermatol. 136, 2015–2017.
Luckett-Chastain L.R., Gallucci R.M. (2009) Interleukin (IL)-6 modulates transforming growth factor-b expression in skin and dermal fibroblasts from IL-6-deficient mice. Br. J. Dermatol. 161, 237–248.
Luckett L.R., Gallucci R.M. (2007) Interleukin-6 (IL-6) modulates migration and matrix metalloproteinase function in dermal fibroblasts from IL-6KO mice. Br. J. Dermatol. 156, 1163–1171.
Heinrich P.C., Behrmann I., Haan S., Hermanns H.M., Müller-Newen G., Schaper F. (2003) Principles of interleukin (IL)-6-type cytokine signalling and its regulation. Biochem. J. 374, 1–20.
Gallucci R.M., Sugawara T., Yucesoy B., Berryann K., Simeonova P.P., Matheson J.M., Luster M.I. (2001) Interleukin-6 treatment augments cutaneous wound healing in immunosuppressed mice. J. Interf. Cytokine Res. 21, 603–609.
Dinarello C.A. (2018) Overview of the IL-1 family in innate inflammation and acquired immunity. Immunol. Rev. 281, 8–27.
Garlanda C., Dinarello C.A., Mantovani A. (2013) The Interleukin-1 family: Back to the future. Immunity. 39, 1003–1018.
Palomo J., Dietrich D., Martin P., Palmer G., Gabay C. (2015) The interleukin (IL)-1 cytokine family – balance between agonists and antagonists in inflammatory diseases. Cytokine. 76, 25–37.
Dripps D.J., Brandhuber J., Thompson R.C., Eisenbergt P. (1991) Interleukin-1 (IL-1) receptor antagonist binds to the 80-kDa IL-1 receptor but does not initiate IL-1 signal transduction. J. Biol. Chem. 266, 10331–10336.
Brannan C.I., Wignall J.M., Jenkins N.A., Grubin C.E., Mosley B., Lupton S.D., Slack J.L., Cosman D., Brunton L.L., McMahan C.J. (2018) A novel IL-1 receptor, cloned from B cells by mammalian expression, is expressed in many cell types. EMBO J. 10, 2821–2832.
Sauder D.N., Kilian P.L., McLane J.A., Quick T.W., Jakubovic H., Davis S.C., Eaglstein W.H., Mertz P.M. (1990) Interleukin-1 enhances epidermal wound healing. Lymphokine Res. 9, 465–473.
Graves D.T., Nooh N., Gillen T., Davey M., Patel S., Cottrell D., Amar S. (2001) IL-1 plays a critical role in oral, but not dermal, wound healing. J. Immunol. 167, 5316–5320.
Ishida Y., Kondo T., Kimura A., Matsushima K., Mukaida N. (2006) Absence of IL-1 receptor antagonist impaired wound healing along with aberrant NF-kB activation and a reciprocal suppression of TGF- signal pathway. J. Immunol. 176, 5598–5606.
Thomay A.A., Daley J.M., Sabo E., Worth P.J., Shelton L.J., Harty M.W., Reichner J.S., Albina J.E. (2009) Disruption of interleukin-1 signaling improves the quality of wound healing. Am. J. Pathol. 174, 2129–2136.
Mirza R.E., Fang M.M., Ennis W.J., Koh T.J. (2013) Blocking interleukin-1β induces a healing-associated wound macrophage phenotype and improves healing in type 2 diabetes. Diabetes. 62, 2579–2587.
Yan C., Gao N., Sun H., Yin J., Lee P., Zhou L., Fan X., Yu F.S. (2016) Targeting imbalance between IL-1β and IL-1 receptor antagonist ameliorates delayed epithelium wound healing in diabetic mouse corneas. Am. J. Pathol. 186, 1466–1480.
Hull S.M., Nutbrown M., Pepall L., Thornton M.J., Randall V.A., Cunliffe W.J. (1991) Immunohistologic and ultrastructural comparison of the dermal papilla and hair follicle bulb from “active” and “normal” areas of alopecia areata. J. Invest. Dermatol. 96, 673–681.
Harmon C.S., Nevins T.D. (1993) IL-1 alpha inhibits human hair follicle growth and hair fiber production in whole-organ cultures. Lymphokine Cytokine Res. 12, 197–203.
Groves R.W., Rauschmayr T., Nakamura K., Sarkar S., Williams I.R., Kupper T.S. (1996) Inflammatory and hyperproliferative skin disease in mice that express elevated levels of the IL-1 receptor (type I) on epidermal keratinocytes. Evidence that IL-1-inducible secondary cytokines produced by keratinocytes in vivo can cause skin disease. J. Clin. Invest. 98, 336–344.
Hoffmann R., Happle R. (1995) Does interleukin-1 induce hair loss? Dermatology. 191, 273–275.
Варюшина Е.А., Москаленко В.В., Лебедева Т.П., Бубнов А.Н., Симбирцев А.С. (2008) Использование интерлейкина-1β для местного лечения гнойно-некротических поражений нижних конечностей. Мед. Иммунол. 10, 439–448.
Lee P., Lee D., Chan C., Chen S., Ch I., Jamora C. (2009) Dynamic expression of epidermal caspase 8 simulates a wound healing response. Nature. 458, 519–523.
Lee P., Gund R., Dutta A., Pincha N., Rana I., Ghosh S., Witherden D.A., Kandyba E., Macleod A.S., Kobielak K., Havran W.L., Jamora C. (2017) Stimulation of hair follicle stem cell proliferation through an IL-1 dependent activation of gdT-cells. Elife. 6, e28875.
Naik S., Larsen S.B., Gomez N.C., Alaverdyan K., Sendoel A., Yuan S., Polak L., Kulukian A., Chai S., Fuchs E. (2017) Inflammatory memory sensitizes skin epithelial stem cells to tissue damage. Nature. 550, 475–480.
Negishi H., Taniguchi T., Yanai H. (2018) The Interferon (IFN) class of cytokines and the IFN regulatory factor (IRF) transcription factor family. Cold Spring Harb. Perspect. Biol. 10, 1–16.
McNab F., Mayer-Barber K., Sher A., Wack A., O’Garra A. (2015) Type I interferons in infectious disease. Nat. Rev. Immunol. 15, 87–103.
Schoenborn J.R., Wilson C.B. (2007) Regulation of interferon-gamma during innate and adaptive immune responses. Adv. Immunol. 96, 41–101.
Blank T., Prinz M. (2017) Type I interferon pathway in CNS homeostasis and neurological disorders. Glia. 65, 1397–1406.
Stout A.J., Gresser I., Thompson W.D. (1993) Inhibition of wound healing in mice by local interferon alpha/beta injection. Int. J. Exp. Pathol. 74, 79–85.
Miles R.H., Paxton T.P., Zacheis D., Dries D.J., Gamelli R.L. (1994) Systemic administration of interferon-gamma impairs wound healing. J. Surg. Res. 56, 288–294.
Ishida Y., Kondo T., Takayasu T., Iwakura Y., Mukaida N. (2004) The essential involvement of cross-talk between IFN-γ and TGF-β in the skin wound-healing process. J. Immunol. 172, 1848–1855.
Gregorio J., Meller S., Conrad C., Di Nardo A., Homey B., Lauerma A., Arai N., Gallo R.L., Digiovanni J., Gilliet M. (2010) Plasmacytoid dendritic cells sense skin injury and promote wound healing through type I interferons. J. Exp. Med. 207, 2921–2930.
Zhang L., Sen G.L., Ward N.L., Johnston A., Chun K., Chen Y., Adase C., Sanford J.A., Gao N., Chensee M., Sato E., Fritz Y., Baliwag J., Williams M.R., Hata T., Gallo R.L. (2016) Antimicrobial peptide LL37 and MAVS signaling drive Interferon- b production by epidermal keratinocytes during skin injury. Immunity. 45, 119–130.
Schaffer M., Bongartz M., Hoffmann W., Viebahn R. (2006) Regulation of nitric oxide synthesis in wounds by IFN-γ depends on TNF-α. J. Investig. Surg. 19, 371–379.
Canesso M.C., Vieira A.T., Castro T.B., Schirmer B.G., Cisalpino D., Martins F.S., Rachid M.A., Nicoli J.R., Teixeira M.M., Barcelos L.S. (2014) Skin wound healing is accelerated and scarless in the absence of commensal microbiota. J. Immunol. 193, 5171–5180.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология