

Молекулярная биология, 2019, T. 53, № 5, стр. 790-798
Микроглия в гомеостазе центральной нервной системы и нейровоспалении
В. С. Гоголева a, *, М. С. Друцкая a, К. С.-Н. Атретханы a, b
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
b Биологический факультет Московского государственного университета им. М.В. Ломоносова
119234 Москва, Россия
* E-mail: violettegogoleva@mail.ru
Поступила в редакцию 26.03.2019
После доработки 26.03.2019
Принята к публикации 15.04.2019
Аннотация
Механизмы развития нейродегенеративных и нейровоспалительных заболеваний привлекают в последнее время большое внимание. Особую роль в развитии нейропатологий отводят взаимодействию нервной и иммунной систем. Считается, что развитию нейровоспалительных и нейродегенеративных заболеваний может способствовать нарушение функций микроглии ‒ клеток иммунной системы, которые выполняют функцию резидентных макрофагов центральной нервной системы (ЦНС) и участвуют в формировании, а также в поддержании гомеостаза ЦНС. Понимание механизмов регуляции микроглии в норме и при патологии необходимо для создания методов эффективной терапии различных неврологических заболеваний. С помощью технологий редактирования генома изучены основные программы развития и функционирования микроглии. В обзоре рассмотрены последние сведения о происхождении микроглии, ее регуляторной роли в ЦНС и о вкладе в развитие нейровоспаления.
ВВЕДЕНИЕ
В настоящее время наблюдается увеличение частоты развития патологий центральной нервной системы (ЦНС), среди которых наиболее распространены такие нейродегенеративные и нейровоспалительные заболевания, как болезнь Альцгеймера и рассеянный склероз. Механизмы, лежащие в основе патогенеза этих заболеваний, различаются, но в обоих случаях наблюдается ухудшение неврологических функций и воспаление [1, 2]. Роль иммунной системы в развитии патологий ЦНС активно изучается. Единственными клетками иммунной системы в паренхиме ЦНС являются клетки микроглии, выполняющие функции резидентных макрофагов [3]. Изучение роли микроглии в развитии и поддержании ЦНС сдерживалось недостатком модельных систем и невозможностью разграничения микроглии от других миелоидных клеток (табл. 1). Так, для изучения микроглии первоначально использовали такие маркеры, как CD11b, CD11c, лизоцим M, экспрессия которых характерна для многих клеток миелоидного происхождения. Позже обнаружили специфические для микроглии маркеры, в частности рецептор хемокина CX3CR1 и фактор транскрипции Sall1. Развитие технологий и появление новых методов регулируемого удаления генов, транскриптомного анализа единичных клеток, а также прижизненной визуализации развития клеточных популяций позволило охарактеризовать основные программы развития и функционирования микроглии в норме и при патологии.
Таблица 1.
Подходы к изучению функций микроглии in vivo с помощью методов обратной генетики
| Маркер микроглии | Система тканеспецифического удаления гена в миелоидных клетках | Ожидаемая модификация генома | Специфичность системы | Ссылка |
|---|---|---|---|---|
| CD11b | Cd11bCre × floxed gene | Делеция фланкированной последовательности в CD11b+ миелоидных клетках | Миелоидные клетки, экспрессирующие CD11b, и микроглия | [4] |
| Lysozyme M | LysMCre × floxed gene | Делеция фланкированной последовательности в LysM+ клетках | Макрофаги, нейтрофилы, а также нейроны и микроглия | [5, 6] |
| CD11c | Cd11cCre × floxed gene | Делеция фланкированной последовательности в CD11c+ клетках | Миелоидные клетки, экспрессирующие CD11c, а также определенная субпопуляция микроглии, экспрессирующая CD11c | [7] |
| Iba1 | Iba1-EGFP | Экспрессия EGFP в Iba1+ клетках для их визуализации | Менингеальные, периваскулярные макрофаги и микроглия | [8, 9] |
| CX3CR1 | Cx3cr1-GFP | Экспрессия GFP в CX3CR1+ клетках для их визуализации | Ранние миелоидные клетки-предшественники, моноциты, дендритные клетки, NK-клетки, резидентные макрофаги и микроглия | [10–12] |
| Ccr2RFP : Cx3cr1GFP | Экспрессия RFP в циркулирующих CCR2+ моноцитах и экспрессия GFP в CX3CR1+ клетках | Отделение периферических моноцитов от резидентных макрофагов и микроглии | [13] | |
| Cx3cr1CreER : iDTR | Экспрессия DTR на поверхности микроглии и других CX3CR1+ клеток при тамоксифен-зависимой активации Cre-рекомбиназы делает клетки чувствительными к дифтерийному токсину | Удаление CX3CR1+ клеток сопровождается увеличением количества астроцитов и цитокиновым штормом | [14] | |
| Cx3cr1Cre × floxed gene | Делеция фланкированной последовательности в CX3CR1+ клетках | Экспрессия CX3CR1 характерна для периферических моноцитов и резидентных макрофагов | [12] | |
| Cx3cr1CreER × floxed gene | Индуцибельная делеция фланкированной последовательности в CX3CR1+ клетках при введении тамоксифена | Таргетное удаление гена в клетках микроглии сохраняется, в то время как в крови происходит обновление популяции CX3CR1+ моноцитов из миелоидных клеток-предшественников костного мозга | [15] | |
| Cx3cr1CreER : R26tdTomato | Индуцибельная экспрессия tdTomato в CX3CR1+ клетках при введении тамоксифена | Разделение периферических моноцитов от резидентных макрофагов и микроглии | [16] | |
| Sall1 | Sall1CreER × floxed gene | Индуцибельная делеция фланкированной последовательности в микроглии | Экспрессия Sall1 характерна только для микроглии | [17, 18] |
ПРОИСХОЖДЕНИЕ, РАЗВИТИЕ И ПОДДЕРЖАНИЕ МИКРОГЛИИ
Долгое время считалось, что клетки микроглии, выполняющие функции макрофагов ЦНС, имеют гемопоэтическое происхождение [19]. Однако на мышах, экспрессирующих белок GFP под промотором маркера микроглии Cx3cr1, показали, что эти клетки возникают из эритромиелоидных клеток-предшественников в желточном мешке в ходе раннего эмбрионального развития [20]. Позже установили, что дифференцировка этих клеток в клетки микроглии зависит от факторов транскрипции PU.1 и IRF8 [21]. Далее клетки-предшественники, имеющие фенотип CD45+c-kit–CX3CR1+, мигрируют в ЦНС (E9.5), после чего происходит формирование гематоэнцефалического барьера (рис. 1а) [22]. В дополнение к этому показано, что циркулирующие моноциты крови не участвуют в поддержании гомеостаза микроглии во взрослом организме [23, 24]. Кроме того, анализ возникновения миелоидных клеток в системе с Cre-индуцибельной экспрессией желтого флуоресцентного белка (YFP) в Kit+ эритромиелоидных клетках-предшественниках желточного мешка или гемопоэтических клетках-предшественниках во взрослом организме показал, что клетки микроглии представляют одну из немногих популяций тканерезидентных макрофагов, развитие которых совершенно не зависит от стволовых кроветворных клеток костного мозга [25].
Рис. 1.
Онтогенез и физиологическая роль микроглии в норме и при патологии. а ‒ Клетки микроглии происходят из эритромиелоидных клеток-предшественников в желточном мешке в раннем эмбриогенезе. Под воздействием факторов PU.1 и IRF8 эритромиелоидные клетки-предшественники дифференцируются в клетки-предшественники микроглии, а затем заселяют ЦНС. б ‒ Микроглия в процессе поддержания гомеостаза ЦНС во взрослом организме может элиминировать синаптические структуры благодаря присутствию на поверхности молекулы CD11b и ремоделировать синапсы путем выделения нейротрофического фактора BDNF (brain-derived neurotrophic factor). Кроме того, микроглия регулирует нейрогенез за счет фагоцитоза погибающих нейронов и запуска механизмов программируемой клеточной гибели. в ‒ При нейровоспалении происходит накопление агрегатов β-амилоида и остатков миелина, что может приводить к активации микроглии. В активированной микроглии наблюдается увеличение фагоцитарной активности, которая зависит от сигнального пути, опосредуемого TREM2 (triggering receptor expressed on myeloid cells). Микроглия, ассоциированная с патологией (DAM), может продуцировать различные цитокины. Рисунок сделан с использованием материалов сайта http://smart.servier.com.
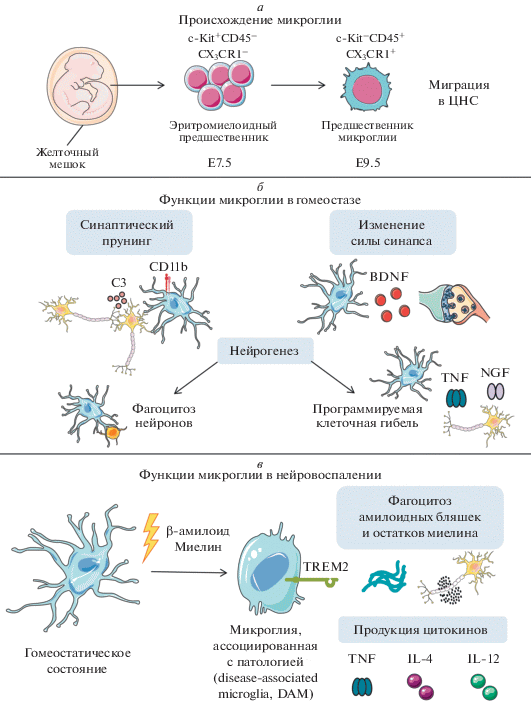
Благодаря появлению транскриптомного анализа одиночных клеток (single-cell seq) установлено, что микроглия представляет собой гетерогенную популяцию клеток. Так, с помощью транскриптомного и эпигенетического анализа выявлено существование трех этапов развития микроглии: раннего (начиная с формирования клеток-предшественников в желточном мешке до E14), премикроглии (начиная с E14 до раннего постнатального периода) и стадии микроглии взрослого организма [26]. При этом каждый этап развития характеризуется конкретными сигнальными каскадами и функциями. Кроме того, на развитие и активацию микроглии могут влиять различные факторы, такие как микробиота и активация иммунной системы во время беременности, что в дальнейшем может приводить к развитию нейропатологий [27, 28].
Клетки микроглии представляют собой долгоживущую популяцию [29]. Для поддержания пролиферации микроглии и ее выживания во взрослом организме необходима активация сигнального пути, опосредуемого CSF1R [17, 30]. Лигандами CSF1R являются CSF1 и IL-34, продуцируемые в ЦНС, в первую очередь, нейронами [31]. Для гомеостатического поддержания микроглии во взрослом организме, но не в эмбриональном развитии необходим именно IL-34 [32, 33]. Кроме того, кондиционное удаление Il1r1 в CX3CR1+ микроглии приводит к ее морфологическим изменениям и уменьшению количества клеток, предположительно, за счет важной роли сигнальной оси IL-1/IL-1R в пролиферации микроглии [14]. При этом пролиферация микроглии взрослого организма обусловлена не клетками-предшественниками, а именно репопуляцией оставшихся клеток микроглии [34].
Таким образом, микроглия представляет собой уникальный тип миелоидных клеток, способных к самоподдержанию и обеспечению гомеостаза ЦНС в постнатальном периоде.
РОЛЬ МИКРОГЛИИ В ГОМЕОСТАЗЕ ЦНС
Микроглия выполняет различные физиологические функции на всех этапах развития ЦНС. Клетки микроглии представляют собой динамичную популяцию [35], взаимодействуя практически со всеми типами клеток в головном мозге, они опосредуют различные процессы развития и поддержания гомеостаза ЦНС.
Во-первых, как в процессе раннего развития ЦНС, так и на более поздних этапах микроглия может контролировать нейрогенез за счет фагоцитоза и элиминации гибнущих нейронов. Микроглия может регулировать не только фагоцитоз, но и программируемую гибель нейронов и их клеток-предшественников путем секреции таких растворимых факторов, как NGF (nerve growth factor) [36] и TNF [37], а также продукции активных форм кислорода [38].
Во-вторых, известно, что по мере развития организма микроглия участвует в таком процессе, как синаптический прунинг (synaptic pruning) [39]. В ходе этого процесса происходит элиминация избыточных синаптических структур, что необходимо для повышения эффективности нейронных взаимодействий. Нарушение прунинга ассоциировано с такими патологическими состояниями, как шизофрения и аутизм [40]. При этом синаптический прунинг происходит комплементзависимым образом за счет взаимодействия CD11b (CR3) на поверхности микроглии и белка C3 в участке синапса [41, 42].
В-третьих, микроглия может модулировать силу синапса, ответственного за взаимодействие нейронов, что лежит в основе формирования памяти и способности к обучению [43]. При этом клетки микроглии способны секретировать различные нейротрофические факторы, в частности BDNF, – важный регулятор синаптического ремоделирования. Так, индуцируемая тамоксифеном делеция BDNF в CX3CR1+ клетках микроглии приводит к снижению экспрессии генов, ответственных за формирование синапса, а также к уменьшению формирования самих синапсов в ответ на обучение [44].
Таким образом, микроглия активно участвует в поддержании гомеостаза ЦНС, существенно влияя на нейрогенез, образование и элиминирование синапсов, а также на формирование когнитивных функций (рис. 1б).
РОЛЬ МИКРОГЛИИ В РАЗВИТИИ НЕЙРОВОСПАЛЕНИЯ
Под воздействием различных стимулов микроглия, физиологическая роль которой заключается в поддержании гомеостаза ЦНС, может приобретать активированный фенотип и способствовать развитию патологических процессов [45, 46]. Ранее считалось, что клетки микроглии подобно обычным макрофагам могут быть как провоспалительными (М1), так и противовоспалительными (М2) [47, 48]. Однако в поздних исследованиях было выяснено, что активированная микроглия может продуцировать как нейропротективные, так и нейротоксичные факторы [49]. В настоящее время принята концепция, согласно которой микроглия переходит из гомеостатического состояния в патологическое с последующим приобретением активированного DAM-фенотипа, ассоциированного с заболеванием. Развитию этого фенотипа способствуют различные факторы, такие как апоптотические тельца, возникающие в результате гибели нейронов, продукты деградации липидов, внеклеточные скопления белковых агрегатов, а также остатки миелина [50].
В нормальном состоянии микроглия участвует в поддержании гомеостаза ЦНС и нейрогенеза за счет фагоцитоза погибающих нейронов. Однако при развитии патологических процессов с последующим приобретением DAM-фенотипа фагоцитарная активность микроглии существенно увеличивается. Согласно последним данным, фагоцитарная активность микроглии в патологическом состоянии зависит от активации сигнального пути, опосредованного TREM2 ‒ рецептором, экспрессия которого характерна для миелоидных клеток и остеокластов [51]. При взаимодействии TREM2 c различными лигандами, в том числе с липополисахаридом, липидами и липопротеинами различной структуры [52], наблюдается активация генов, ответственных за фагоцитоз и реорганизацию цитоскелета [53]. При этом в гене, кодирующем TREM2, выявлены генетические полиморфизмы, ассоциированные с повышенным риском болезни Альцгеймера [54]. Известно, что болезнь Альцгеймера характеризуется прогрессирующей нейродегенерацией, сопровождающейся накоплением в тканях мозга амилоидных бляшек и нейрофибриллярных клубков [55]. Оказалось, что экспрессия TREM2 в модели нейродегенерации необходима для эффективного фагоцитоза амилоидных бляшек клетками микроглии [56, 57]. Другой генетический фактор риска болезни Альцгеймера ‒ полиморфизм гена, кодирующего аполипопротеин Е (ApoE) [58]. АроЕ регулирует транспорт липидов за счет связывания со своими рецепторами из семейства LDLR (low-density lipoprotein receptors). На клетках микроглии TREM2 может связываться с комплексами амилоидных бляшек и ApoE, что говорит о протективной роли TREM2 [41]. Таким образом, экспрессия TREM2 на клетках микроглии является не только одной из характеристик DAM-фенотипа, но и ассоциирована с фагоцитарной функцией микроглии в развитии нейровоспаления.
Нарушение регуляции провоспалительных цитокинов и хемокинов характерно для различных патологических состояний ЦНС [59]. При этом микроглия может продуцировать in vitro такие про- и противовоспалительные цитокины, как TNF, IL-6, IL-1β, IL-12, IL-10 [60]. Продукция микроглией цитокинов in vivo изучается в основном в различных моделях демиелинизации и нейродегенерации. Однако интерпретацию данных о вкладе цитокинов, продуцируемых микроглией, в развитие нейровоспалительных заболеваний затрудняет присутствие периферических моноцитов, инфильтрирующих ЦНС при разрушении гематоэнцефалического барьера и способных к секреции этих цитокинов. Поэтому разработаны различные экспериментальные подходы, позволяющие разделить вклад периферических моноцитов и микроглии в развитие патологического процесса.
Во-первых, микроглия представляет собой резидентные клетки ЦНС негемопоэтического происхождения, поэтому она обладает устойчивостью к радиоактивному излучению в отличие от моноцитов крови. Создание костномозговых химер позволяет разделить вклад цитокинов из различных миелоидных источников в зависимости от их радиорезистентности. Так, удаление субъединицы p40, общей для цитокинов IL-12 и IL-23 из резидентных клеток ЦНС, приводило к уменьшению накопления амилоидных бляшек у трансгенных мышей, моделирующих болезнь Альцгеймера [61]. С использованием костномозговых химер показано, что делеция IL-4, необходимого для дифференцировки Т-клеток по Th2-пути, в резидентных клетках ЦНС приводит к усилению клинических симптомов у мышей с экспериментальным аутоиммунным энцефаломиелитом – моделью рассеянного склероза [62].
Во-вторых, на основе системы Cre-loxP разработан подход, позволяющий индуцибельно удалять ген интереса только из клеток микроглии. В этой системе ген, кодирующий Cre-рекомбиназу, сшит с лигандсвязывающим доменом рецептора эстрогена, для активации которого необходим антагонист эстрогена – тамоксифен. Такая конструкция контролируется промотором Cx3cr1, активным в моноцитах крови и резидентных макрофагах. При введении тамоксифена Cre-рекомбиназа активируется как в моноцитах, так и в клетках микроглии, однако уже через 3‒4 недели благодаря дифференцировке миелоидных клеток-предшественников костного мозга происходит полная репопуляция моноцитов крови [12]. При скрещивании таких мышей (Cx3cr1CreER) с мышами, несущими фланкированную loxP-сайтами последовательность, происходит Cre-зависимое вырезание нужного гена, индуцированное тамоксифеном [15]. Такой подход позволил показать, что делеция TNF в CX3CR1+ микроглии не влияет на развитие экспериментального аутоиммунного энцефаломиелита у мышей [63]. При этом установлено, что в этой модели рассеянного склероза рецептор TNF (TNFR2), экспрессирующийся на микроглии, играет протективную роль [64] (табл. 1).
На данном этапе поиск молекулярных паттернов, ассоциированных с приобретением микроглией активированного фенотипа, необходим для создания новых подходов к терапии и скринингу патологий ЦНС. Так, недавно осуществлен транскриптомный анализ микроглии в норме и при нейровоспалении у мышей с экспериментальным аутоиммунным энцефаломиелитом. Обнаружено, что одним из ключевых отличий гомеостатической микроглии от патологической является экспрессия белка MD-1 (Ly86), участвующего в реакциях врожденного иммунитета. Такая микроглия характеризовалась повышенной пролиферацией и экспрессией хемокинов CXCL10 и CCL2 [16]. Кроме того, на данном этапе активно проводится транскриптомный анализ одиночных клеток микроглии, выделенной из образцов головного мозга человека. Так, микроглия человека, ассоциированная с гомеостатическим состоянием, представлена неоднородной популяцией клеток. Гетерогенность субпопуляций микроглии сохраняется и при патологических состояниях, в частности, при рассеянном склерозе. При этом субпопуляции микроглии классифицируют по маркерам SPP1, CTSD, CD74, что коррелирует с данными, полученными в модели демиелинизации у мышей [65].
Таким образом, под воздействием различных факторов микроглия способна переходить из гомеостатического состояния в патологическое с увеличением фагоцитарной активности и продукции цитокинов (рис. 1в). При этом с помощью современных методов удалось выявить сигнатуры микроглии, ассоциированные с патологическим состоянием.
ЗАКЛЮЧЕНИЕ
Несмотря на ключевую роль микроглии в функционировании ЦНС, изучение молекулярных механизмов, лежащих в основе патогенеза нейровоспалительных заболеваний, началось сравнительно недавно после создания подходящих модельных систем. С помощью методов обратной генетики и транскриптомного анализа ведется поиск молекулярных маркеров, ассоциированных с приобретением микроглией активированного фенотипа. Кроме того, благодаря активному развитию технологий редактирования генома получены новые линии мышей, позволяющие проводить избирательную модификацию клеток микроглии для более детального изучения ее функций (табл. 1). Так, например, ранее считалось, что микроглия может участвовать в реактивации Т-клеток и локальном представлении антигена в ЦНС мышей с экспериментальным аутоиммунным энцефаломиелитом [66]. Однако недавно с использованием более современных систем показано, что именно CD11c+ дендритные клетки, но не микроглия ответственны за представление антигена в модели нейровоспаления и демиелинизации [18, 67]. При этом использование регулируемой системы удаления гена интереса позволило разграничить вклад различных цитокинов, секретируемых либо тканерезидентными, либо периферическими миелоидными клетками.
Таким образом, использование методов обратной генетики и транскриптомного анализа позволило детально изучить происхождение микроглии, ее функции в гомеостазе ЦНС, а также функциональные особенности при патологических процессах. Дальнейшие исследования с использованием новых технологий позволят выявить DAM-паттерны и установить их функциональную значимость для понимания механизмов развития заболеваний и разработки новых подходов к терапии и скринингу патологий ЦНС.
Авторы выражают признательность С.А. Недоспасову за ценные замечания.
Работа поддержана грантом Российского научного фонда (№ 19-75-30032).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
De Strooper B., Karran E. (2016) The cellular phase of Alzheimer’s disease. Cell. 164, 603‒615.
Baecher-Allan C., Kaskow B.J., Weiner H.L. (2018) Multiple sclerosis: mechanisms and immunotherapy. Neuron. 97, 742‒768.
Kierdorf K., Prinz M. (2017) Microglia in steady state. J. Clin. Invest. 127, 3201‒3209.
Boillee S., Yamanaka K., Lobsiger C.S., Copeland N.G., Jenkins N.A., Kassiotis G., Kollias G., Cleveland D.W. (2006) Onset and progression in inherited ALS determined by motor neurons and microglia. Science. 312, 1389‒1392.
Cho I.H., Hong J., Suh E.C., Kim J.H., Lee H., Lee J.E., Lee S., Kim C.H., Kim D.W., Jo E.K., Lee K.E., Karin M., Lee S.J. (2008) Role of microglial IKKbeta in kainic acid-induced hippocampal neuronal cell death. Brain. 131, 3019‒3033.
Orthgiess J., Gericke M., Immig K., Schulz A., Hirrlinger J., Bechmann I., Eilers J. (2016) Neurons exhibit Lyz2 promoter activity in vivo: implications for using LysM-Cre mice in myeloid cell research. Eur. J. Immunol. 46, 1529‒1532.
Wlodarczyk A., Holtman I.R., Krueger M., Yogev N., Bruttger J., Khorooshi R., Benmamar-Badel A., de Boer-Bergsma J.J., Martin N.A., Karram K., Kramer I., Boddeke E.W., Waisman A., Eggen B.J., Owens T. (2017) A novel microglial subset plays a key role in myelinogenesis in developing brain. EMBO J. 36, 3292‒3308.
Hirasawa T., Ohsawa K., Imai Y., Ondo Y., Akazawa C., Uchino S., Kohsaka S. (2005) Visualization of microglia in living tissues using Iba1-EGFP transgenic mice. J. Neurosci. Res. 81, 357‒362.
Goldmann T., Wieghofer P., Jordao M.J., Prutek F., Hagemeyer N., Frenzel K., Amann L., Staszewski O., Kierdorf K., Krueger M., Locatelli G., Hochgerner H., Zeiser R., Epelman S., Geissmann F., Priller J., Ros-si F.M., Bechmann I., Kerschensteiner M., Linnarsson S., Jung S., Prinz M. (2016) Origin, fate and dynamics of macrophages at central nervous system interfaces. Nat. Immunol. 17, 797‒805.
Jung S., Aliberti J., Graemmel P., Sunshine M.J., Kreutzberg G.W., Sher A., Littman D.R. (2000) Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell Biol. 20, 4106‒4114.
Liu K., Victora G.D., Schwickert T.A., Guermonprez P., Meredith M.M., Yao K., Chu F.F., Randolph G.J., Rudensky A.Y., Nussenzweig M. (2009) In vivo analysis of dendritic cell development and homeostasis. Science. 324, 392‒397.
Yona S., Kim K.W., Wolf Y., Mildner A., Varol D., Breker M., Strauss-Ayali D., Viukov S., Guilliams M., Misharin A., Hume D.A., Perlman H., Malissen B., Zelzer E., Jung S. (2013) Fate mapping reveals origins and dynamics of monocytes and tissue macrophages under homeostasis. Immunity. 38, 79‒91.
Yamasaki R., Lu H., Butovsky O., Ohno N., Rietsch A.M., Cialic R., Wu P.M., Doykan C.E., Lin J., Cotleur A.C., Kidd G., Zorlu M.M., Sun N., Hu W., Liu L., Lee J.C., Taylor S.E., Uehlein L., Dixon D., Gu J., Floruta C.M., Zhu M., Charo I.F., Weiner H.L., Ransohoff R.M. (2014) Differential roles of microglia and monocytes in the inflamed central nervous system. J. Exp. Med. 211, 1533‒1549.
Bruttger J., Karram K., Wortge S., Regen T., Marini F., Hoppmann N., Klein M., Blank T., Yona S., Wolf Y., Mack M., Pinteaux E., Muller W., Zipp F., Binder H., Bopp T., Prinz M., Jung S., Waisman A. (2015) Genetic cell ablation reveals clusters of local self-renewing microglia in the mammalian central nervous system. Immunity. 43, 92‒106.
Goldmann T., Wieghofer P., Muller P.F., Wolf Y., Varol D., Yona S., Brendecke S.M., Kierdorf K., Staszewski O., Datta M., Luedde T., Heikenwalder M., Jung S., Prinz M. (2013) A new type of microglia gene targeting shows TAK1 to be pivotal in CNS autoimmune inflammation. Nat. Neurosci. 16, 1618‒1626.
Jordao M.J.C., Sankowski R., Brendecke S.M., Sagar, Locatelli G., Tai Y.H., Tay T.L., Schramm E., Armbruster S., Hagemeyer N., Gross O., Mai D., Cicek O., Falk T., Kerschensteiner M., Grun D., Prinz M. (2019) Single-cell profiling identifies myeloid cell subsets with distinct fates during neuroinflammation. Science. 363, eaat7554.
Buttgereit A., Lelios I., Yu X., Vrohlings M., Krakoski N.R., Gautier E.L., Nishinakamura R., Becher B., Greter M. (2016) Sall1 is a transcriptional regulator defining microglia identity and function. Nat. Immunol. 17, 1397–1406.
Mundt S., Mrdjen D., Utz S.G., Greter M., Schreiner B., Becher B. (2019) Conventional DCs sample and present myelin antigens in the healthy CNS and allow parenchymal T cell entry to initiate neuroinflammation. Sci. Immunol. 4, eaau8380.
Hickey W.F., Kimura H. (1988) Perivascular microglial cells of the CNS are bone marrow-derived and present antigen in vivo. Science. 239, 290‒292.
Ginhoux F., Greter M., Leboeuf M., Nandi S., See P., Gokhan S., Mehler M.F., Conway S.J., Ng L.G., Stanley E.R., Samokhvalov I.M., Merad M. (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science. 330, 841‒845.
Kierdorf K., Erny D., Goldmann T., Sander V., Schulz C., Perdiguero E.G., Wieghofer P., Heinrich A., Riemke P., Holscher C., Muller D.N., Luckow B., Brocker T., Debowski K., Fritz G., Opdenakker G., Diefenbach A., Biber K., Heikenwalder M., Geissmann F., Rosenbauer F., Prinz M. (2013) Microglia emerge from erythromyeloid precursors via Pu.1- and Irf8-dependent pathways. Nat. Neurosci. 16, 273‒280.
Li Q., Barres B.A. (2018) Microglia and macrophages in brain homeostasis and disease. Nat. Rev. Immunol. 18, 225‒242.
Ajami B., Bennett J.L., Krieger C., Tetzlaff W., Rossi F.M. (2007) Local self-renewal can sustain CNS microglia maintenance and function throughout adult life. Nat. Neurosci. 10, 1538‒1543.
Hashimoto D., Chow A., Noizat C., Teo P., Beasley M.B., Leboeuf M., Becker C.D., See P., Price J., Lucas D., Greter M., Mortha A., Boyer S.W., Forsberg E.C., Tanaka M., van Rooijen N., Garcia-Sastre A., Stanley E.R., Ginhoux F., Frenette P.S., Merad M. (2013) Tissue-resident macrophages self-maintain locally throughout adult life with minimal contribution from circulating monocytes. Immunity. 38, 792‒804.
Sheng J., Ruedl C., Karjalainen K. (2015) Most tissue-resident macrophages except microglia are derived from fetal hematopoietic stem cells. Immunity. 43, 382‒393.
Matcovitch-Natan O., Winter D.R., Giladi A., Vargas Aguilar S., Spinrad A., Sarrazin S., Ben-Yehuda H., David E., Zelada Gonzalez F., Perrin P., Keren-Shaul H., Gury M., Lara-Astaiso D., Thaiss C.A., Cohen M., Bahar Halpern K., Baruch K., Deczkowska A., Lorenzo-Vivas E., Itzkovitz S., Elinav E., Sieweke M.H., Schwartz M., Amit I. (2016) Microglia development follows a stepwise program to regulate brain homeostasis. Science. 353, aad8670.
Thion M.S., Low D., Silvin A., Chen J., Grisel P., Schulte-Schrepping J., Blecher R., Ulas T., Squarzoni P., Hoeffel G., Coulpier F., Siopi E., David F.S., Scholz C., Shihui F., Lum J., Amoyo A.A., Larbi A., Poidinger M., Buttgereit A., Lledo P.M., Greter M., Chan J.K.Y., Amit I., Beyer M., Schultze J.L., Schlitzer A., Pettersson S., Ginhoux F., Garel S. (2018) Microbiome influences prenatal and adult microglia in a sex-specific manner. Cell. 172, 500‒516.
Mattei D., Ivanov A., Ferrai C., Jordan P., Guneykaya D., Buonfiglioli A., Schaafsma W., Przanowski P., Deuther-Conrad W., Brust P., Hesse S., Patt M., Sabri O., Ross T.L., Eggen B.J.L., Boddeke E., Kaminska B., Beule D., Pombo A., Kettenmann H., Wolf S.A. (2017) Maternal immune activation results in complex microglial transcriptome signature in the adult offspring that is reversed by minocycline treatment. Transl. Psychiatry. 7, e1120.
Lawson L.J., Perry V.H., Gordon S. (1992) Turnover of resident microglia in the normal adult mouse brain. Neuroscience. 48, 405‒415.
Elmore M.R., Najafi A.R., Koike M.A., Dagher N.N., Spangenberg E.E., Rice R.A., Kitazawa M., Matusow B., Nguyen H., West B.L., Green K.N. (2014) Colony-stimulating factor 1 receptor signaling is necessary for microglia viability, unmasking a microglia progenitor cell in the adult brain. Neuron. 82, 380‒397.
Chitu V., Gokhan S., Nandi S., Mehler M.F., Stanley E.R. (2016) Emerging roles for CSF-1 receptor and its ligands in the nervous system. Trends Neurosci. 39, 378‒393.
Wang Y., Szretter K.J., Vermi W., Gilfillan S., Rossini C., Cella M., Barrow A.D., Diamond M.S., Colonna M. (2012) IL-34 is a tissue-restricted ligand of CSF1R required for the development of Langerhans cells and microglia. Nat. Immunol. 13, 753‒760.
Greter M., Lelios I., Pelczar P., Hoeffel G., Price J., Leboeuf M., Kundig T.M., Frei K., Ginhoux F., Merad M., Becher B. (2012) Stroma-derived interleukin-34 controls the development and maintenance of langerhans cells and the maintenance of microglia. Immunity. 37, 1050‒1060.
Huang Y., Xu Z., Xiong S., Sun F., Qin G., Hu G., Wang J., Zhao L., Liang Y.X., Wu T., Lu Z., Humayun M.S., So K.F., Pan Y., Li N., Yuan T.F., Rao Y., Peng B. (2018) Repopulated microglia are solely derived from the proliferation of residual microglia after acute depletion. Nat. Neurosci. 21, 530‒540.
Nimmerjahn A., Kirchhoff F., Helmchen F. (2005) Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science. 308, 1314‒1318.
Frade J.M., Barde Y.A. (1998) Microglia-derived nerve growth factor causes cell death in the developing retina. Neuron. 20, 35‒41.
Sedel F., Bechade C., Vyas S., Triller A. (2004) Macrophage-derived tumor necrosis factor alpha, an early developmental signal for motoneuron death. J. Neurosci. 24, 2236‒2246.
Marin-Teva J.L., Dusart I., Colin C., Gervais A., van Rooijen N., Mallat M. (2004) Microglia promote the death of developing Purkinje cells. Neuron. 41, 535‒547.
Paolicelli R.C., Bolasco G., Pagani F., Maggi L., Scianni M., Panzanelli P., Giustetto M., Ferreira T.A., Guiducci E., Dumas L., Ragozzino D., Gross C.T. (2011) Synaptic pruning by microglia is necessary for normal brain development. Science. 333, 1456‒1458.
Sellgren C.M., Gracias J., Watmuff B., Biag J.D., Thanos J.M., Whittredge P.B., Fu T., Worringer K., Brown H.E., Wang J., Kaykas A., Karmacharya R., Goold C.P., Sheridan S.D., Perlis R.H. (2019) Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 22, 374‒385.
Stevens B., Allen N.J., Vazquez L.E., Howell G.R., Christopherson K.S., Nouri N., Micheva K.D., Mehalow A.K., Huberman A.D., Stafford B., Sher A., Litke A.M., Lambris J.D., Smith S.J., John S.W., Barres B.A. (2007) The classical complement cascade mediates CNS synapse elimination. Cell. 131, 1164‒1178.
Schafer D.P., Lehrman E.K., Kautzman A.G., Koyama R., Mardinly A.R., Yamasaki R., Ransohoff R.M., Greenberg M.E., Barres B.A., Stevens B. (2012) Microglia sculpt postnatal neural circuits in an activity and complement-dependent manner. Neuron. 74, 691‒705.
Rogers J.T., Morganti J.M., Bachstetter A.D., Hudson C.E., Peters M.M., Grimmig B.A., Weeber E.J., Bickford P.C., Gemma C. (2011) CX3CR1 deficiency leads to impairment of hippocampal cognitive function and synaptic plasticity. J. Neurosci. 31, 16241‒16250.
Parkhurst C.N., Yang G., Ninan I., Savas J.N., Yates J.R., 3rd, Lafaille J.J., Hempstead B.L., Littman D.R., Gan W.B. (2013) Microglia promote learning-dependent synapse formation through brain-derived neurotrophic factor. Cell. 155, 1596‒1609.
Frank-Cannon T.C., Alto L.T., McAlpine F.E., Tan-sey M.G. (2009) Does neuroinflammation fan the flame in neurodegenerative diseases? Mol. Neurodegener. 4, 47.
Kierdorf K., Prinz M. (2013) Factors regulating microglia activation. Front. Cell Neurosci. 7, 44.
Michelucci A., Heurtaux T., Grandbarbe L., Morga E., Heuschling P. (2009) Characterization of the microglial phenotype under specific pro-inflammatory and anti-inflammatory conditions: Effects of oligomeric and fibrillar amyloid-beta. J. Neuroimmunol. 210, 3‒12.
Beers D.R., Henkel J.S., Zhao W., Wang J., Huang A., Wen S., Liao B., Appel S.H. (2011) Endogenous regulatory T lymphocytes ameliorate amyotrophic lateral sclerosis in mice and correlate with disease progression in patients with amyotrophic lateral sclerosis. Brain. 134, 1293‒1314.
Chiu I.M., Morimoto E.T., Goodarzi H., Liao J.T., O’Keeffe S., Phatnani H.P., Muratet M., Carroll M.C., Levy S., Tavazoie S., Myers R.M., Maniatis T. (2013) A neurodegeneration-specific gene-expression signature of acutely isolated microglia from an amyotrophic lateral sclerosis mouse model. Cell Rep. 4, 385‒401.
Deczkowska A., Keren-Shaul H., Weiner A., Colonna M., Schwartz M., Amit I. (2018) Disease-associated microglia: a universal immune sensor of neurodegeneration. Cell. 173, 1073‒1081.
Keren-Shaul H., Spinrad A., Weiner A., Matcovitch-Natan O., Dvir-Szternfeld R., Ulland T.K., David E., Baruch K., Lara-Astaiso D., Toth B., Itzkovitz S., Colonna M., Schwartz M., Amit I. (2017) A unique microglia type associated with restricting development of Alzheimer’s disease. Cell. 169, 1276‒1290 e1217.
Wolfe C.M., Fitz N.F., Nam K.N., Lefterov I., Koldamova R. (2018) The Role of APOE and TREM2 in Alzheimer’s disease-current understanding and perspectives. Int. J. Mol Sci. 20, 81.
Colonna M., Wang Y. (2016) TREM2 variants: new keys to decipher Alzheimer disease pathogenesis. Nat. Rev. Neurosci. 17, 201‒207.
Jonsson T., Stefansson H., Steinberg S., Jonsdottir I., Jonsson P.V., Snaedal J., Bjornsson S., Huttenlocher J., Levey A.I., Lah J.J., Rujescu D., Hampel H., Giegling I., Andreassen O.A., Engedal K., Ulstein I., Djurovic S., Ibrahim-Verbaas C., Hofman A., Ikram M.A., van Duijn C.M., Thorsteinsdottir U., Kong A., Stefansson K. (2013) Variant of TREM2 associated with the risk of Alzheimer’s disease. N. Engl. J. Med. 368, 107‒116.
Masters C.L., Bateman R., Blennow K., Rowe C.C., Sperling R.A., Cummings J.L. (2015) Alzheimer’s disease. Nat. Rev. Dis. Primers. 1, 15056.
Wang Y., Cella M., Mallinson K., Ulrich J.D., Young K.L., Robinette M.L., Gilfillan S., Krishnan G.M., Sudhakar S., Zinselmeyer B.H., Holtzman D.M., Cirrito J.R., Colonna M. (2015) TREM2 lipid sensing sustains the microglial response in an Alzheimer’s disease model. Cell. 160, 1061‒1071.
Yeh F.L., Wang Y., Tom I., Gonzalez L.C., Sheng M. (2016) TREM2 binds to apolipoproteins, including APOE and CLU/APOJ, and thereby facilitates uptake of amyloid-beta by microglia. Neuron. 91, 328‒340.
Liu C.C., Liu C.C., Kanekiyo T., Xu H., Bu G. (2013) Apolipoprotein E and Alzheimer disease: risk, mechanisms and therapy. Nat. Rev. Neurol. 9, 106‒118.
Heppner F.L., Ransohoff R.M., Becher B. (2015) Immune attack: the role of inflammation in Alzheimer disease. Nat. Rev. Neurosci. 16, 358‒372.
Lee Y.B., Nagai A., Kim S.U. (2002) Cytokines, chemokines, and cytokine receptors in human microglia. J. Neurosci. Res. 69, 94‒103.
Vom Berg J., Prokop S., Miller K.R., Obst J., Kalin R.E., Lopategui-Cabezas I., Wegner A., Mair F., Schipke C.G., Peters O., Winter Y., Becher B., Heppner F.L. (2012) Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nat. Med. 18, 1812‒1819.
Ponomarev E.D., Maresz K., Tan Y., Dittel B.N. (2007) CNS-derived interleukin-4 is essential for the regulation of autoimmune inflammation and induces a state of alternative activation in microglial cells. J. Neurosci. 27, 10714‒10721.z
Wolf Y., Shemer A., Polonsky M., Gross M., Mildner A., Yona S., David E., Kim K.W., Goldmann T., Amit I., Heikenwalder M., Nedospasov S., Prinz M., Friedman N., Jung S. (2017) Autonomous TNF is critical for in vivo monocyte survival in steady state and inflammation. J. Exp. Med. 214, 905‒917.
Gao H., Danzi M.C., Choi C.S., Taherian M., Dalby-Hansen C., Ellman D.G., Madsen P.M., Bixby J.L., Lemmon V.P., Lambertsen K.L., Brambilla R. (2017) Opposing functions of microglial and macrophagic TNFR2 in the pathogenesis of experimental autoimmune encephalomyelitis. Cell Rep. 18, 198‒212.
Masuda T., Sankowski R., Staszewski O., Bottcher C., Amann L., Scheiwe C., Nessler S., Kunz P., van Loo G., Coenen V.A., Reinacher P.C., Michel A., Sure U., Gold R., Priller J., Stadelmann C., Prinz M. (2019) Spatial and temporal heterogeneity of mouse and human microglia at single-cell resolution. Nature. 566, 388‒392.
Waisman A., Johann L. (2018) Antigen-presenting cell diversity for T cell reactivation in central nervous system autoimmunity. J. Mol. Med. (Berl). 96, 1279‒1292.
Wolf Y., Shemer A., Levy-Efrati L., Gross M., Kim J.S., Engel A., David E., Chappell-Maor L., Grozovski J., Rotkopf R., Biton I., Eilam-Altstadter R., Jung S. (2018) Microglial MHC class II is dispensable for experimental autoimmune encephalomyelitis and cuprizone-induced demyelination. Eur. J. Immunol. 48, 1308‒1318.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология