

Молекулярная биология, 2019, T. 53, № 5, стр. 799-814
Мышиные модели сепсиса и септического шока
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
* E-mail: kirkorneev@gmail.com
Поступила в редакцию 09.04.2019
После доработки 09.04.2019
Принята к публикации 18.04.2019
Аннотация
Разветвленная сеть регуляции системного воспаления превращает создание воспроизводимой экспериментальной модели сепсиса в непростую задачу. Не существует одной единственной мышиной модели, которая могла бы охватить все клинические аспекты этой сложной патологии. Тем не менее, комбинирование существующих подходов может иметь большое значение для анализа конкретных механизмов развития сепсиса и разработки новых терапевтических подходов. В этом обзоре рассмотрены популярные мышиные модели сепсиса и септического шока, а также их ограничения и стратегии развития.
ВВЕДЕНИЕ
Согласно современным представлениям, сепсис (от древнегреч. σῆψις – гниение) – это угрожающая жизни дисфункция органов, возникающая при нерегулируемом ответе организма на инфекцию. Под септическим шоком подразумевают разновидность сепсиса, при котором метаболические, клеточные и гемодинамические нарушения существенно повышают вероятность смертельного исхода [1]. По данным Всемирной организации здравоохранения септические осложнения возникают ежегодно у 30 млн человек, и 6 млн из них погибают, но и эти данные могут оказаться заниженными из-за плохого учета заболевания в странах с низким и средним уровнем дохода [2, 3]. Большое количество новых случаев сепсиса приходится на развитые страны. В больницах США, например, каждая третья смерть происходит на фоне септических осложнений [4], причем одной из основных причин возникновения сепсиса являются госпитальные инфекции, возбудители которых приобретают устойчивость к антибактериальной терапии [5, 6]. Обычно сепсис возникает на фоне инфекций в барьерных тканях, вызванных патогенными штаммами Escherichia coli, Staphylococcus aureus, Klebsiella pneumoniae, Acinetobacter baumannii, Pseudomonas aeruginosa, Streptococcus spp., однако нарушение границ барьерных тканей компонентами нормальной микробиоты также может привести к индукции сепсиса [7‒9]. Наиболее чувствительны к развитию сепсиса люди старше 65 лет, младенцы, а также лица с ослабленной иммунной системой и/или хроническими заболеваниями (аутоиммунные и опухолевые патологии, болезни почек, легких и др.) [10].
Ключевое событие в индукции сепсиса ‒ активация врожденного иммунного ответа после распознавания организмом молекулярных образов патогенности (PAMP) с помощью паттерн-распознающих рецепторов (PRR), таких как Толл-подобные рецепторы (TLR), NOD- и RIG-подобные рецепторы и так называемые рецепторы-мусорщики (scavenger) [11, 12]. Активация этих рецепторов приводит к системной продукции провоспалительных цитокинов и хемокинов. PRR могут распознавать молекулярные образы опасности (DAMP), высвобождающиеся из поврежденных клеток хозяина, что приводит к чрезмерной активации иммуноцитов и эндотелиоцитов. Следствием такого дисбаланса является сверхпродукция цитокинов (цитокиновый шторм) [13]. Сепсис имеет двухфазную природу: за бурно развивающейся провоспалительной фазой следует компенсаторная противовоспалительная, которая часто приводит к иммунной супрессии [14, 15]. Ранняя гипердинамическая фаза характеризуется повышенным сердечным выбросом (объем крови, перекачиваемый сердцем за единицу времени) и низким системным сосудистым сопротивлением, в то время как последующая гиподинамическая фаза характеризуется пониженным сердечным выбросом при низком системном сосудистом сопротивлении [8]. Помимо этого, при сепсисе наблюдается усиление коагуляции, ослабление фибринолиза, полиорганная недостаточность и другие патологические изменения, что превращает создание воспроизводимой животной модели в нетривиальную задачу [16].
ЭКСПЕРИМЕНТАЛЬНЫЕ МОДЕЛИ СЕПСИСА И ИХ СРАВНИТЕЛЬНАЯ ХАРАКТЕРИСТИКА
Выделяют три основных типа индукции экспериментального сепсиса: 1) введение токсических агентов (липополисахарид (ЛПС), CpG, зимозан и другие лиганды PRR); 2) введение в организм живых патогенов (бактерий или содержимого кишечника, индукция пневмонии, менингита, уросепсиса и др.); 3) нарушение целостности барьерной ткани организма (перфорация кишечника, раневые модели сепсиса и др.) [14]. Первые две группы представлены преимущественно малоинвазивными нехирургическими моделями, в то время как для моделирования сепсиса из третьей группы необходимо хирургическое вмешательство. Характеристика наиболее распространенных экспериментальных моделей индукции сепсиса в мышах представлена в табл. 1.
Таблица 1.
Сравнительная характеристика экспериментальных моделей сепсиса
| Модель | Преимущества | Недостатки |
|---|---|---|
| Индукция сепсиса путем введения в организм токсических агентов (нехирургические методы) |
||
| Системное введение ЛПС [17‒21] | • Легкая, малоинвазивная, контролируемая, стандартизированная и воспроизводимая модель. • Воспроизводит протекание острой фазы грамотрицательного сепсиса. • Регуляция развития сепсиса за счет изменения количества ЛПС или его биоактивности. • Несколько альтернативных способов введения ЛПС. • Не требует работы с патогенами |
• Плохо имитирует гемодинамические, иммунологические и метаболические особенности
сепсиса. • Не воспроизводит развитие полимикробного сепсиса. • ЛПС обычно активирует кратковременный иммунный ответ. • Различается внутри- и межвидовой уровень восприимчивости к ЛПС |
| Токсичность, индуцированная ЛПС и D-GalN [22‒25] | • Все вышеперечисленное. • Требуется меньше ЛПС, поскольку D-GalN повышает чувствительность к ЛПС |
• Все вышеперечисленное. • Быстрое развитие септического шока и ранняя гибель |
| Индукция сепсиса с помощью введения в организм живых патогенов (в основном нехирургические методы) |
||
| Введение в организм бактерий [20, 22, 26‒36] | • Легкая, воспроизводимая и малоинвазивная модель. • Имитирует экстремальный клинический сепсис. • Подходит для изучения иммунного ответа на конкретный бактериальный штамм. • Альтернативные способы введения бактерий. • Возможность использования клинически релевантных патогенных штаммов бактерий. • Регуляция развития сепсиса путем изменения количества и состава вводимых бактерий |
• Плохо имитирует гемодинамические, иммунологические и метаболические особенности
сепсиса. • Развитие эндотоксемии при системном введении большого количества бактерий. • Некоторые бактерии не индуцируют сепсис из-за низкого персистентного потенциала. • Часто используют только один штамм бактерии, в то время как сепсис обычно имеет полимикробную природу |
| Индукция сепсиса на фоне пневмонии [14, 37‒50] | • Простота и воспроизводимость. • Имитирует развитие сепсиса на фоне диффузной пневмонии. • Регуляция развития сепсиса за счет изменения состава вводимых бактерий, а также антибактериальной терапии. • Альтернативные малоинвазивные способы введения бактерий |
• Не всегда приводит к развитию сепсиса. • Гемодинамические фазы не выражены. • Работа с патогенными штаммами бактерий. • Необходимость анестезии при внутрибронхиальном заражении. • Инвертированный ответ на антицитокиновую терапию |
| Внутрибрюшинное введение содержимого слепой кишки или растворенных фекалий [51‒57] | • Простота и воспроизводимость. • Имитирует развитие полимикробного перитонита. • Малоинвазивный метод. • Регуляция динамики развития сепсиса путем изменения количества вводимого вещества |
• Не всегда отражает метаболические, гемодинамические и иммунологические особенности
сепсиса. • Сложность стандартизации из-за варибельности состава микробиоты и пробоподготовки. • Возможная толерантность к собственной микробиоте |
| Имплантация содержащего бактерии фибринового сгустка в брюшную полость [14, 44, 58, 59] | • Имитирует развитие микробного перитонита и проще в исполнении, чем CLP или CASP. • Не индуцирует раннюю смертность. • Подходит для изучения моноинфекций и их терапии с помощью антибиотиков. • Регуляция развития сепсиса путем изменения концентрации бактерий и плотности сгустка |
• Проблемы с воспроизводимостью из-за сложной стандартизации приготовления сгустков
с бактериями и техники операции. • Хирургическое повреждение тканей. • Часто используют только один штамм бактерии, в то время как сепсис обычно полимикробный. • Не работает на новорожденных мышах |
| Индукция сепсиса путем нарушения целостности барьерной ткани организма (хирургические методы) |
||
| Лигирование и прокол слепой кишки (CLP) [20, 60‒75] | • Имитирует развитие полимикробного перитонита с ишемией тканей. • Активация как провоспалительного, так и противовоспалительного иммунного ответа. • Отсутствие пробоподготовки (максимальное сохранение разнообразия микробиоты). • Регуляция развития сепсиса путем изменения диаметра прокола, длины лигируемого участка, а также удаления некротической части кишки |
• Плохая воспроизводимость из-за сложной стандартизации. • Хирургическое повреждение тканей. • Формирование абсцесса вокруг прокола. • Системный воспалительный ответ слабее, чем при CASP. • Не работает на новорожденных мышах |
| Стентирование восходящей ободочной кишки (CASP) [71, 73, 76‒78] | • Лучше CLP имитирует острый диффузный полимикробный перитонит. • Регуляция развития сепсиса с помощью изменения диаметра стента или его удаления. • Не происходит образование абсцесса, в отличие от CLP. • Отсутствие пробоподготовки (максимальное сохранение разнообразия микробиоты) |
• Наиболее сложная модель. • Гемодинамика, иммунные и метаболические изменения хуже изучены, чем у CLP. • Плохо воспроизводит двухфазный иммунный ответ. • Хирургическое повреждение тканей. • Не работает на новорожденных мышах |
ИНДУКЦИЯ СЕПСИСА С ПОМОЩЬЮ НАРУШЕНИЯ ЦЕЛОСТНОСТИ БАРЬЕРНОЙ ТКАНИ ОРГАНИЗМА
На сегодняшний день клинически наиболее релевантные модели сепсиса имитируют полимикробный перитонит путем нарушения целостности кишечника и последующего попадания микробиоты в перитонеальную полость [19].
Лигирование и прокол слепой кишки (cecal ligation and puncture, CLP) воспроизводит клиническую картину внутрибрюшинного абсцесса и полимикробного перитонита при аппендиците или девертикулите с ишемией тканей, поскольку присутствует очаг инфекции с постепенным выходом бактерий в брюшную полость [61]. Модель имеет и другие признаки сепсиса, такие как активация не только провоспалительного, но и противовоспалительного иммунного ответа, ранняя гипердинамическая и поздняя гиподинамическая фазы, полиорганная дисфункция, гипотермия и метаболические изменения, а также продукция DAMP и сходная кинетика цитокинового ответа [60, 66, 68]. В то же время вокруг прокола нередко формируется абсцесс, изолирующий выход содержимого слепой кишки в брюшную полость, поэтому у некоторых мышей сепсис не развивается в септический шок, а острый воспалительный ответ переходит в хронический, который может длиться несколько месяцев [69, 73]. Для имитации развития сепсиса у пациентов с хроническим заболеванием почек возможно проведение CLP после предварительного введения фолиевой кислоты [75]. Методика CLP не подразумевает введения токсинов или живых патогенов, а отсутствие пробоподготовки приводит к максимальному сохранению разнообразия микробиоты кишечника по сравнению с индукцией сепсиса методом внутрибрюшинного введения содержимого кишечника. Модель позволяет регулировать динамику развития сепсиса путем изменения длины лигируемого участка слепой кишки, диаметра иглы (18-25G) и количества проколов (в меньшей степени), а также с помощью инфузионной терапии, применения антибиотиков и имитации аппендэктомии – удаления омертвевшей части слепой кишки при проведении второй хирургической операции [61]. Необходимо отметить, что эта модель достаточно воспроизводима только при большой выборке экспериментальных животных в силу сложности стандартизации метода из-за вариабельности параметров оперативного вмешательства: типа анестезии, способа лапаротомии, длины лигирования слепой кишки, размера иглы и количества проколов, а также зависимости от генетической линии мышей, их возраста, пола, состава микробиоты и условий содержания [20, 65, 72].
Стентирование восходящей ободочной кишки (colon ascendens stent peritonitis, CASP) воспроизводит клиническую картину полимикробного острого диффузного перитонита, причем CASP лучше имитирует патофизиологические изменения, чем CLP: уровни бактериальной диссеминации и системного воспаления постоянно увеличиваются, как и продукция цитокинов, возникает полиорганная недостаточность, кроме того не образуется абсцесс, препятствующий выходу содержимого кишечника в брюшную полость [76, 78]. Методика CASP позволяет регулировать динамику развития сепсиса путем изменения диаметра стента (14-22G) или с помощью удаления стента и сшивания перфорации в кишечнике при повторном хирургическом вмешательстве [77]. Однако эта модель более сложная в исполнении, чем CLP, ее гемодинамика и метаболические изменения хуже охарактеризованы, а противовоспалительный цитокиновый ответ возникает практически одновременно с провоспалительным, что не воспроизводит двухфазный иммунный ответ при сепсисе [71, 73]. К ограничениям обеих моделей следует отнести невозможность проведения экспериментов на новорожденных мышах, а также индукции сепсиса с помощью клинически релевантных штаммов бактерий [71].
Относительно недавно разработан метод лигирования и разреза слепой кишки (cecal ligation and incision, CLI), который имитирует более острое начало сепсиса, по сравнению c CLP, однако эта модель не используется широко, а ее метаболические, гемодинамические и иммунологические ответы пока недостаточно охарактеризованы [79‒81].
ИНДУКЦИЯ СЕПСИСА ЗА СЧЕТ ВВЕДЕНИЯ ЖИВЫХ ПАТОГЕНОВ
Введение в организм грамположительных (Streptococcus pneumoniae, S. aureus) или грамотрицательных (E. coli, Bаcteroides fragilis, K. pneumoniae, A. baumannii, P. aeruginosa) бактерий ‒ воспроизводимый и малоинвазивный способ индукции сепсиса, подходящий для изучения механизмов активации иммунного ответа на определенный патоген без проведения хирургического вмешательства [20, 22]. Разные бактерии в зависимости от того, являются ли они клиническими изолятами или лабораторными штаммами, могут дифференциально активировать PRR, например, вследствие различий в биологической активности их ЛПС [21, 82, 83]. В целом модель слабо отражает клиническую картину развития сепсиса, поскольку в организме отсутствует локальный очаг инфекции, откуда непрерывно и дозировано распространяются бактерии (происходит единовременное массированное бактериальное заражение). Кроме того, профиль экспрессии цитокинов имеет ускоренную кинетику, особенно на начальных этапах развития сепсиса [30]. Часто используют только один штамм бактерии, в то время как сепсис обычно имеет полимикробную природу [84]. С другой стороны, использование патогенных штаммов позволяет имитировать внутрибольничные инфекции, которые часто возникают в результате монобактериального заражения: при внутрибрюшинном введении модель отчасти воспроизводит клиническую картину перитонита, при внутривенном – заражение крови через внутрисосудистый катетер, причем в отличие от монотоксических моделей активируются сразу несколько PRR. Модель подразумевает альтернативные пути доставки бактерий в организм: внутривенный (i.v.), внутрибрюшинный (i.p.), подкожный (s.c.) и др. В зависимости от места введения патогенов такой подход позволяет моделировать менингококкемию [85] или, например, уросепсис [86]. Изменения в составе вводимых бактерий, а также применение антибиотиков позволяют регулировать динамику развития сепсиса [19]. При системном введении много бактерий погибает вследствие активации системы комплемента, вызывая бурное развитие эндотоксемии, приводящей к ранней гибели организма и ограничению полномасштабного развития сепсиса [30, 87]. Для предотвращения развития эндотоксемии необходимо снижение количества вводимых бактерий, что возможно при использовании высоковирулентных штаммов или дополнительных адъювантов (например, стерилизованных фекалий). Сепсис может иметь различающиеся механизмы развития в зависимости от штамма бактерии: так, IFNγ способствует выживанию при инфицировании P. aeruginosa и S. pneumoniae, но снижает выживаемость при заражении S. aureus и E. coli [30, 88, 89]. Способ введения патогенов в организм также может влиять на механизм патогенеза: при внутрибрюшинном введении бактерий IL-10 играет защитную роль, однако при индукции бактериальной пневмонии IL-10 будет способствовать прогрессии болезни [90, 91]. Бактерии могут дифференциально инфицировать разные организмы: например, Salmonella typhimurium заражает мышей с большей эффективностью, чем людей [92].
Имплантация содержащего бактерии фибринового сгустка в брюшную полость требует проведения хирургического вмешательства под общим наркозом и приводит к непрерывному высвобождению бактерий, что лучше имитирует распространение патогенов из очага инфекции, чем системное введение бактерий в организм [59]. Гемодинамические и метаболические изменения, а также кинетика цитокинового ответа воспроизводят клиническую картину микробного перитонита [58]. С одной стороны, модель лучше подходит для изучения моноинфекций, чем системное введение бактерий, поскольку является многодневной (отсутствует ранняя смертность вследствие эндотоксемии), но, с другой стороны, предполагается использование одного штамма бактерий, в то время как сепсис обычно имеет полимикробный компонент [84]. Метод хорошо подходит для изучения раннего применения антибиотиков при развитии патологии. Модель воспроизводима только при унифицированной подготовке сгустков с бактериями и отработанной технике лапаротомии [93]. Она позволяет регулировать скорость развития сепсиса путем изменения плотности фибринового сгустка и подбора необходимой концентрации бактерий [22]. Кроме того, существует возможность остановить прогрессию перитонита путем дополнительного хирургического вмешательства и удаления сгустка [20].
Внутрибрюшинное введение растворенных фекалий или содержимого слепой кишки (intraperitoneal cecal slurry injection model, CS) считаются более простыми в исполнении и малоинвазивными способами индукции полимикробного сепсиса, однако они требуют предварительной пробоподготовки и стандартизации бактериального состава вводимого материала [52, 57, 94]. В отличие от классического сепсиса массовое введение содержимого кишечника может индуцировать сильный воспалительный ответ, приводящий либо к быстрой гибели, либо к полному выздоровлению [54]. Модели плохо воспроизводят гемодинамические и метаболические изменения, а также имеют отличающиеся профили экспрессии релевантных генов и продукции цитокинов [56]. Динамика развития сепсиса регулируется путем изменения количества вводимого содержимого кишечника, которое характеризуется меньшим разнообразием микробиоты, чем в моделях CLP и CASP, поскольку некоторые бактерии (в первую очередь анаэробные) погибают в ходе пробоподготовки. Замораживание вводимого материала отчасти решает проблему стандартизации смеси для разных экспериментов, однако приводит к гибели отдельных чувствительных штаммов [20]. Иногда у мышей может развиться толерантность к собственной микробиоте – в этом случае необходимо использовать дополнительный адъювант, например сульфат бария [95].
Индукция сепсиса на фоне пневмонии является клинически значимой моделью, поскольку инфекции дыхательных путей нередко приводят к вторичной инфекции и последующему острому респираторному дистресс-синдрому, бактериемии, поражению легких и полиорганной недостаточности [20, 96, 97]. Модель отличается относительной простотой, воспроизводимостью и подразумевает наличие альтернативных способов введения бактерий в организм, таких как интраназальное (i.n.), внутритрахейное (i.t.), внутрибронхиальное (i.b.), аэрозольное распыление и др. (надо отметить, что i.t. или i.b. способы заражения требуют применения анестезии, которая может повлиять на развитие иммунного ответа) [50]. При использовании патогенных штаммов S. pneumoniae или S. pyogenes модель позволяет имитировать развитие внебольничной пневмонии, при введении P. aeruginosa, K. pneumoniae, S. aureus и A. baumannii – внутрибольничной пневмонии, и даже воспроизводить клиническую картину развития внутрибольничных инфекций на фоне перитонита в случае комбинирования с моделями CLP и CASP [14, 50]. Модель часто характеризуется инвертированным ответом на терапию в мышах: ингибирование противовоспалительных цитокинов может повышать выживаемость, а ингибирование провоспалительных цитокинов снижает ее [43]. Метод характеризуется разной чувствительностью к различным патогенам. Например, для развития пневмонии, индуцируемой P. aeruginosa, необходимо инокулировать в легкие большое количество бактерий, причем пневмония будет развиваться в течение суток, а не нескольких дней, что плохо отражает динамику развития болезни [44].
ИНДУКЦИЯ СЕПСИСА С ПОМОЩЬЮ ВВЕДЕНИЯ ТОКСИНОВ
Под малоинвазивными методами индукции сепсиса с помощью токсических агентов обычно подразумевают введение в организм лигандов PRR: зимозана, CpG, пептидогликана, липотейхоевых кислот и др. [30]. Наиболее распространены прямая модель ЛПС-токсичности (системное введение ЛПС) и модель острой гепатотоксичности (системное введение ЛПС в комбинации с D-GalN) [93]. Эти контролируемые и воспроизводимые модели существенно упрощают многостадийную клиническую картину развития сепсиса и скорее воспроизводят такие признаки эндотоксемии или острой фазы грамотрицательного сепсиса, как отсутствие очага инфекции, появление гиподинамии без предварительного гипердинамического состояния, лактоацидоз, мощная, но кратковременная продукция провоспалительных цитокинов, повышение продукции DAMP (например, HMGB-1), мощная активация врожденного иммунного ответа [19, 25, 30]. Введение токсинов не имитирует развитие полимикробного сепсиса, а также взаимодействие патогена с организмом хозяина, поскольку иммунной системе не нужно элиминировать патоген [17]. В то же время, высокие уровни ЛПС в крови обнаруживаются при менингококкемии, бактериемии и антибактериальной терапии, что подчеркивает клиническую значимость этих моделей [98]. Основными индукторами сепсиса в этих моделях являются провоспалительные цитокины, экспрессирующиеся при активации сигнального пути TLR4, причем уровень продукции провоспалительных цитокинов в этих моделях, как и у пациентов, коррелирует с тяжестью протекания сепсиса [56, 99, 100]. Эти подходы широко применяют для изучения сигнальных путей TLR4 и в доклинических исследованиях блокаторов медиаторов воспаления (например, провоспалительных цитокинов), однако, они редко оказываются эффективными при клинических испытаниях. Существует несколько альтернативных способов доставки токсина в организм (i.v., i.p. и др.), в том числе путем пролонгированного непрерывного введения, благодаря стабильности молекулы ЛПС [22]. Вариабельность вводимой дозы и возможность применения ЛПС с различающейся биологической активностью позволяет регулировать уровень иммунного ответа [21, 83]. Необходимо отметить, что уровень восприимчивости к ЛПС может сильно различаться у разных видов. Например, люди на несколько порядков чувствительнее к действию ЛПС, чем мыши [18].
Сенсибилизация с использованием D-GalN не приводит к гибели мышей, однако позволяет на несколько порядков снизить количество вводимого ЛПС, необходимого для индукции сепсиса, по сравнению с прямой моделью ЛПС-токсичности [24]. К преимуществам модели острой гепатотоксичности, индуцированной ЛПС/D-GalN, следует отнести не только низкую стоимость, но и простоту, хорошую воспроизводимость и легкость стандартизации экспериментов. Введение ЛПС/D-GalN приводит к активации TLR4 в резидентных макрофагах печени – клетках Купфера, и последующей продукции провоспалительных цитокинов, в первую очередь TNF, индуцируя воспаление и печеночную недостаточность, причем ингибирование NF-κB в клетках Купфера снижает повреждение печени [25, 101‒103]. D-GalN метаболизируется исключительно в гепатоцитах, поэтому введение D-GalN повышает чувствительность печени к TNF, активация сигнального пути TNF-RI запускает апоптоз в гепатоцитах [104‒106], возникает острая печеночная недостаточность, ассоциированная со значительным усилением продукции трансаминаз (АЛТ, АСТ), TNF и интерлейкинов в кровь, что в конечном итоге приводит к гибели организма [107, 108]. Установлено, что киназы и трансферазы, превращающие глюкозу в UDP-глюкозу и галактозу в UDP-галактозу, участвуют также в превращении D-GalN в UDP-D-GalN, поэтому при достаточно высокой концентрации D-GalN в организме уридин будет секвестрироваться путем включения в состав UDP-D-GalN. Трансфераза, переносящая галактозильную группу UDP-галактозы на белок с образованием галактозилированных белков и восстановлением UTP, не может осуществить аналогичное превращение UDP-D-GalN, поэтому в гепатоцитах происходит истощение UTP и, как следствие, ингибирование транскрипции [23]. Нарушение транскрипции приводит к прекращению синтеза антиапоптотических белков (Bcl-2, Bcl-xL) и активации каспазного каскада [25, 109]. Если в течение этого периода в организм ввести ЛПС или TNF, то возникнет септический шок, однако введение уридина защищает мышей от токсичности ЛПС/D-GalN [110]. Поврежденные гепатоциты будут выделять DAMP и алармины, что дополнительно будет стимулировать активацию макрофагов [111].
Интересно, что в клетках Купфера, экспрессирующих трансмембранный TNF (tmTNF), нарушен баланс про- и противовоспалительных цитокинов, что способствует развитию воспаления, усилению повреждения печени, а также индукции апоптоза через tmTNF и экспрессию проапоптотического фактора FasL [112]. TNF является активатором нейтрофилов и моноцитов, он индуцирует экспрессию молекул адгезии ICAM-1, VCAM-1 и селектинов в эндотелиоцитах и гепатоцитах, продукцию хемокинов CXCL2, CXCL8, CCL2 гепатоцитами, что способствует миграции нейтрофилов и моноцитов в капилляры синусов печени с последующей эмиграцией в паренхиму печени и усилению воспаления путем продукции провоспалительных медиаторов [113‒117]. Механизм влияния апоптоза гепатоцитов на индукцию воспаления не до конца изучен, однако известно, что ингибирование апоптоза приводит к отмене эмиграции нейтрофилов в печень и их активации [25]. Терапевтическое применение дофамина, ингибиторов циклооксигеназ и растительных флавоноидов (алпинетин, изовитексин и др.) приводит к снижению продукции провоспалительных цитокинов и улучшению выживаемости мышей в этой модели [118‒122]. Общая схема развития индуцированной ЛПС/D-GalN острой гепатотоксичности представлена на рис. 1.
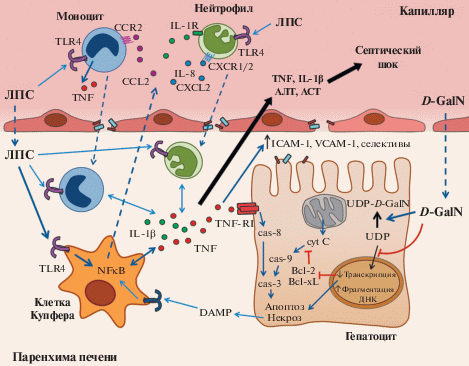
ЭКСПЕРИМЕНТАЛЬНЫЕ МОДЕЛИ СЕПСИСА: ОГРАНИЧЕНИЯ И СТРАТЕГИЯ РАЗВИТИЯ
С 1980-х годов проведено несколько десятков клинических исследований различных препаратов (стероиды, блокаторы цитокинов и др.) против сепсиса, и лишь немногие из них смогли повысить выживаемость пациентов, в то время как большинство препаратов не влияли или даже ухудшали течение болезни [19]. Вот лишь некоторые примеры фармакологических препаратов, показавших эффективность в терапии экспериментального сепсиса у животных, но не прошедших клинические испытания (фазы III и IV) на людях: селепрессин (ClinicalTrials.gov Identifier: NCT02508649), анакинра (IL-1RA), блокаторы TNF (этанерцепт, CDP571 и др.), антитела к эндотоксину, TAK-242, тифакогин, TCV-309, ленерцепт, ингибиторы NO, антитромбин, BB-882, BN 5021, щелочные фосфатазы, дротрекогин альфа (активированный), метилпреднизолона натрия сукцинат [19, 123]. Очевидным объяснением этому служит ограниченная клиническая релевантность доклинических моделей сепсиса на мышах, поскольку они не могут полностью имитировать метаболические, гемодинамические и иммунологические изменения в организме больных сепсисом или септическим шоком. Мыши отличаются от людей по некоторым иммунологическим свойствам, влияющим на патогенез сепсиса. Мыши на несколько порядков устойчивее к действию токсинов (ЛПС, дифтерийный токсин), чем человек, у них меньше общая доля нейтрофилов в крови, причем их ферментативная способность ниже, система комплемента менее активна, отличаются также наборы пентраксинов, участвующих в воспалительном процессе, отсутствуют гены, кодирующие важные компоненты иммунной системы: IL-8, IL-37, TLR10, ICAM-3 и др. [72, 124, 125]. Комбинирование нескольких экспериментальных моделей сепсиса, каждая из которых в отдельности не является универсальной, может способствовать лучшему пониманию молекулярных механизмов развития сепсиса. Необходимо учитывать также вариабельность патогенеза различных септических осложнений, а этиологические неоднородности должны быть отражены в стратегии доклинических исследований терапевтических препаратов [126].
Лабораторные мыши содержатся в апатогенных условиях (SPF-виварий) и обычно имеют не вполне зрелую иммунную систему с дефицитом Т-клеток памяти [127], что может способствовать лучшей имитации развития сепсиса у новорожденных, но не у взрослых людей. Такие мыши могут иметь ограниченное разнообразие микробиоты, которая непосредственно влияет на иммунную систему и развитие патологий [128]. Помимо этого, на фоне септических осложнений у людей, но не у SPF-мышей, часто активируются персистирующие вирусные инфекции (например, герпесвирусы), которые могут изменять устойчивость организма к бактериальным коинфекциям [72, 96, 97]. Использование в экспериментах “грязных” мышей может способствовать лучшей имитации развития патологии у людей [129]. Кроме того, подавляющее большинство исследований проводится на инбредных линиях мышей, однако человеческая популяция гетерогенна, что свидетельствует о важности работы на межлинейных гибридах, аутбредных и нелинейных мышах.
Использование в доклинических исследованиях сепсиса небольших животных ограничивает применение инвазивного мониторинга гемодинамики, а также затрудняет активное проведение поддерживающего лечения (вентиляция легких, инфузионная терапия, заместительная почечная терапия, парентеральное питание и др.). Кроме того, экспериментальные модели сепсиса нередко игнорируют проведение сопутствующей антибактериальной и вазопрессорной поддержки, которую всегда оказывают пациентам [44, 84, 130]. Использование в моделировании сепсиса более крупных животных (например, нечеловекообразных приматов) существенно увеличит экономические затраты, но позволит эффективнее проводить инвазивный мониторинг и поддерживающую терапию [131].
Обычно в экспериментах используют молодых (8‒12 недель) здоровых мышей, однако необходимым представляется изучение патогенеза сепсиса и способов его терапии на старых мышах, поскольку именно пожилые люди наиболее подвержены септическим осложнениям [132‒135]. Помимо этого, интерпретация доклинических исследований затруднена из-за частого использования мышей одного пола, в то время как половые гормоны и релевантные гены, сцепленные с половыми хромосомами, могут влиять на дифференциальную предрасположенность к развитию сепсиса [136].
Мышиные модели далеко не всегда точно имитируют воспалительные заболевания у человека [137], однако очевидно, что избежать экспериментальной работы на мышах невозможно в силу изученности этих объектов, сходства профилей экспрессии генов во время развития воспаления у мышей и человека, пересечения их физиологических, генетических и биохимических особенностей, а также низкой стоимости, простоты постановки экспериментов и минимальных этических проблем [138‒140]. Кроме того, наличие обширной панели генетически модифицированных мышей с измененными компонентами иммунной системы дает дополнительные возможности для изучения молекулярных механизмов патогенеза сепсиса. Важным представляется также расширение использования гуманизированных линий мышей для постановки экспериментального сепсиса, поскольку это позволяет лучше воспроизводить развитие патологии у человека [134, 141‒145]. Однако стоит отметить, что гуманизации подвергаются, в первую очередь, компоненты иммунной системы, в то время как важные для развития сепсиса нервная система, эпителий, эндотелий, метаболические пути остаются мышиными [72, 146‒149].
Важно не только разрабатывать новые, но и стандартизировать существующие экспериментальные модели сепсиса [150]. Необходимо также стандартизировать многопараметрическую систему оценки тяжести септического состояния на животных моделях [151]. В доклинических исследованиях важно комбинировать экспериментальные модели с различным типом индукции сепсиса, поскольку использование только одной модели может привести к неверной интерпретации роли факторов, участвующих в патогенезе сепсиса, или эффективности терапевтических подходов. Так, в одних исследованиях показана необходимость сигнального пути TLR4 для развития полимикробного сепсиса и ЛПС- или ЛПС/D-GalN-индуцированной токсичности [152‒154], тогда как в других работах с использованием моделей CLP и СASP выявлен минорный вклад TLR4 в патогенез сепсиса [155‒157]. В качестве другого примера можно привести цитокин IL-12, который участвует в развитии сепсиса в CASP-модели [158], но не играет роли в CLP-модели [156].
Очевидно, что разработка фармакологических препаратов для терапии сепсиса будет продолжаться. Перспективным представляется изучение блокаторов системы комплемента (в первую очередь, C5b-9, C5a и его рецепторов) [159, 160], а также иммунологических чекпойнтов (PD-1, CTLA-4, TIM-3 и др.), влияющих на активацию врожденного и адаптивного иммунных ответов [161]. Терапевтический интерес имеет модулирование сигнальных путей цитокинов-регуляторов патогенеза сепсиса и септического шока: IFNγ, GM-CSF, MIF, IL-7, IL-8, IL-15, IL-17, IL-27, IL-33 и др. [162‒166]. Важна также разработка фармакологических ингибиторов HMGB1 [167] и микроРНК (miR-132, miR-146a, miR-150, miR-155 и miR-223) [168–170], чья экспрессия изменяется во время развития сепсиса. Активно изучают терапевтические свойства кортикостероидов, β-блокаторов, тромбомодулина и мезенхимных стволовых клеток [16, 171‒173]. Применение биотелеметрических подходов позволит более качественно определять физиологические изменения, происходящие в организме во время развития сепсиса, и снизить стоимость экспериментов из-за сокращения размеров опытных групп [64, 174].
Список литературы
Singer M., Deutschman C.S., Seymour C.W., Shankar-Hari M., Annane D., Bauer M., Bellomo R., Bernard G.R., Chiche J.D., Coopersmith C.M., Hotchkiss R.S., Levy M.M., Marshall J.C., Martin G.S., Opal S.M., Rubenfeld G.D., van der Poll T., Vincent J.L., Angus D.C. (2016) The Third International Consensus definitions for sepsis and septic shock (Sepsis-3). J. Am. Med. Assoc. 315, 801‒810.
Reinhart K., Daniels R., Kissoon N., Machado F.R., Schachter R.D., Finfer S. (2017) Recognizing sepsis as a global health priority ‒ a WHO resolution. N. Engl. J. Med. 377, 414‒417.
Fleischmann C., Scherag A., Adhikari N.K., Hartog C.S., Tsaganos T., Schlattmann P., Angus D.C., Reinhart K., International Forum of Acute Care T. (2016) Assessment of global incidence and mortality of hospital-treated sepsis. Current estimates and limitations. Am. J. Respir. Crit. Care Med. 193, 259‒272.
Rhee C., Dantes R., Epstein L., Murphy D.J., Seymour C.W., Iwashyna T.J., Kadri S.S., Angus D.C., Danner R.L., Fiore A.E., Jernigan J.A., Martin G.S., Septimus E., Warren D.K., Karcz A., Chan C., Menchaca J.T., Wang R., Gruber S., Klompas M., Program C.D.C.P.E. (2017) Incidence and trends of sepsis in US hospitals using clinical vs claims data, 2009‒2014. JAMA. 318, 1241‒1249.
Angus D.C., van der Poll T. (2013) Severe sepsis and septic shock. N. Engl. J. Med. 369, 840‒851.
Denstaedt S.J., Singer B.H., Standiford T.J. (2018) Sepsis and nosocomial infection: patient characteristics, mechanisms, and modulation. Front. Immunol. 9, 2446.
Ramachandran G. (2014) Gram-positive and gram-negative bacterial toxins in sepsis: a brief review. Virulence. 5, 213‒218.
Chen P., Stanojcic M., Jeschke M.G. (2014) Differences between murine and human sepsis. Surg. Clin. North Am. 94, 1135‒1149.
Ranieri V.M., Thompson B.T., Barie P.S., Dhainaut J.F., Douglas I.S., Finfer S., Gardlund B., Marshall J.C., Rhodes A., Artigas A., Payen D., Tenhunen J., Al-Khalidi H.R., Thompson V., Janes J., Macias W.L., Vangerow B., Williams M.D., Group P.-S.S. (2012) Drotrecogin alfa (activated) in adults with septic shock. N. Engl. J. Med. 366, 2055‒2064.
Gotts J.E., Matthay M.A. (2016) Sepsis: pathophysiology and clinical management. Brit. Med J. 353, i1585.
Raymond S.L., Holden D.C., Mira J.C., Stortz J.A., Loftus T.J., Mohr A.M., Moldawer L.L., Moore F.A., Larson S.D., Efron P.A. (2017) Microbial recognition and danger signals in sepsis and trauma. Biochim. Biophys. Acta. Mol. Basis Dis. 1863, 2564‒2573.
Ozment T.R., Ha T., Breuel K.F., Ford T.R., Ferguson D.A., Kalbfleisch J., Schweitzer J.B., Kelley J.L., Li C., Williams D.L. (2012) Scavenger receptor class a plays a central role in mediating mortality and the development of the pro-inflammatory phenotype in polymicrobial sepsis. PLoS Pathog. 8, e1002967.
Rittirsch D., Flierl M.A., Ward P.A. (2008) Harmful molecular mechanisms in sepsis. Nat. Rev. Immunol. 8, 776‒787.
Kingsley S.M., Bhat B.V. (2016) Differential paradigms in animal models of sepsis. Curr. Infect. Dis. Rep. 18, 26.
Ward N.S., Casserly B., Ayala A. (2008) The compensatory anti-inflammatory response syndrome (CARS) in critically ill patients. Clin. Chest. Med. 29, 617‒625, viii.
Wannemuehler T.J., Manukyan M.C., Brewster B.D., Rouch J., Poynter J.A., Wang Y., Meldrum D.R. (2012) Advances in mesenchymal stem cell research in sepsis. J. Surg. Res. 173, 113‒126.
Copeland S., Warren H.S., Lowry S.F., Calvano S.E., Remick D., Inflammation, the Host Response to Injury I. (2005) Acute inflammatory response to endotoxin in mice and humans. Clin. Diagn. Lab. Immunol. 12, 60‒67.
Warren H.S., Fitting C., Hoff E., Adib-Conquy M., Beasley-Topliffe L., Tesini B., Liang X., Valentine C., Hellman J., Hayden D., Cavaillon J.M. (2010) Resilience to bacterial infection: difference between species could be due to proteins in serum. J. Infect. Dis. 201, 223‒232.
Fink M.P. (2014) Animal models of sepsis. Virulence. 5, 143‒153.
Lewis A.J., Seymour C.W., Rosengart M.R. (2016) Current murine models of sepsis. Surg. Infect. (Larchmt). 17, 385‒393.
Корнеев К.В., Кондакова А.Н., Арбатский Н.П., Новотоцкая-Власова К.А., Ривкина Е.М., Анисимов А.П., Круглов А.А., Купраш Д.В., Недоспасов С.А., Книрель Ю.А., Друцкая М.С. (2014) Различия в биологической активности липополисахаридов в зависимости от степени ацилирования липида А из мутантных штаммов Yersinia pestis и бактерий рода Psychrobacter. Биохимия. 79, 1629–1635.
Poli-de-Figueiredo L.F., Garrido A.G., Nakagawa N., Sannomiya P. (2008) Experimental models of sepsis and their clinical relevance. Shock. 30(Suppl .1), 53‒59.
Silverstein R. (2004) D-galactosamine lethality model: scope and limitations. J. Endotoxin. Res. 10, 147‒162.
Lehmann V., Freudenberg M.A., Galanos C. (1987) Lethal toxicity of lipopolysaccharide and tumor necrosis factor in normal and D-galactosamine-treated mice. J. Exp. Med. 165, 657‒663.
Maes M., Vinken M., Jaeschke H. (2016) Experimental models of hepatotoxicity related to acute liver failure. Toxicol. Appl. Pharmacol. 290, 86‒97.
van Zoelen M.A., Schouten M., de Vos A.F., Flor-quin S., Meijers J.C., Nawroth P.P., Bierhaus A., van der Poll T. (2009) The receptor for advanced glycation end products impairs host defense in pneumococcal pneumonia. J. Immunol. 182, 4349‒4356.
Achouiti A., de Vos A.F., de Beer R., Florquin S., van’t Veer C., van der Poll T. (2013) Limited role of the receptor for advanced glycation end products during Streptococcus pneumoniae bacteremia. J. Innate. Immun. 5, 603‒612.
Achouiti A., van der Meer A.J., Florquin S., Yang H., Tracey K.J., van 't Veer C., de Vos A.F., van der Poll T. (2013) High-mobility group box 1 and the receptor for advanced glycation end products contribute to lung injury during Staphylococcus aureus pneumonia. Crit. Care. 17, R296.
Achouiti A., Van’t Veer C., de Vos A.F., van der Poll T. (2015) The receptor for advanced glycation end products promotes bacterial growth at distant body sites in Staphylococcus aureus skin infection. Microbes Infect. 17, 622‒627.
Buras J.A., Holzmann B., Sitkovsky M. (2005) Animal models of sepsis: setting the stage. Nat. Rev. Drug Discov. 4, 854‒865.
van Zoelen M.A., Schmidt A.M., Florquin S., Meijers J.C., de Beer R., de Vos A.F., Nawroth P.P., Bierhaus A., van der Poll T. (2009) Receptor for advanced glycation end products facilitates host defense during Escherichia coli-induced abdominal sepsis in mice. J. Infect. Dis. 200, 765‒773.
Ramsgaard L., Englert J.M., Manni M.L., Milutinovic P.S., Gefter J., Tobolewski J., Crum L., Coud-riet G.M., Piganelli J., Zamora R., Vodovotz Y., Enghild J.J., Oury T.D. (2011) Lack of the receptor for advanced glycation end-products attenuates E. coli pneumonia in mice. PLoS One. 6, e20132.
Tadie J.M., Bae H.B., Banerjee S., Zmijewski J.W., Abraham E. (2012) Differential activation of RAGE by HMGB1 modulates neutrophil-associated NADPH oxidase activity and bacterial killing. Am. J. Physiol. Cell. Physiol. 302, C249‒256.
Achouiti A., de Vos A.F., van’t Veer C., Florquin S., Tanck M.W., Nawroth P.P., Bierhaus A., van der Poll T., van Zoelen M.A. (2016) Receptor for advanced glycation end products (RAGE) serves a protective role during Klebsiella pneumoniae ‒ induced pneumonia. PLoS One. 11, e0141000.
Noto M.J., Becker K.W., Boyd K.L., Schmidt A.M., Skaar E.P. (2017) RAGE-mediated suppression of interleukin-10 results in enhanced mortality in a murine model of Acinetobacter baumannii sepsis. Infect. Immun. 85, e00954-16.
van Zoelen M.A., Achouiti A., Schmidt A.M., Yang H., Florquin S., Tracey K.J., van der Poll T. (2010) Ligands of the receptor for advanced glycation end products, including high-mobility group box 1, limit bacterial dissemination during Escherichia coli peritonitis. Crit. Care Med. 38, 1414‒1422.
Witteveen E., Wieske L., Manders E., Verhamme C., Ottenheijm C.A.C., Schultz M.J., van Schaik I.N., Horn J. (2019) Muscle weakness in a S. pneumoniae sepsis mouse model. Ann. Transl. Med. 7, 9.
Van Den Boogaard F.E., Brands X., Schultz M.J., Levi M., Roelofs J.J., Van’t Veer C., Van Der Poll T. (2011) Recombinant human tissue factor pathway inhibitor exerts anticoagulant, anti-inflammatory and antimicrobial effects in murine pneumococcal pneumonia. J. Thromb. Haemost. 9, 122‒132.
Schouten M., van’t Veer C., van den Boogaard F.E., Gerlitz B., Grinnell B.W., Roelofs J.J., Levi M., van der Poll T. (2010) Therapeutic recombinant murine activated protein C attenuates pulmonary coagulopathy and improves survival in murine pneumococcal pneumonia. J. Infect. Dis. 202, 1600‒1607.
Coopersmith C.M., Stromberg P.E., Dunne W.M., Davis C.G., Amiot D.M., 2nd, Buchman T.G., Karl I.E., Hotchkiss R.S. (2002) Inhibition of intestinal epithelial apoptosis and survival in a murine model of pneumonia-induced sepsis. J. Am. Med. Assoc. 287, 1716‒1721.
Coopersmith C.M., Amiot D.M., 2nd, Stromberg P.E., Dunne W.M., Davis C.G., Osborne D.F., Husain K.D., Turnbull I.R., Karl I.E., Hotchkiss R.S., Buchman T.G. (2003) Antibiotics improve survival and alter the inflammatory profile in a murine model of sepsis from Pseudomonas aeruginosa pneumonia. Shock. 19, 408‒414.
Muenzer J.T., Davis C.G., Dunne B.S., Unsinger J., Dunne W.M., Hotchkiss R.S. (2006) Pneumonia after cecal ligation and puncture: a clinically relevant “two-hit” model of sepsis. Shock. 26, 565‒570.
Knapp S., Schultz M.J., van der Poll T. (2005) Pneumonia models and innate immunity to respiratory bacterial pathogens. Shock. 24 (Suppl. 1), 12‒18.
van der Poll T. (2012) Preclinical sepsis models. Surg. Infect. (Larchmt). 13, 287‒292.
Chiavolini D., Pozzi G., Ricci S. (2008) Animal models of Streptococcus pneumoniae disease. Clin. Microbiol. Rev. 21, 666‒685.
Murakami K., Bjertnaes L.J., Schmalstieg F.C., McGuire R., Cox R.A., Hawkins H.K., Herndon D.N., Traber L.D., Traber D.L. (2002) A novel animal model of sepsis after acute lung injury in sheep. Crit. Care Med. 30, 2083‒2090.
Sordi R., Menezes-de-Lima O., Della-Justina A.M., Rezende E., Assreuy J. (2013) Pneumonia-induced sepsis in mice: temporal study of inflammatory and cardiovascular parameters. Int. J. Exp. Pathol. 94, 144‒155.
Robertson C.M., Perrone E.E., McConnell K.W., Dunne W.M., Boody B., Brahmbhatt T., Diacovo M.J., Van Rooijen N., Hogue L.A., Cannon C.L., Buchman T.G., Hotchkiss R.S., Coopersmith C.M. (2008) Neutrophil depletion causes a fatal defect in murine pulmonary Staphylococcus aureus clearance. J. Surg. Res. 150, 278‒285.
Torres A., Rello J. (2010) Update in community-acquired and nosocomial pneumonia 2009. Am. J. Respir. Crit. Care Med. 181, 782‒787.
Müller-Redetzky H., Suttorp N., Witzenrath M. (2012) Experimental models of pneumonia-induced sepsis. Drug Discov. Today: Disease Models. 9, e23‒e32.
Sam A.D., 2nd, Sharma A.C., Law W.R., Ferguson J.L. (1997) Splanchnic vascular control during sepsis and endotoxemia. Front. Biosci. 2, e72‒92.
Starr M.E., Steele A.M., Saito M., Hacker B.J., Evers B.M., Saito H. (2014) A new cecal slurry preparation protocol with improved long-term reproducibility for animal models of sepsis. PLoS One. 9, e115705.
Gentile L.F., Nacionales D.C., Lopez M.C., Vanzant E., Cuenca A., Cuenca A.G., Ungaro R., Szpila B.E., Larson S., Joseph A., Moore F.A., Leeuwenburgh C., Baker H.V., Moldawer L.L., Efron P.A. (2014) Protective immunity and defects in the neonatal and elderly immune response to sepsis. J. Immunol. 192, 3156‒3165.
Gentile L.F., Nacionales D.C., Lopez M.C., Vanzant E., Cuenca A., Szpila B.E., Cuenca A.G., Joseph A., Moore F.A., Leeuwenburgh C., Baker H.V., Moldawer L.L., Efron P.A. (2014) Host responses to sepsis vary in different low-lethality murine models. PLoS One. 9, e94404.
Brealey D., Karyampudi S., Jacques T.S., Novelli M., Stidwill R., Taylor V., Smolenski R.T., Singer M. (2004) Mitochondrial dysfunction in a long-term rodent model of sepsis and organ failure. Am. J. Physiol. Regul. Integr. Comp. Physiol. 286, R491‒497.
Rittirsch D., Hoesel L.M., Ward P.A. (2007) The disconnect between animal models of sepsis and human sepsis. J. Leukoc. Biol. 81, 137‒143.
Bernardshaw S., Hetland G., Grinde B., Johnson E. (2006) An extract of the mushroom Agaricus blazei Murill protects against lethal septicemia in a mouse model of fecal peritonitis. Shock. 25, 420‒425.
Mathiak G., Szewczyk D., Abdullah F., Ovadia P., Feuerstein G., Rabinovici R. (2000) An improved clinically relevant sepsis model in the conscious rat. Crit. Care Med. 28, 1947‒1952.
Toky V., Sharma S., Arora B.B., Chhibber S. (2003) Establishment of a sepsis model following implantation of Klebsiella pneumoniae-infected fibrin clot into the peritoneal cavity of mice. Folia Microbiol. (Praha). 48, 665‒669.
Rittirsch D., Huber-Lang M.S., Flierl M.A., Ward P.A. (2009) Immunodesign of experimental sepsis by cecal ligation and puncture. Nat. Protoc. 4, 31‒36.
Mishra S.K., Choudhury S. (2018) Experimental protocol for cecal ligation and puncture model of polymicrobial sepsis and assessment of vascular functions in mice. Methods Mol. Biol. 1717, 161‒187.
Wichterman K.A., Baue A.E., Chaudry I.H. (1980) Sepsis and septic shock ‒ a review of laboratory models and a proposal. J. Surg. Res. 29, 189‒201.
Herrmann I.K., Castellon M., Schwartz D.E., Hasler M., Urner M., Hu G., Minshall R.D., Beck-Schimmer B. (2013) Volatile anesthetics improve survival after cecal ligation and puncture. Anesthesiology. 119, 901‒906.
Lewis A.J., Yuan D., Zhang X., Angus D.C., Rosengart M.R., Seymour C.W. (2016) Use of biotelemetry to define physiology-based deterioration thresholds in a murine cecal ligation and puncture model of sepsis. Crit. Care Med. 44, e420‒431.
Dejager L., Pinheiro I., Dejonckheere E., Libert C. (2011) Cecal ligation and puncture: the gold standard model for polymicrobial sepsis? Trends Microbiol. 19, 198‒208.
Tao W., Deyo D.J., Traber D.L., Johnston W.E., Sherwood E.R. (2004) Hemodynamic and cardiac contractile function during sepsis caused by cecal ligation and puncture in mice. Shock. 21, 31‒37.
Hubbard W.J., Choudhry M., Schwacha M.G., Kerby J.D., Rue L.W., 3rd, Bland K.I., Chaudry I.H. (2005) Cecal ligation and puncture. Shock. 24 (Suppl. 1), 52‒57.
Xiao H., Siddiqui J., Remick D.G. (2006) Mechanisms of mortality in early and late sepsis. Infect Immun. 74, 5227‒5235.
Nacionales D.C., Szpila B., Ungaro R., Lopez M.C., Zhang J., Gentile L.F., Cuenca A.L., Vanzant E., Mathias B., Jyot J., Westerveld D., Bihorac A., Joseph A., Mohr A., Duckworth L.V., Moore F.A., Baker H.V., Leeuwenburgh C., Moldawer L.L., Bra-kenridge S., Efron P.A. (2015) A detailed characterization of the dysfunctional immunity and abnormal myelopoiesis induced by severe shock and trauma in the aged. J. Immunol. 195, 2396‒2407.
Delano M.J., Scumpia P.O., Weinstein J.S., Coco D., Nagaraj S., Kelly-Scumpia K.M., O’Malley K.A., Wynn J.L., Antonenko S., Al-Quran S.Z., Swan R., Chung C.S., Atkinson M.A., Ramphal R., Gabrilovich D.I., Reeves W.H., Ayala A., Phillips J., Laface D., Heyworth P.G., Clare-Salzler M., Moldawer L.L. (2007) MyD88-dependent expansion of an immature GR-1+CD11b+ population induces T cell suppression and Th2 polarization in sepsis. J. Exp. Med. 204, 1463‒1474.
Maier S., Traeger T., Entleutner M., Westerholt A., Kleist B., Huser N., Holzmann B., Stier A., Pfeffer K., Heidecke C.D. (2004) Cecal ligation and puncture versus colon ascendens stent peritonitis: two distinct animal models for polymicrobial sepsis. Shock. 21, 505‒511.
Кавайон Ж.М. (2018) Новые методы лечения при сепсисе: модели на животных “не работают” (обзор). Общая реаниматология. 14, 46‒53.
Schabbauer G. (2012) Polymicrobial sepsis models: CLP versus CASP. Drug Discov. Today: Disease Models. 9, e17‒e21.
Nakagawa N.K., Jukemura J., Aikawa P., Nogueira R.A., Poli-de-Figueiredo L.F., Sannomiya P. (2007) In vivo observation of mesenteric leukocyte-endothelial interactions after cecal ligation/puncture and surgical sepsis source control. Clinics (Sao Paulo). 62, 321‒326.
Doi K., Leelahavanichkul A., Hu X., Sidransky K.L., Zhou H., Qin Y., Eisner C., Schnermann J., Yuen P.S., Star R.A. (2008) Pre-existing renal disease promotes sepsis-induced acute kidney injury and worsens outcome. Kidney Int. 74, 1017‒1025.
Traeger T., Koerner P., Kessler W., Cziupka K., Diedrich S., Busemann A., Heidecke C.D., Maier S. (2010) Colon ascendens stent peritonitis (CASP) ‒ a standardized model for polymicrobial abdominal sepsis. J. Vis. Exp. (46), e2299. https://doi.org/10.3791/2299
Zantl N., Uebe A., Neumann B., Wagner H., Sie-wert J.R., Holzmann B., Heidecke C.D., Pfeffer K. (1998) Essential role of gamma interferon in survival of colon ascendens stent peritonitis, a novel murine model of abdominal sepsis. Infect. Immun. 66, 2300‒2309.
Gomez H.G., Gonzalez S.M., Londono J.M., Hoyos N.A., Nino C.D., Leon A.L., Velilla P.A., Rugeles M.T., Jaimes F.A. (2014) Immunological characterization of compensatory anti-inflammatory response syndrome in patients with severe sepsis: a longitudinal study*. Crit. Care Med. 42, 771‒780.
Scheiermann P., Hoegl S., Revermann M., Ahluwalia D., Zander J., Boost K.A., Nguyen T., Zwissler B., Muhl H., Hofstetter C. (2009) Cecal ligation and incision: an acute onset model of severe sepsis in rats. J. Surg. Res. 151, 132‒137.
Fink T., Heymann P., Taha-Melitz S., Taha A., Wolf B., Rensing H., Volk T., Mathes A.M. (2013) Dobutamine pretreatment improves survival, liver function, and hepatic microcirculation after polymicrobial sepsis in rat. Shock. 40, 129‒135.
Fink T., Glas M., Wolf A., Kleber A., Reus E., Wolff M., Kiefer D., Wolf B., Rensing H., Volk T., Mathes A.M. (2014) Melatonin receptors mediate improvements of survival in a model of polymicrobial sepsis. Crit. Care Med. 42, e22‒31.
Korneev K.V., Arbatsky N.P., Molinaro A., Palmigiano A., Shaikhutdinova R.Z., Shneider M.M., Pier G.B., Kondakova A.N., Sviriaeva E.N., Sturiale L., Garozzo D., Kruglov A.A., Nedospasov S.A., Drutskaya M.S., Knirel Y.A., Kuprash D.V. (2015) Structural Relationship of the lipid a acyl groups to activation of murine Toll-Like receptor 4 by lipopolysaccharides from pathogenic strains of Burkholderia mallei, Acinetobacter baumannii, and Pseudomonas aeruginosa. Front. Immunol. 6, 595.
Korneev K.V., Kondakova A.N., Sviriaeva E.N., Mitkin N.A., Palmigiano A., Kruglov A.A., Telegin G.B., Drutskaya M.S., Sturiale L., Garozzo D., Nedospasov S.A., Knirel Y.A., Kuprash D.V. (2018) Hypoacylated LPS from foodborne pathogen Campylobacter jejuni induces moderate TLR4-mediated inflammatory response in murine macrophages. Front. Cell. Infect. Microbiol. 8, 58.
Stortz J.A., Raymond S.L., Mira J.C., Moldawer L.L., Mohr A.M., Efron P.A. (2017) Murine models of sepsis and trauma: can we bridge the Gap? ILAR J. 58, 90‒105.
Ribes S., Domenech A., Cabellos C., Tubau F., Linares J., Viladrich P.F., Gudiol F. (2003) Experimental meningitis due to a high-level cephalosporin-resistant strain of Streptococcus pneumoniae serotype 23F. Enferm. Infect. Microbiol. Clin. 21, 329‒333.
Svensson M., Yadav M., Holmqvist B., Lutay N., Svanborg C., Godaly G. (2011) Acute pyelonephritis and renal scarring are caused by dysfunctional innate immunity in mCxcr2 heterozygous mice. Kidney Int. 80, 1064‒1072.
Cross A.S., Opal S.M., Sadoff J.C., Gemski P. (1993) Choice of bacteria in animal models of sepsis. Infect. Immun. 61, 2741‒2747.
Sasaki S., Nishikawa S., Miura T., Mizuki M., Yamada K., Madarame H., Tagawa Y.I., Iwakura Y., Nakane A. (2000) Interleukin-4 and interleukin-10 are involved in host resistance to Staphylococcus aureus infection through regulation of gamma interferon. Infect. Immun. 68, 2424‒2430.
Rubins J.B., Pomeroy C. (1997) Role of gamma interferon in the pathogenesis of bacteremic pneumococcal pneumonia. Infect. Immun. 65, 2975‒2977.
van der Poll T., Marchant A., Keogh C.V., Goldman M., Lowry S.F. (1996) Interleukin-10 impairs host defense in murine pneumococcal pneumonia. J. Infect. Dis. 174, 994‒1000.
van der Poll T., Marchant A., Buurman W.A., Berman L., Keogh C.V., Lazarus D.D., Nguyen L., Goldman M., Moldawer L.L., Lowry S.F. (1995) Endogenous IL-10 protects mice from death during septic peritonitis. J. Immunol. 155, 5397‒5401.
Mittrucker H.W., Kaufmann S.H. (2000) Immune response to infection with Salmonella typhimurium in mice. J. Leukoc. Biol. 67, 457‒463.
Nemzek J.A., Hugunin K.M., Opp M.R. (2008) Modeling sepsis in the laboratory: merging sound science with animal well-being. Comp. Med. 58, 120‒128.
Gonnert F.A., Recknagel P., Seidel M., Jbeily N., Dahlke K., Bockmeyer C.L., Winning J., Losche W., Claus R.A., Bauer M. (2011) Characteristics of clinical sepsis reflected in a reliable and reproducible rodent sepsis model. J. Surg. Res. 170, e123‒134.
Ko J.J., Mann F.A. (2014) Barium peritonitis in small animals. J. Vet. Med. Sci. 76, 621‒628.
Свиряева Е.Н., Корнеев К.В., Друцкая М.С., Купраш Д.В. (2016) Механизмы перестройки иммунного ответа при вирусно-бактериальных коинфекциях дыхательных путей. Биохимия. 81, 1593–1603.
Свиряева Е.Н., Корнеев К.В., Друцкая М.С., Недоспасов С.А., Купраш Д.В. (2016) Моделирование вирусно-бактериальных коинфекций на молекулярном уровне с использованием агонистов рецепторов врожденного иммунитета. Докл. Акад. Наук. 471, 112–114.
Lepper P.M., Held T.K., Schneider E.M., Bolke E., Gerlach H., Trautmann M. (2002) Clinical implications of antibiotic-induced endotoxin release in septic shock. Intensive Care Med. 28, 824‒833.
Ben Ari Z., Avlas O., Pappo O., Zilbermints V., Cheporko Y., Bachmetov L., Zemel R., Shainberg A., Sharon E., Grief F., Hochhauser E. (2012) Reduced hepatic injury in Toll-like receptor 4-deficient mice following D-galactosamine/lipopolysaccharide-induced fulminant hepatic failure. Cell Physiol. Biochem. 29, 41‒50.
Kuzmich N.N., Sivak K.V., Chubarev V.N., Porozov Y.B., Savateeva-Lyubimova T.N., Peri F. (2017) TLR4 signaling pathway modulators as potential therapeutics in inflammation and sepsis. Vaccines. (Basel). 5, 34.
Wheeler M.D., Kono H., Yin M., Nakagami M., Uesugi T., Arteel G.E., Gabele E., Rusyn I., Yamashina S., Froh M., Adachi Y., Iimuro Y., Bradford B.U., Smutney O.M., Connor H.D., Mason R.P., Goyert S.M., Peters J.M., Gonzalez F.J., Samulski R.J., Thurman R.G. (2001) The role of Kupffer cell oxidant production in early ethanol-induced liver disease. Free Radic. Biol. Med. 31, 1544‒1549.
Lu J.W., Wang H., Yan-Li J., Zhang C., Ning H., Li X.Y., Zhang H., Duan Z.H., Zhao L., Wei W., Xu D.X. (2008) Differential effects of pyrrolidine dithiocarbamate on TNF-alpha-mediated liver injury in two different models of fulminant hepatitis. J. Hepatol. 48, 442‒452.
Hoffmann F., Sass G., Zillies J., Zahler S., Tiegs G., Hartkorn A., Fuchs S., Wagner J., Winter G., Coes-ter C., Gerbes A.L., Vollmar A.M. (2009) A novel technique for selective NF-kappaB inhibition in Kupffer cells: contrary effects in fulminant hepatitis and ischaemia-reperfusion. Gut. 58, 1670‒1678.
Decker K., Keppler D. (1974) Galactosamine hepatitis: key role of the nucleotide deficiency period in the pathogenesis of cell injury and cell death. Rev. Physiol. Biochem. Pharmacol. 71, 77‒106.
Leist M., Gantner F., Bohlinger I., Tiegs G., Germann P.G., Wendel A. (1995) Tumor necrosis factor-induced hepatocyte apoptosis precedes liver failure in experimental murine shock models. Am. J. Pathol. 146, 1220‒1234.
Zhou B.R., Gumenscheimer M., Freudenberg M., Galanos C. (2003) A striking correlation between lethal activity and apoptotic DNA fragmentation of liver in response of D-galactosamine-sensitized mice to a non-lethal amount of lipopolysaccharide. Acta Pharmacol. Sin. 24, 193‒198.
Mignon A., Rouquet N., Fabre M., Martin S., Pages J.C., Dhainaut J.F., Kahn A., Briand P., Joulin V. (1999) LPS challenge in D-galactosamine-sensitized mice accounts for caspase-dependent fulminant hepatitis, not for septic shock. Am. J. Respir. Crit. Care Med. 159, 1308‒1315.
Kawaguchi K., Kikuchi S., Hasegawa H., Maruyama H., Morita H., Kumazawa Y. (1999) Suppression of lipopolysaccharide-induced tumor necrosis factor-release and liver injury in mice by naringin. Eur. J. Pharmacol. 368, 245‒250.
Nakama T., Hirono S., Moriuchi A., Hasuike S., Nagata K., Hori T., Ido A., Hayashi K., Tsubouchi H. (2001) Etoposide prevents apoptosis in mouse liver with D-galactosamine/lipopolysaccharide-induced fulminant hepatic failure resulting in reduction of lethality. Hepatology. 33, 1441‒1450.
Silverstein R., Norimatsu M., Morrison D.C. (1997) Fundamental differences during Gram-positive versus Gram-negative sepsis become apparent during bacterial challenge of D-galactosamine-treated mice. J. Endotoxin Res. 4, 173‒181.
Tsutsui H., Imamura M., Fujimoto J., Nakanishi K. (2010) The TLR4/TRIF-mediated activation of NLRP3 inflammasome underlies endotoxin-induced liver injury in mice. Gastroenterol. Res. Pract. 2010, 641865.
Yang P., Zhou W., Li C., Zhang M., Jiang Y., Jiang R., Ba H., Li C., Wang J., Yin B., Gong F., Li Z. (2016) Kupffer-cell-expressed transmembrane TNF-alpha is a major contributor to lipopolysaccharide and D-galactosamine-induced liver injury. Cell Tissue Res. 363, 371‒383.
Lawson J.A., Burns A.R., Farhood A., Lynn Bajt M., Collins R.G., Smith C.W., Jaeschke H. (2000) Pathophysiologic importance of E- and L-selectin for neutrophil-induced liver injury during endotoxemia in mice. Hepatology. 32, 990‒998.
Bajt M.L., Farhood A., Jaeschke H. (2001) Effects of CXC chemokines on neutrophil activation and sequestration in hepatic vasculature. Am. J. Physiol. Gastrointest. Liver Physiol. 281, G1188‒1195.
Dorman R.B., Gujral J.S., Bajt M.L., Farhood A., Jaeschke H. (2005) Generation and functional significance of CXC chemokines for neutrophil-induced liver injury during endotoxemia. Am. J. Physiol. Gastrointest Liver Physiol. 288, G880‒886.
Jaeschke H. (2006) Mechanisms of liver injury. II. Mechanisms of neutrophil-induced liver cell injury during hepatic ischemia-reperfusion and other acute inflammatory conditions. Am. J. Physiol. Gastrointest Liver Physiol. 290, G1083‒1088.
Zimmermann H.W., Trautwein C., Tacke F. (2012) Functional role of monocytes and macrophages for the inflammatory response in acute liver injury. Front. Physiol. 3, 56.
Liong E.C., Xiao J., Lau T.Y., Nanji A.A., Tipoe G.L. (2012) Cyclooxygenase inhibitors protect D-galactosamine/lipopolysaccharide induced acute hepatic injury in experimental mice model. Food Chem. Toxicol. 50, 861‒866.
Zhou H., Tang L., Yang Y., Lin L., Dai J., Ge P., Ai Q., Jiang R., Zhang L. (2018) Dopamine alleviated acute liver injury induced by lipopolysaccharide/D-galactosamine in mice. Int. Immunopharmacol. 61, 249‒255.
Wang Y.Y., Diao B.Z., Zhong L.H., Lu B.L., Cheng Y., Yu L., Zhu L.Y. (2018) Maslinic acid protects against lipopolysaccharide/d-galactosamine-induced acute liver injury in mice. Microb. Pathog. 119, 49‒53.
Hu J.J., Wang H., Pan C.W., Lin M.X. (2018) Isovitexin alleviates liver injury induced by lipopolysaccharide/D-galactosamine by activating Nrf2 and inhibiting NF-kappaB activation. Microb. Pathog. 119, 86‒92.
Liu T.G., Sha K.H., Zhang L.G., Liu X.X., Yang F., Cheng J.Y. (2019) Protective effects of alpinetin on lipopolysaccharide/d-Galactosamine-induced liver injury through inhibiting inflammatory and oxidative responses. Microb. Pathog. 126, 239‒-244.
Pickkers P., Mehta R.L., Murray P.T., Joannidis M., Molitoris B.A., Kellum J.A., Bachler M., Hoste E.A.J., Hoiting O., Krell K., Ostermann M., Rozendaal W., Valkonen M., Brealey D., Beishuizen A., Meziani F., Murugan R., de Geus H., Payen D., van den Berg E., Arend J., Investigators S.-A. (2018) Effect of human recombinant alkaline phosphatase on 7-day creatinine clearance in patients with sepsis-associated acute kidney injury: A randomized clinical trial. J. Am. Med. Assoc. 320, 1998‒2009.
Ong G.L., Mattes M.J. (1989) Mouse strains with typical mammalian levels of complement activity. J. Immunol. Methods. 125, 147‒158.
Ratelade J., Verkman A.S. (2014) Inhibitor(s) of the classical complement pathway in mouse serum limit the utility of mice as experimental models of neuromyelitis optica. Mol. Immunol. 62, 104‒113.
Lewis A.J., Rosengart M.R. (2018) Bench-to-bedside: A translational perspective on murine models of sepsis. Surg. Infect. (Larchmt). 19, 137‒141.
Beura L.K., Hamilton S.E., Bi K., Schenkel J.M., Odumade O.A., Casey K.A., Thompson E.A., Fraser K.A., Rosato P.C., Filali-Mouhim A., Sekaly R.P., Jenkins M.K., Vezys V., Haining W.N., Jameson S.C., Masopust D. (2016) Normalizing the environment recapitulates adult human immune traits in laboratory mice. Nature. 532, 512‒516.
Turner P.V. (2018) The role of the gut microbiota on animal model reproducibility. Animal. Model Exp. Med. 1, 109‒115.
Masopust D., Sivula C.P., Jameson S.C. (2017) Of mice, dirty mice, and men: using mice to understand human immunology. J. Immunol. 199, 383‒388.
Seboxa T., Amogne W., Abebe W., Tsegaye T., Azazh A., Hailu W., Fufa K., Grude N., Henriksen T.H. (2015) High mortality from blood stream infection in Addis Ababa, ethiopia, is due to antimicrobial resistance. PLoS One. 10, e0144944.
Chen L., Welty-Wolf K.E., Kraft B.D. (2019) Nonhuman primate species as models of human bacterial sepsis. Lab. Anim. (NY). 48, 57‒65.
Saito H., Sherwood E.R., Varma T.K., Evers B.M. (2003) Effects of aging on mortality, hypothermia, and cytokine induction in mice with endotoxemia or sepsis. Mech. Ageing Dev. 124, 1047‒1058.
Howell G.M., Gomez H., Collage R.D., Loughran P., Zhang X., Escobar D.A., Billiar T.R., Zuckerbraun B.S., Rosengart M.R. (2013) Augmenting autophagy to treat acute kidney injury during endotoxemia in mice. PLoS One. 8, e69520.
Ernst W., Zimara N., Hanses F., Mannel D.N., Seelbach-Gobel B., Wege A.K. (2013) Humanized mice, a new model to study the influence of drug treatment on neonatal sepsis. Infect. Immun. 81, 1520‒1531.
Turnbull I.R., Clark A.T., Stromberg P.E., Dixon D.J., Woolsey C.A., Davis C.G., Hotchkiss R.S., Buchman T.G., Coopersmith C.M. (2009) Effects of aging on the immunopathologic response to sepsis. Crit. Care Med. 37, 1018‒1023.
Mege J.L., Bretelle F., Leone M. (2018) Sex and bacterial infectious diseases. New Microbes New Infect. 26, S100‒S103.
Seok J., Warren H.S., Cuenca A.G., Mindrinos M.N., Baker H.V., Xu W., Richards D.R., McDonald-Smith G.P., Gao H., Hennessy L., Finnerty C.C., Lopez C.M., Honari S., Moore E.E., Minei J.P., Cuschieri J., Bankey P.E., Johnson J.L., Sperry J., Nathens A.B., Billiar T.R., West M.A., Jeschke M.G., Klein M.B., Gamelli R.L., Gibran N.S., Brownstein B.H., Miller-Graziano C., Calvano S.E., Mason P.H., Cobb J.P., Rahme L.G., Lowry S.F., Maier R.V., Moldawer L.L., Herndon D.N., Davis R.W., Xiao W., Tompkins R.G., Inflammation, Host Response to Injury L.S.C.R.P. (2013) Genomic responses in mouse models poorly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA. 110, 3507‒3512.
Osuchowski M.F., Remick D.G., Lederer J.A., Lang C.H., Aasen A.O., Aibiki M., Azevedo L.C., Bahrami S., Boros M., Cooney R., Cuzzocrea S., Jiang Y., Junger W.G., Hirasawa H., Hotchkiss R.S., Li X.A., Radermacher P., Redl H., Salomao R., Soebandrio A., Thiemermann C., Vincent J.L., Ward P., Yao Y.M., Yu H.P., Zingarelli B., Chaudry I.H. (2014) Abandon the mouse research ship? Not just yet! Shock. 41, 463‒475.
Takao K., Miyakawa T. (2015) Genomic responses in mouse models greatly mimic human inflammatory diseases. Proc. Natl. Acad. Sci. USA. 112, 1167‒1172.
Efron P.A., Mohr A.M., Moore F.A., Moldawer L.L. (2015) The future of murine sepsis and trauma research models. J. Leukoc. Biol. 98, 945‒952.
Laudanski K., Lapko N., Zawadka M., Zhou B.X., Danet-Desnoyers G., Worthen G.S. (2017) The clinical and immunological performance of 28 days survival model of cecal ligation and puncture in humanized mice. PLoS One. 12, e0180377.
Parker D. (2017) Humanized mouse models of Staphylococcus aureus infection. Front Immunol. 8, 512.
Schlieckau F., Schulz D., Fill Malfertheiner S., Entleutner K., Seelbach-Goebel B., Ernst W. (2018) A novel model to study neonatal Escherichia coli sepsis and the effect of treatment on the human immune system using humanized mice. Am. J. Reprod. Immunol. 80, e12859.
Lapko N., Zawadka M., Polosak J., Worthen G.S., Danet-Desnoyers G., Puzianowska-Kuznicka M., Laudanski K. (2017) Long-term monocyte dysfunction after sepsis in humanized mice is related to persisted activation of macrophage-colony stimulation factor (M-CSF) and demethylation of PU.1, and it can be reversed by blocking M-CSF in vitro or by transplanting naive autologous stem cells in vivo. Front. Immunol. 8, 401.
Unsinger J., McDonough J.S., Shultz L.D., Ferguson T.A., Hotchkiss R.S. (2009) Sepsis-induced human lymphocyte apoptosis and cytokine production in “humanized” mice. J. Leukoc. Biol. 86, 219‒227.
Laudanski K., Stentz M., DiMeglio M., Furey W., Steinberg T., Patel A. (2018) Potential pitfalls of the humanized mice in modeling sepsis. Int. J. Inflam. 2018, 6563454.
Deutschman C.S., Tracey K.J. (2014) Sepsis: current dogma and new perspectives. Immunity. 40, 463‒475.
Melican K., Michea Veloso P., Martin T., Bruneval P., Dumenil G. (2013) Adhesion of Neisseria meningitidis to dermal vessels leads to local vascular damage and purpura in a humanized mouse model. PLoS Pathog. 9, e1003139.
Rongvaux A., Willinger T., Martinek J., Strowig T., Gearty S.V., Teichmann L.L., Saito Y., Marches F., Halene S., Palucka A.K., Manz M.G., Flavell R.A. (2014) Development and function of human innate immune cells in a humanized mouse model. Nat. Biotechnol. 32, 364‒372.
Osuchowski M.F., Thiemermann C., Remick D.G. (2017) Sepsis-3 on the block: What does it mean for preclinical sepsis modeling? Shock. 47, 658‒660.
Shrum B., Anantha R.V., Xu S.X., Donnelly M., Haeryfar S.M., McCormick J.K., Mele T. (2014) A robust scoring system to evaluate sepsis severity in an animal model. BMC Res. Notes. 7, 233.
Poltorak A., He X., Smirnova I., Liu M.Y., Van Huffel C., Du X., Birdwell D., Alejos E., Silva M., Galanos C., Freudenberg M., Ricciardi-Castagnoli P., Layton B., Beutler B. (1998) Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 282, 2085‒2088.
Daubeuf B., Mathison J., Spiller S., Hugues S., Herren S., Ferlin W., Kosco-Vilbois M., Wagner H., Kirschning C.J., Ulevitch R., Elson G. (2007) TLR4/MD-2 monoclonal antibody therapy affords protection in experimental models of septic shock. J. Immunol. 179, 6107‒6114.
Cao C., Chai Y., Shou S., Wang J., Huang Y., Ma T. (2018) Toll-like receptor 4 deficiency increases resistance in sepsis-induced immune dysfunction. Int. Immunopharmacol. 54, 169‒176.
Weighardt H., Kaiser-Moore S., Vabulas R.M., Kirschning C.J., Wagner H., Holzmann B. (2002) Cutting edge: myeloid differentiation factor 88 deficiency improves resistance against sepsis caused by polymicrobial infection. J. Immunol. 169, 2823‒2827.
Echtenacher B., Freudenberg M.A., Jack R.S., Mannel D.N. (2001) Differences in innate defense mechanisms in endotoxemia and polymicrobial septic peritonitis. Infect. Immun. 69, 7271‒7276.
Feterowski C., Emmanuilidis K., Miethke T., Gerauer K., Rump M., Ulm K., Holzmann B., Weig-hardt H. (2003) Effects of functional Toll-like receptor-4 mutations on the immune response to human and experimental sepsis. Immunology. 109, 426‒431.
Entleutner M., Traeger T., Westerholt A., Holzmann B., Stier A., Pfeffer K., Maier S., Heidecke C.D. (2006) Impact of interleukin-12, oxidative burst, and iNOS on the survival of murine fecal peritonitis. Int. J. Colorectal Dis. 21, 64‒70.
Napier B.A., Brubaker S.W., Sweeney T.E., Monette P., Rothmeier G.H., Gertsvolf N.A., Puschnik A., Carette J.E., Khatri P., Monack D.M. (2016) Complement pathway amplifies caspase-11-dependent cell death and endotoxin-induced sepsis severity. J. Exp. Med. 213, 2365‒2382.
Ward P.A., Fattahi F. (2019) New strategies for treatment of infectious sepsis. J. Leukoc. https://doi.org/10.1002/JLB.4MJR1118-425R
Patil N.K., Guo Y., Luan L., Sherwood E.R. (2017) Targeting immune cell checkpoints during sepsis. Int. J. Mol. Sci. 18, E2413.
Shin J., Jin M. (2017) Potential immunotherapeutics for immunosuppression in sepsis. Biomol. Ther. (Seoul). 25, 569‒577.
Brown K.A., Brown G.A., Lewis S.M., Beale R., Treacher D.F. (2016) Targeting cytokines as a treatment for patients with sepsis: A lost cause or a strategy still worthy of pursuit? Int. Immunopharmacol. 36, 291‒299.
Guo Y., Luan L., Patil N.K., Wang J., Bohannon J.K., Rabacal W., Fensterheim B.A., Hernandez A., Sherwood E.R. (2017) IL-15 enables septic shock by maintaining NK cell integrity and function. J. Immunol. 198, 1320‒1333.
Alves-Filho J.C., Sonego F., Souto F.O., Freitas A., Verri W.A., Jr., Auxiliadora-Martins M., Basile-Filho A., McKenzie A.N., Xu D., Cunha F.Q., Liew F.Y. (2010) Interleukin-33 attenuates sepsis by enhancing neutrophil influx to the site of infection. Nat. Med. 16, 708‒712.
Limongi D., D’Agostini C., Ciotti M. (2016) New sepsis biomarkers. Asian Pacific J. Trop. Biomed. 6, 516‒519.
Ueno T., Ikeda T., Yokoyama T., Kihara Y., Konno O., Nakamura Y., Iwamoto H., Shimizu T., McGrath M.M., Chandraker A. (2016) Reduction in circulating level of HMGB-1 following continuous renal replacement therapy in sepsis. Cytokine. 83, 206‒209.
Nahid M.A., Satoh M., Chan E.K. (2011) Mechanistic role of microRNA-146a in endotoxin-induced differential cross-regulation of TLR signaling. J. Immunol. 186, 1723‒1734.
Wang J.F., Yu M.L., Yu G., Bian J.J., Deng X.M., Wan X.J., Zhu K.M. (2010) Serum miR-146a and miR-223 as potential new biomarkers for sepsis. Biochem. Biophys. Res. Commun. 394, 184‒188.
Pop-Began V., Paunescu V., Grigorean V., Pop-Began D., Popescu C. (2014) Molecular mechanisms in the pathogenesis of sepsis. J. Med. Life. 7(2), 38‒41.
Heming N., Lamothe L., Ambrosi X., Annane D. (2016) Emerging drugs for the treatment of sepsis. Expert Opin. Emerg. Drugs. 21, 27‒37.
McIntyre L.A., Stewart D.J., Mei S.H.J., Courtman D., Watpool I., Granton J., Marshall J., Dos Santos C., Walley K.R., Winston B.W., Schlosser K., Fergusson D.A., Canadian Critical Care Trials G., Canadian Critical Care Translational Biology G. (2018) Cellular immunotherapy for septic shock. A phase I clinical trial. Am. J. Respir. Crit. Care Med. 197, 337‒347.
Krasnodembskaya A., Samarani G., Song Y., Zhuo H., Su X., Lee J.W., Gupta N., Petrini M., Matthay M.A. (2012) Human mesenchymal stem cells reduce mortality and bacteremia in gram-negative sepsis in mice in part by enhancing the phagocytic activity of blood monocytes. Am. J. Physiol. Lung Cell Mol. Physiol. 302, L1003‒1013.
Lewis A., Zuckerbraun B., Griepentrog J., Zhang X., Rosengart M. (2017) Reducing animal use with a biotelemetry-enhanced murine model of sepsis. Sci. Rep. 7, 6622.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология