

Молекулярная биология, 2019, T. 53, № 5, стр. 815-829
Математическое моделирование внутриклеточной регуляции иммунных процессов
Д. С. Гребенников a, b, Д. О. Донец a, О. Г. Орлова a, J. Argilaguet c, A. Meyerhans c, d, Г. А. Бочаров b, *
a Московский физико-технический институт (Национальный исследовательский университет)
141701 Московская обл., Долгопрудный, Россия
b Институт вычислительной математики им. Г.И. Марчука Российской академии наук
119333 Москва, Россия
c Infection Biology Laboratory, Department of Experimental and Health Sciences, Universitat Pompeu Fabra
08003 Barcelona, Spain
d ICREA
08010 Barcelona, Pg. Lluís Companys 23, Spain
* E-mail: bocharov@m.inm.ras.ru
Поступила в редакцию 05.03.2019
После доработки 16.04.2019
Принята к публикации 18.04.2019
Аннотация
Современный этап развития исследований в иммунологии можно охарактеризовать значительным углублением знаний о структурных характеристиках иммунной системы, регуляции активности ее различных компонент, функционирующих как целостная распределенная система. Математическое моделирование – аналитический инструмент для описания, анализа и предсказания динамики иммунных реакций в рамках редукционистского подхода. Построение математических моделей иммунной системы человека, отражающих достигнутое понимание ее структуры, и описывающих процессы, определяющие ее функционирование, представляет актуальную задачу для современной системной иммунологии и ее новой междисциплинарной области – математической иммунологии. С этой целью необходимо систематическое развитие многомасштабных математических моделей, описывающих развитие иммунных реакций на следующих уровнях детализации: 1) внутриклеточной регуляции активности компонент иммунной системы, 2) популяционной динамики клеток в органах и 3) системных иммунофизиологических процессов во всем организме. Нами рассмотрены основные работы в области моделирования внутриклеточных регуляторных сетей в контексте задач многомасштабного моделирования. Эти регуляторные процессы определяют основные функции компонент иммунной системы: активацию, деление, дифференцировку, апоптоз, и миграцию иммунных клеток. С учетом сложности и большой размерности регуляторных сетей, задача выделения минимальных описаний сигнальных путей и регуляторных контуров, функционирующих внутри клеток, актуальна для современной математической иммунологии.
ВВЕДЕНИЕ
Иммунная система обеспечивает защиту организма от чужеродных патогенов, развития опухолей и активно участвует в регенерации органов и тканей, при этом отклонения от нормального функционирования приводят к развитию заболеваний различной природы, включая аутоиммунные и злокачественные процессы. Современный этап развития исследований в иммунологии характеризует значительное углубление знаний о структурных характеристиках иммунной системы, регуляции активности ее различных компонент, функционирующих как целостная распределенная система. Методы многомасштабного глобального скрининга и детальной визуализации процессов в органах и тканях, используемые в современной иммунологии, привели к формированию беспрецедентной по степени разрешения и детализации системы представлений о структурной организации и регуляторных сетях, имеющих место в функционирующей иммунной системе. Это создает эмпирическую основу для перехода от статических представлений об иммунной системе, полученных на основе больших данных, к динамическим моделям, описывающим реакции иммунной системы на внешние возмущения различной природы.
Построение математических моделей иммунной системы человека, отражающих достигнутое понимание ее структуры и рассматривающих процессы, определяющие ее функционирование, представляет актуальную задачу для современной системной иммунологии и ее новой междисциплинарной области – математической иммунологии. Разработку математических моделей в иммунологии осуществляют в контексте фундаментальной задачи математической иммунологии, сформулированной выдающимся иммунологом У. Полом: “…иммунная система ставит сложные задачи перед исследователями в области биоматематики. Прежде всего – это вопросы количественного предсказания результатов воздействий на иммунную систему, в которых мы ожидаем непосредственного участия наших коллег-математиков и специалистов по моделированию” [1]. Однако до настоящего времени основной фронт исследований в математической иммунологии основан на построении и анализе математических моделей с малым или средним числом переменных и параметров, описывающих процессы, как правило, на одном уровне детализации. Существует лишь одна исследовательская группа Д. Киршнер (http://malthus.micro.med.umich.edu/), которая систематически развивает многомасштабный подход к моделированию иммунных процессов, связанных с инфекциями верхних дыхательных путей. Необходимость перехода к методам интегративного моделирования иммунной системы с высоким уровнем детализации процессов отмечена в концептуальной работе Р. Джермейна [2]. Чтобы решить задачи многомасштабного и гибридного моделирования пространственно-временной динамики иммунных процессов, необходимо систематическое развитие базового семейства математических описаний для следующих уровней реализации процессов функционирования иммунной системы. Это: 1) внутриклеточная регуляция активности клеток иммунной системы, на основе стохастических и детерминистических моделей, отражающих механизмы регуляции деления, дифференцировки, подвижности и гибели клеток под действием цитокинов, хемокинов и вирусных компонент; 2) индивидуальная и популяционная динамика клеток иммунной системы и гуморальных факторов в расчетных областях, отражающих анатомически корректную структуру лимфоидных органов; 3) системный уровень иммунных процессов в сети лимфоидных органов. Обзоры исследований по моделям системного уровня описания иммунных процессов можно найти в последних работах [3, 4]. Результаты в области моделирования пространственной организации и динамики иммунных процессов рассмотрены в работах [5, 6]. В данной статье нами проанализированы основные работы по моделированию внутриклеточной регуляции иммунных процессов.
МОДЕЛИ ВНУТРИКЛЕТОЧНОЙ РЕГУЛЯЦИИ ИММУННЫХ ПРОЦЕССОВ
Антигениндуцированное деление и дифференцировка лимфоцитов
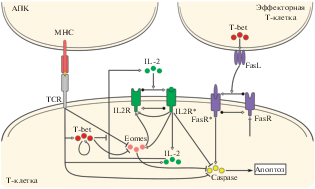
Кинетику развития иммунного ответа на антигенные возмущения различной природы определяет интенсивность процессов пролиферации, дифференцировки и гибели лимфоцитов. Развитие адаптивных иммунных реакций Т-лимфоцитов, в частности, цитотоксических СD8+ T-клеток (CTL) – важнейшее звено, определяющее исход инфекционного заболевания в случае вирусных инфекций. Систематическое исследование регуляторных механизмов, определяющих пространственно-временную динамику CTL в лимфатическом узле, с помощью многомасштабных моделей представлено в работах [7–9]. В моделях рассмотрены процессы перемещения и взаимодействия клеток в рамках двумерной пространственной постановки на основе клеточных моделей Поттса. Описано поле концентраций интерлейкина-2 (IL-2), действие которого в сочетании с уровнем активации Т-клеточных рецепторов антигенпрезентирующими клетками индуцирует внутриклеточные молекулярные регуляторные сети, определяющие окончательную реакцию популяций антигенспецифических Т-клеток на стимуляцию антигеном и данным цитокином. Схема молекулярных процессов, регулирующих отклик отдельной клетки, представлена на рис. 1. В блоке многомасштабной модели, описывающем внутриклеточные регуляторные сети, представлены следующие факторы: цитокины (IL-2), транскрипционные факторы (Tbet, Eomes), рецепторы IL-2, рецепторы Fas и каспазы, участвующие в индукции апоптоза (обозначаемые объединенной переменной “Caspase”). Семейство каспаз содержит 14 белков, которые разделены на две группы – инициаторные и эффекторные каспазы. Поскольку синтез каспаз изначально происходит в неактивной форме, функция инициаторных каспаз состоит в активации эффекторных каспаз путем протеолитического расщепления, опосредуемого через цепочку молекулярных взаимодействий внутри клетки. Однако при интерпретации оценок параметров модели следует учесть, что непосредственный медиатор процесса апоптоза – каспаза-3. В рассматриваемой модели все каспазы описаны одной обобщенной переменной.
Рис. 1.
Схема молекулярной регуляторной сети в CD8+ Т-клетке, определяющей реакцию на антигенную стимуляцию с учетом концентрации IL-2. Активации соответствуют стрелки, а ингибированию – линия с Т-образным окончанием. На этой и последующих схемах не указаны (для упрощения) процессы естественной деградации компонент, которые имеют место в отношении всех из них. IL2R – рецептор IL-2, FasR, FasL – Fas-рецептор и лиганд соответственно, * – активированное состояние.
Изменение концентрации рецепторов IL-2 ([IL2R]) можно описать следующим уравнением:
Изменение концентрации активированных рецепторов, связанных с IL-2 ([IL2R*]), описывает следующее уравнение:
Изменение пространственного распределения IL-2, синтезируемого Т-клетками, описывает нестационарное реакционно-диффузионное уравнение:
Регуляция внутриклеточной концентрации белков: транскрипционных факторов (Tbet, Eomes), каспаз, участвующих в апоптозе (Cas), и активированного Fas-рецептора к лиганду Fas (CD95L или FasL) в мембранной форме – описывается соответственно следующими феноменологическими уравнениями временной динамики.
Изменение концентрации транскрипционного фактора Tbet, обозначенного $[{{T}_{b}}]$ описывает уравнение
Изменение концентрации транскрипционного фактора Eomes, обозначенного $[E]$ описывает уравнение
Изменение концентрации каспаз, участвующих в апоптозе, обозначенных $[Cas]$, определяет уравнение
Изменение концентрации активированных рецепторов Fas, обозначенных $[Fs*]$, выражает уравнение
Отметим, что в данной модели взаимодействие Fas : FasL учтено неявно. Индуцируемая Fas-лигандом активация Caspase описана с помощью переменной [Tbcm], представляющей собой сумму внутриклеточных концентраций Tbet во всех эффекторных Т-клетках, находящихся в контакте с рассматриваемой клеткой.
Модель калибровали по эмпирическим данным, и все ее параметры приведены в работе [9]. Выполнена оценка параметров, определяющих силу обратных связей в регуляторных контурах, скорости деградации молекулярных факторов регуляции, скорости прямых и обратных реакций при формировании комплексов лиганд–рецептор и коэффициенты полунасыщения. Представляют интерес оценки пороговых концентраций внутриклеточных факторов, определяющих в модели переходы клеток из состояния покоя к началу деления, в фазу дифференцировки в эффекторы, в состояние клеток памяти и начало апоптоза.
• Так, для активации клетки, необходимо, чтобы число активированных рецепторов IL-2 превышало порог величиной порядка 63 моль/л, что представляет условие начала деления клеток.
• Если уровень транскрипционного фактора Tbet достигает пороговой величины 17 моль/л, то имеет место дифференцировка активированной CD8+ T-клетки в эффекторную клетку.
• В случае, если концентрация транскрипционного фактора Eomes превышает 16 моль/л, то клетка переходит в состояние клетки памяти.
• Наконец, если уровень каспазы 3 (Caspase-3), основного функционального компонента виртуальной переменной Caspases, превышает величину порядка 19 моль/л, происходит апоптоз клетки.
Данная гибридная модель описывает популяционную динамику клеток, перемещающихся по двумерной пространственной решетке (макро-уровень). При этом для каждой CD8+ Т-клетки с помощью системы обыкновенных дифференциальных уравнений можно описать внутриклеточную регуляцию фенотипического состояния клетки (микроуровень), в зависимости от поля концентрации внеклеточного цитокина (IL-2). Эта многомасштабная модель разработана для исследования влияния асимметрии клеточного деления в ходе ответа на антигенную стимуляцию на фенотипическую структуру Т-клеточной популяции, в частности, на процесс дифференцировки СD8+ T-клеток в эффекторные клетки и клетки памяти. В качестве референтных данных для верификации модели использовали экспериментальные данные по динамике трансгенных клеток в селезенке мышей, инфицированных вирусом осповакцины. С помощью модели показано, что учет неравномерности распределения внутриклеточных молекулярных факторов между дочерними клетками в ходе деления – достаточный компонент модели для получения неоднородной фенотипической структуры антигенспецифической Т-клеточной популяции. Данная структура динамически регулируется от начала антигенной стимуляции до завершения острой фазы и формирования иммунной памяти, т.е. от исходной однородной популяции наивных клеток до гетерогенной популяции, представляющей собой суперпозицию активированных делящихся клеток, эффекторных Т-лимфоцитов и клеток памяти. Наконец, путем исследования чувствительности модели к вариациям параметров предсказано, что для генерации максимального количества клеток памяти степень асимметрии деления должна находиться в пределах 10%. Асимметрия деления реализована в модели случайным образом. При этом разделение молекулярного содержимого между дочерними клетками для рассматриваемых в модели регуляторных факторов лежит в пределах 40–60%, за исключением Tbet, для которого диапазон варьирует от 0 до 100%. Последнее обстоятельство отражает позиционные различия в делении клетки, контактирующей с АПК, когда из одна из двух дочерних клеток теряет контакт с ней.
В отличие от предыдущей модели развития Т‑клеточного иммунного ответа, в которой не учтена кинетика размножения патогена, в многомасштабной модели ВИЧ-инфекции, разработанной в работах [10, 11], эта важная динамическая переменная принята во внимание. Необходимость рассмотрения кинетики репликации вирусов связана с тем, что, согласно концепции “регулируемого баланса пролиферации и дифференцировки лимфоцитов” Гроссмана–Поля [12], иммунная система реагирует на возмущение антигенного гомеостаза, и скорость нарастания антигенного возмущения – важная детерминанта силы реакции. При этом имеет место динамическая парадигма анализа исхода инфекции, так называемая “игра чисел”, постулированная Р.М. Цинкернагелем [13, 14] и характеризующая взаимоотношение параметров вирусной репликации и иммунной защиты хозяина в определении фенотипа инфекционного заболевания.
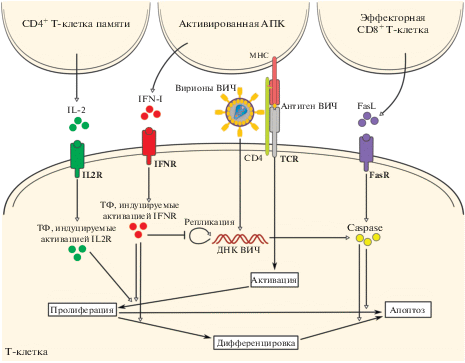
Модель иммунного ответа при ВИЧ-инфекции рассматривает процессы на трех различных масштабах детализации: 1) на системном уровне – вирусная нагрузка, численность CD4+ Т-клеток (неинфицированных и инфицированных) и CD8+ Т‑клеток в крови; 2) в лимфатических узлах – пространственные плотности распределения АПК, клеток-мишеней и инфицированных CD4+ Т-клеток, CD8+ Т-лимфоцитов, цитокинов (интерферона первого типа IFN-I, IL-2, FasL) и ВИЧ; 3) на внутриклеточном уровне – число вирусных геномов в клетке, концентрация факторов, индуцируемых сигнальными путями, инициируемыми связыванием IL-2, IFN-I и FasL с соответствующими рецепторами Т-лимфоцитов. Схема внутриклеточной регуляторной сети процессов, определяющей переход лимфоцитов между различными состояниями, представлена на рис. 2.
Рис. 2.
Схема молекулярной регуляторной сети в Т-лимфоцитах, определяющей реакцию на антигенную стимуляцию/вирусную инфекцию, с учетом внеклеточных полей цитокинов IFN-I, IL-2, FasL. В модели рассмотрено взаимодействие CD4+ Т-лимфоцитов, СD8+ T-лимфоцитов и антигенпрезентирующих клеток, опосредованное приведенными на схеме цитокинами. ТФ – факторы транскрипции.
Состояние каждой из Т-клеток в модели зависит от локальной концентрации внеклеточных сигнальных факторов в области расположения клетки ${{C}_{{e,j}}}({\mathbf{x}},t)$, пространственное распределение которых описывает уравнение реакции-диффузии следующего вида:
В модели связывание IL-2, IFN-I и FasL с соответствующими рецепторами Т-лимфоцитов, предположительно, приводит к активации сигнального пути продукции транскрипционных факторов соответствующих генов, кинетику концентрации которых описывают следующие уравнения. В случае внутриклеточных факторов, индуцируемых активацией клеточных рецепторов к IL-2,
Гибель CD4+ Т-лимфоцитов определяет суперпозиция процессов, индуцирующих генерацию (посредством протеолитического расщепления) внутриклеточных каспаз, т.е. числом активированных рецепторов Fas, наличием вирусных компонент внутри клетки и цитолитическими факторами, выделяемыми антигенспецифическими CD8+ Т-лимфоцитами-эффекторами при контакте с клеткой-мишенью соответственно:
Наконец, изменение числа внутриклеточных вирусных частиц описывает уравнение
В правой части уравнения первое слагаемое моделирует процесс заражения клетки свободными вирусами, второе – синтез вирусных частиц, подавляемый IFN-I, и баланс вирусных частиц при контакте с клетями-мишенями (расход на заражение и проникновение из контактирующих зараженных клеток).
Для калибровки модели были использованы опубликованные оценки параметров элементарных процессов или данные их кинетики, что описано в работах [10, 11]. Как показано на рис. 2, судьбу активированного Т-лимфоцита определяет соотношение внутриклеточных концентраций рассматриваемых транскрипционных факторов, и она сопряжена с введением нескольких порогов активации транскрипционных факторов для разных генов и временной последовательности процессов активации.
• Если уровень активационных факторов, индуцированных IL-2, превышает пороговую величину $C_{{IL{\text{2}}}}^{{division}}$ в момент времени окончания клеточного цикла, т.е. ${{C}_{{fIL{\text{2}}}}}({{t}^{{ecc}}}) \geqslant C_{{IL{\text{2}}}}^{{division}}$, то клетка поделится.
• Если уровень сигнальных факторов, индуцированных активацией рецептора IFN-I, превышает пороговую величину $C_{{IFN}}^{{differetiation}}$ в момент времени начала клеточного цикла, т.е. ${{C}_{{fIFN}}}({{t}^{{bcc}}}) \geqslant C_{{IFN}}^{{differentiation}}$, а уровень факторов, индуцированных IL-2, меньше пороговой величины $C_{{IL{\text{2}}}}^{{division}}$ в момент времени окончания клеточного цикла, т.е. ${{C}_{{fIL{\text{2}}}}}({{t}^{{ecc}}}) < C_{{IL{\text{2}}}}^{{division}}$, то клетка идет по пути дифференцировки в эффектор.
• Если уровень сигнальных факторов, индуцированных активацией рецептора к IFN-I, меньше пороговой величины $C_{{IFN}}^{{differetiation}}$ в начале клеточного цикла, т.е. ${{C}_{{fIFN}}}({{t}^{{bcc}}}) < C_{{IFN}}^{{differentiation}}$, и уровень факторов, индуцированных IL-2, меньше пороговой величины $C_{{IL{\text{2}}}}^{{division}}$ в момент времени окончания клеточного цикла, т.е. ${{C}_{{fIL{\text{2}}}}}({{t}^{{ecc}}}) < C_{{IL{\text{2}}}}^{{division}}$, то происходит апоптоз Т-клетки.
• Если уровень внутриклеточных протеаз, индуцированных внутри клетки, превышает пороговую величину $C_{{Caspase}}^{{apoptosis}}$ в момент начала клеточного цикла, т.е. ${{C}_{{fFas}}}({{t}^{{bcc}}}) \geqslant C_{{Caspase}}^{{apoptosis}}$, то клетка гибнет.
Многомасштабная модель использована для анализа закономерностей пространственной регуляции противовирусного иммунного ответа, обусловленной различиями в локализации активационных сигналов. Показано, что нарушение баланса пролиферации и дифференцировки клеток, приводящее к истощению антигенспецифического иммунного ответа, может быть обусловлено изменениями в пространственной локализации сигнальных полей цитокинов. Так, совмещение пространственной локализации полей антигенной стимуляции и поля концентрации IFN-I при отсутствии пространственного пересечения с полем IL-2 приведет к формированию клона терминально дифференцированных Т-клеток. Заметим, что пространственную локализацию полей определяет положение и миграция различных субпопуляций клеток врожденного и адаптивного иммунитета, а также характеристики диффузии и деградации цитокинов [15–18].
Регуляция клеток врожденного иммунитета
Наиболее систематические исследования в области многомасштабного моделирования иммунных процессов связаны с построением математических моделей иммунных реакций при туберкулезе группой Д. Киршнер [19‒21]. В большой серии работ изучено влияние на динамику данной инфекции различных компонент врожденного и адаптивного иммунитета, иммунотерапии и хемотерапии. Поскольку в предыдущих разделах нами уже рассмотрены типовые модели регуляции активности антигенспецифичных клеток иммунной системы, в данном разделе мы остановимся подробнее на моделировании регуляции реакций клеток врожденного иммунитета.
Туберкулез ‒ широко распространенное заболевание, вызываемое бактериями Mycobacterium tuberculosis. В результате развития иммунной реакции на внедрение бактерий в легких формируются особые структуры – гранулемы. Туберкулезная гранулема имеет специфическую пространственную и клеточную структуру, которая сформирована в результате иммунной реакции на данный патоген. Функционирование гранулемы зависит от большого числа факторов, определяющих баланс провоспалительных и антивоспалительных реакций, и, следовательно, уровень бактериальной нагрузки, активации макрофагов и апоптоза клеток. Чтобы исследовать зависимости трех вышеперечисленных метрик активности гранулемы, была построена многомасштабная агентная модель динамики клеток иммунной системы (макрофагов, цитотоксических Т-лимфоцитов, активированных Т-лимфоцитов, секретирующих интерферон гамма, и регуляторных Т‑клеток), цитокинов (провоспалительного TNF и противоспалительного IL-10) и хемокинов (CCL2, CCL5, CXCL9). Математические модели данного класса (см. работу [20]) отличает высокая степень детализации регуляторных компонент иммунного ответа, включающая процессы хемотаксиса, определяемые градиентами концентраций хемокинов CCL2, CCL5, CXCL9 и цитокина TNF. Многомасштабная модель описывает популяционную динамику клеток иммунной системы и бактерий в легких, в частности, нескольких субпопуляций макрофагов (покоящихся и активированных, неинфицированных и инфицированных), внутриклеточную регуляцию активности макрофагов и внеклеточную динамику хемокинов и цитокинов.
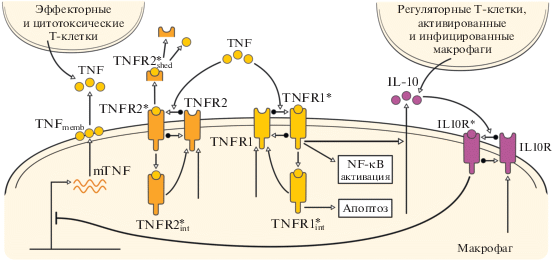
В модели внутриклеточную регуляцию иммунных процессов рассматривают как следствие совместной динамики активированных рецепторов к провоспалительному цитокину TNF с активированными рецепторами к противовоспалительному цитокину IL-10 и индуцируемых ими регуляторных сетей, как схематически показано на рис. 3. Рецептор TNF (TNFRSF1A, обозначенный как TNFR1) активирует фактор транскрипции NF-κB и индуцирует апоптоз.
Рис. 3.
Схема внутриклеточной регуляции активности рецептора TNF, синтеза IL-10, соответствующих клеточных рецепторов и клеточного апоптоза в макрофагах. Рецепторы TNFRSF1A и TNFRSF1B обозначены как TNFR1 и TNFR2 соответственно.
Модель предполагает, что TNF синтезируют активированные и инфицированные макрофаги, цитотоксические Т-лимфоциты и активированные Т-лимфоциты, секретирующие интерферон гамма. Дифференциальные уравнения для скорости изменения концентрации свободного TNF, TNF в мембранной форме и форме матричной РНК, определяющей синтез TNF, последовательно приведены ниже:
Здесь kTACE – скорость секреции TNF с поверхности мембраны клетки во внеклеточное пространство; kon1, koff1 (kon2, koff2) – скорости ассоциации и диссоциации свободного TNF с рецепторами TNFR1 (TNFR2), kmRNA-Mod и ktrans – скорость синтеза мРНК TNF и скорость ее последующей трансляции соответственно. Параметры ρ и NAv осуществляют переход от внутриклеточных к внеклеточным концентрациям.
Изменения концентрации свободных и активированных рецепторов TNF (TNFRSF1A и TNFRSF1B) описывают следующие уравнения:
Связанные с TNF мембранные рецепторы второго типа могут переходить в растворимую форму, что описывает следующие уравнение:
Кроме того, часть связанных с TNF мембранных рецепторов интернализируется, далее либо происходит их деградация, либо рециклирование и повторная экспрессия на поверхности клетки в соответствии с приведенными ниже уравнениями динамики концентраций:
Модель предполагает, что IL-10 синтезируют активированные и инфицированные макрофаги, а также регуляторные Т-клетки. Дифференциальные уравнения для скорости изменения концентрации IL-10, свободных и связанных активированных рецепторов к нему приведены ниже:
Связывание IL-10 с соответствующими рецепторами на поверхности макрофага приводит к подавлению транскрипции мРНК TNF, и, следовательно, скорости синтеза TNF в активированных макрофагах в соответствии со следующей нелинейной зависимостью:
Связывание растворимых рецепторов TNF c соответствующим рецептором на поверхности макрофага приводит к активации синтеза IL-10 в активированных макрофагах, описываемого уравнением Михаэлиса–Ментен для скорости процесса:
Регуляция состояния макрофагов, связанная либо с их активацией, выраженной появлением транскрипционных факторов NF-kB, либо апоптозом, зависит в модели соответственно от концентрации связанных рецепторов TNF и интернализированных рецепторов TNF. Вероятности этих событий описаны в виде случайного процесса Пуассона, в соответствии с уравнениями:
В работе [20] приведены оценки всех 115 параметров многомасштабной модели иммунного ответа на инфекцию бактериями туберкулеза. В результате выполненного исследования показано, что с точки зрения минимизации бактериальной нагрузки, уровня активации макрофагов и апоптоза покоящихся макрофагов – их оценивает величина предложенного в работе обобщенного индекса тяжести инфекционного процесса – необходим оптимальный баланс данных цитокинов в области гранулемы, который можно рассчитать с помощью модели.
В завершение данного раздела заметим, что динамика функциональной поляризации макрофагов определяет характер развития иммунных процессов по воспалительному или противовоспалительному путям. Изучению внутриклеточных регуляторных сетей, определяющих программирование фенотипа макрофагов М1, М2a,b,c, посвящены работы [22, 23]. Установлены структурные связи в виде логических моделей регуляторных сетей, которые отражают взаимодействие между сигнальными путями, молекулярными факторами и транскрипционными факторами, позволяющие воспроизводить наблюдаемую поляризацию фенотипа макрофагов в зависимости от характера активирующих сигналов и состояния окружающих клеток. Данные исследования отличаются анализом весьма разнообразного числа цитокинов, рецепторов, сигнальных и транскрипционных факторов.
Система интерферонов
Система IFN-I ‒ ключевое звено врожденного противовирусного иммунитета. На внутриклеточном уровне регуляцию IFN-I координируют аутокринные и паракринные сигнальные каскады реакций, приводящие к экспрессии IFN-I и активации сотен генов, стимулируемых интерфероном (interferon-stimulated genes, ISGs) [24, 25]. Активация ISGs ограничивает репликацию вирусов на разных стадиях, например, фактор TRIM5α блокирует процесс обратной транскрипции, APOBEG3G препятствует интеграции провирусной ДНК, Tetherin (фактор рестрикции вирусных инфекций) ингибирует отпочковывание созревших вирусных частиц. В то же время геномы таких вирусов, как ВИЧ, кодируют белки, нейтрализующие действие данных защитных факторов, в частности, белок Vif ингибирует действие APOBEG3G, белок Vpu – действие Tetherin [26, 27].
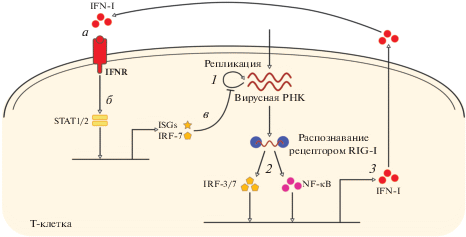
Экспрессия IFN-I и активация ISGs проявляются на клеточно-популяционном уровне гетерогенно и обладают временной вариабельностью: индукция происходит только в части инфицированных клеток, а момент индукции варьирует от клетки к клетке. Это можно объяснить изменчивостью уровней экспрессии сигнальных молекул и транскрипционных факторов, участвующих во внутриклеточном регулировании системы IFN-I [28]. Чтобы исследовать гетерогенность индукции IFN-I в клетках, в работе [28] предложена стохастическая гибридная математическая модель регуляции системы IFN-I. Схема блока внутриклеточных процессов модели представлена на рис. 4. Вирусная РНК при попадании в клетку реплицируется (состояние 1). Рецепторы распознавания патогена (pattern recognition receptors), такие как RIG-I, распознают вирусную РНК и запускают сигнальный путь, приводящий к активации латентных транскрипционных факторов NF-κB и IRF-3/7 и их транслокации в ядро (состояние 2), где они индуцируют экспрессию IFN-I, который затем секретируется в межклеточное пространство (состояние 3). При связывании внеклеточного IFN-I с рецептором происходит активация сигнальных путей STAT1/2 (переход из состояния а в состояние б), что приводит к индукции ISGs и IRF-7, ингибирующих репликацию вирусов (состояние в). Блок внутриклеточных процессов реализуется с помощью стохастического алгоритма Гиллеспи. Подробное описание алгоритма и параметров модели приведены в работе [28]. В каждый момент времени каждая клетка может находиться в одном из 9-ти состояний, соответствующем комбинации промежуточных стадий процессов аутокринного сигнального пути индукции IFN-I (1-2-3) и паракринного сигнального пути активации ISGs (а-б-в), представленных на рис. 4. В начальный момент времени вирус инфицирует клетки, но сигнальные пути системы IFN-I еще не активны, что соответствует состоянию 1а. Процент продуктивно инфицированных клеток и начальное количество вирусных РНК в этих клетках выбирают случайным образом в соответствии с экспериментально заданным титром дозы вирусов. Концентрацию внеклеточного IFN-I в культуре описывает обыкновенное дифференциальное уравнение в предположении мгновенного перемешивания клеток и IFN-I.
Рис. 4.
Схема молекулярной регуляторной сети IFN-I. Промежуточные стадии процессов аутокринного сигнального пути: 1 – внутриклеточная репликация вирусной РНК, 2 – активация транскрипционных факторов NF-κB и IRF-3/7, вызванная распознаванием вирусной РНК рецептором RIG-I, 3 – индукция экспрессии IFN-I. Промежуточные стадии процессов паракринного сигнального пути: а – связывание внеклеточного IFN-I рецептором IFNR, б – активация сигнальных путей STAT1/2, в – активация ISGs и IRF-7, ингибирующих репликацию вирусов.
Калибровку параметров модели проводили по экспериментальным гистограммам межклеточной изменчивости длительности транслокации NF-κB/IRF-7 в ядро и времени до экспрессии IFN-I после инфицирования, а также по данным кинетики вирусной нагрузки, внеклеточной концентрации IFN-I, процента клеток, экспрессирующих IFN-I и IRF-7 при высокой дозе заражения вирусом болезни Ньюкасла. Предсказания модели при малых дозах вирусного заражения (валидированные соответствующими экспериментальными данными) показывают, что в случае низкой вирусной нагрузки эффективный противовирусный интерфероновый ответ развивается благодаря паракринному переносу сигнала, он устойчив по отношению к изменчивости, связанной с вариабельностью уровней экспрессии сигнальных молекул и транскрипционных факторов, а также скоростей процессов в отдельных клетках. Стоит заметить, что для более корректного учета эффектов паракринного сигнального пути необходим переход к многомасштабной модели, учитывающей пространственно-временную динамику клеток и внеклеточного IFN-I.
Отрицательная регуляция активности лимфоцитов
В иммунной системе функционируют многочисленные регуляторные процессы клеточного уровня, определяющие степень функциональной активности лимфоцитов. Один из них связан с экспрессией и активацией мембранного белка ‒ рецептора PD-1 [29], который исторически назван рецептором программируемой клеточной гибели, механизм действия которого принципиально отличатся от Fas. Связывание PD-1 с соответствующими лигандами PD-L1 и PD-L2 снижает активацию Т-клеток. Это снижение активности происходит посредством нескольких механизмов, включая ослабление реакции Т-клеток на антигенную стимуляцию (изменение порога активации с участием Т-клеточных рецепторов, снижение пролиферации, уровня секреции цитокинов) и активацию апоптоза. С одной стороны, это ингибирование активности Т-лимфоцитов предотвращает развитие аутоиммунных реакций, но, с другой стороны, вызывает функциональное и физическое истощение Т-лимфоцитов и представляет механизм формирования хронических инфекционных заболеваний и развития злокачественных процессов [30]. Один из механизмов подавления активности антигенспецифичных Т-лимфоцитов связан с развитием апоптоза. В настоящее время исследованию возможностей коррекции активности Т-клеток с применением анти-PD1-терапии посвящено большое число работ в области иммунологии инфекционных заболеваний и иммуноонкологии [31, 32].
В работе [33] проведен систематический анализ литературных данных и реконструирована минимальная сеть внутриклеточных молекулярных взаимодействий, регулирующих экспрессию PD-1. Схема сети представлена на рис. 5. Во время острой стадии инфекции имеет место первоначальная экспрессия PD-1 на поверхности СD8+ T-клеток, которая через некоторое время снижается. Экспрессия PD-1 происходит посредством стимуляции рецептора TCR антигеном, что запускает сигнальный путь, приводящий к активации фактора NFATc1 (Nuclear Factor of Activated T cells, cytoplasmic 1), являющегося активатором транскрипции гена pdcd1. Последующее снижение экспрессии PD-1 обусловлено индукцией белка Blimp-1 (B Lymphocyte-Induced Maturation Protein 1) при более поздней дифференцировке эффекторных CD8+ T-клеток. Белок Blimp-1 ингибирует экспрессию PD-1 явно и опосредованно через регуляцию экспрессии NFATc1. Известно также, что Blimp-1 и протоонкоген Bcl-6 (B cell lymphoma 6 transcription factor) ингибируют транскрипцию друг друга, а Bcl-6 обеспечивает саморегуляцию, ингибируя собственную транскрипцию. Экспрессию Bcl-6 также ограничивает действие фактора IRF4 (Interferon Regulatory Factor 4), который активируется напрямую сигнальным путем TCR, и опосредованно через стимуляцию NF-κB. IRF4 и Blimp-1 взаимно активируют экспрессию друг друга. IRF4 также участвует в регуляторной цепи, обеспечивающей положительную саморегуляцию посредством связывания с собственным промотором.
Рис. 5.
Схема регуляции экспрессии PD-1 на поверхности CD8+ T-клетки. Комплексы PD-1:PD-L1 обозначены как PD-1*.
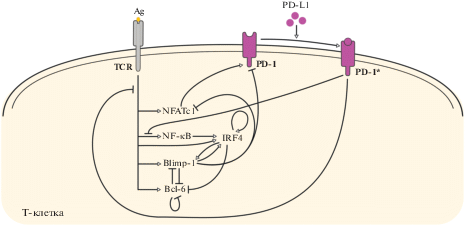
Базовая модель регуляции экспрессии PD-1 описывает динамику взаимодействий четырех переменных: PD-1 (P), IRF4 (I), Blimp-1 (B), Bcl-6 (C). Факторы NFATc1 и NF-κB не описаны в модели явно, NFATc1 включен в обобщенную переменную P, NF-κB – в переменную I. Входные параметры модели ‒ уровень активности стимулируемого антигенами рецептора TCR (u) и доля рецепторов PD-1, связанных лигандом PD-L1 (L). В данной модели использована функция U для описания снижения экспрессии TCR за счет ко-локализации комплексов PD-1:PD-L1 (LP) на поверхности клетки, параметризированного функцией Михаэлиса–Ментен
а также функция ΦL для описания супрессии сигнального пути NF-κB активироваными комплексами PD-1:PD-L1 (LP), параметризованной функцией Хилла:
Изменение обобщенной переменной P описывает уравнение
учитывающее производство и деградацию рецепторов PD1 со скоростями σp и μp соответственно, усиление экспрессии PD1 под влиянием стимуляции рецептора TCR антигеном (U), а также обусловленное индукцией белка Blimp-1 (B) снижение экспрессии PD-1.
Изменение обобщенной переменной I определяет уравнение
учитывающее производство и деградацию IRF4 со скоростями σi и μi соответственно, TCR- и Blimp-1-зависимую активацию IRF4, положительную саморегуляцию IRF4, а также ограничение на скорость активации IRF4 посредством функции ΦL.
Изменение концентрации Blimp-1 (B) описывает уравнение
учитывающее деградацию Blimp-1 со скоростью μb, TCR- и IRF4-зависимую активацию Blimp-1, а также ограничение на скорость активации Blimp-1 за счет ингибирующего действия Bcl-6.
Изменение концентрации Bcl-6 (C) выражает уравнение
учитывающее деградацию Bcl-6 со скоростью μс, TCR-зависимую активацию Bcl-6, а также ингибирующие влияния Blimp-1, IRF4 и Bcl-6 на активацию Bcl-6.
Сеть молекулярных взаимодействий состоит из двух контуров, обладающих свойством контура регуляторной связи с опережением. Вход для обоих контуров ‒ уровень антигенов, стимулирующих TCR, а общие элементы контуров ‒ NF-κB/IRF4. Первый контур включает в себя также Blimp-1 и NFATc1/PD-1, а уровень экспрессии PD-1 ‒ выходная переменная. Второй контур состоит также из фактора Bcl-6, являющегося выходной переменной контура. При увеличении антигенного уровня экспрессия выходов обоих контуров, PD-1 и Bcl-6, увеличивается, а затем падает, однако пороговые значения этой зависимости для PD-1 и Bcl-6 отличны, что обеспечивает модель свойством бифазовости поведения. Это позволило объяснить фенотипические различия в развитии СD8+ T-клеточного иммунного ответа против вируса гриппа в легких, противоопухолевого ответа внутри микросреды опухоли меланомы, взаимодействия опухоли и инфекции, блокады рецепторов PD-1. С этой целью было выполнено расширение базовой модели для описания различных контекстов реализации иммунного ответа в соответствии с деталями каждого сценария.
Миграция лимфоцитов
Корректное воспроизведение миграции клетки в многомасштабных моделях ‒ условие применимости моделей для количественного предсказания инфекционных и онкологических заболеваний, поскольку характеристики подвижности клеток иммунной системы в лимфоидных органах и тканях оказывают существенное влияние на эффективность специфического иммунного ответа [34]. Подвижность клеток и направленную миграцию определяют градиенты полей хемокинов и цитокинов, активирующих хемочувствительные рецепторы лимфоцитов. Простейшая схема модели регуляции активной миграции клеток, представленной в работе [35], приведена на рис. 6. В соответствии с принятой схемой хемочувствительные рецепторы расположены на лидирующем и хвостовом краях поляризованной клетки, динамика каждого кластера рецепторов описывается одинаковыми уравнениями:
Рис. 6.
Простейшая схема регуляции миграции клетки, движущейся в поле хемокина L. Направление миграции определяет разность активных комплексов лиганд–рецептор LR* на двух полюсах поляризованной клетки.
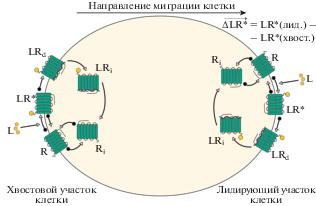
C помощью модели показано, что десенсибилизация рецепторов обеспечивает миграцию клеток вдоль дальнодействующих градиентов хемокинов, не отклоняясь на более сильные, но локальные градиенты других хемокинов. Дальнейшее развитие модели в работах [36‒39] позволило описать аспекты миграции T-лимфоцитов и B-клеток, связанные с движением по градиентам хемокинов (CCL19, CCL21, CXCL12, CXCL13) в лимфатическом узле.
ЗАКЛЮЧЕНИЕ
Чтобы понять механизмы реакции иммунной системы на различные внешние возмущения, необходимо развитие системного подхода, сочетающего эмпирические исследования, теоретические обобщения и математическое моделирование. Перевод совокупности знаний об иммунной системе от статической картины к динамическому рассмотрению ‒ сложнейшая научная задача. В связи с этим необходимо развитие адекватных методов математического моделирования, позволяющих количественно описывать как единую систему совокупность многомасштабных и многоуровневых процессов с учетом большого числа регуляторных связей, которые определяют функционирование иммунной системы [40, 41]. Доминирующее направление исследований в математической иммунологии связано с построением и исследованием математических моделей малой размерности в пределах одного уровня детализации процессов. В последние годы развивают интегративный подход к моделированию заболеваний, обусловленных неэффективным функционированием иммунной системы, с использованием мета-данных различной природы, характеризующих развитие иммунных процессов человека и животных на клеточном, органном и системном уровнях регуляции. Разработка компонент соответствующих гибридных моделей, описывающих регуляторные процессы в клетках, лежит в основе практической реализации данного подхода.
Построение математических моделей регуляции иммунных реакций различной степени детализации связано с редукцией сложности иммунной системы. С этой целью необходимо: 1) использовать упрощающие предположения о наиболее существенных, с точки зрения конкретной задачи исследования, процессов; 2) рассматривать обобщенные переменные, в которых не учтена реальная фенотипическая и функциональная разнородность клеточных и гуморальных компонентов иммунной системы; 3) применять монокомпартментное описание пространственной организации процессов. Чтобы обеспечить биологический реализм и верификацию соответствующих математических моделей, необходимо тесное междисциплинарное взаимодействие математиков с экспериментальными и клиническими иммунологами на этапе постановки задачи и построения модели. Рассмотренные нами в данном обзоре работы отличает подобный характер проведения исследования.
Нами рассмотрены основные работы в области моделирования внутриклеточных регуляторных сетей в контексте задач многомасштабного моделирования, существующие в настоящее время. Эти регуляторные процессы определяют основные функции компонент иммунной системы: активацию, деление, дифференцировку, апоптоз, миграцию. С учетом сложности и большой размерности регуляторных сетей задача выделения минимальных описаний регуляторных контуров чрезвычайно актуальна. Чтобы ее решить, используют математические, инженерные, биохимические методы и подходы [42‒46]. Предстоят дальнейшие исследования по выделению инвариантных (универсальных) внутриклеточных сетей передачи сигналов и активации генов, реализующих в зависимости от контекста управление состоянием клетки на основе системы обратных связей и регуляций с упреждением. Необходима интеграция математических моделей в экспериментальные исследования в качестве одного из аналитических инструментов, обладающего свойством верифицируемости и возможностью установить границы применимости каждой конкретной модели. Это позволит перейти к решению задач моделирования оптимальной терапии иммуноассоциированных заболеваний на основе комбинированного мультитаргетного воздействия на клетки иммунной системы.
Авторы благодарны Е.Н. Богачевой, Д.В. Купрашу и С.А. Недоспасову за критические замечания и конструктивные рекомендации по улучшению обзора.
Работа выполнена при поддержке Российского научного фонда (грант № 18-11-00171), материал раздела “Система интерферонов” подготовлен при поддержке Российского фонда фундаментальных исследований (грант № 17-01-00636).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Paul W.E. (2012) The immune system – complexity exemplified. Math. Mod. Nat. Phen. 7, 4–6.
Germain R.N. (2018) Will systems biology deliver its promise and contribute to the development of new or improved vaccines? What really constitutes the study of “Systems Biology” and how might such an approach facilitate vaccine design. Cold Spring Harb. Perspect. Biol. 10, a033308.
Eftimie R, Gillard J.J., Cantrell D.A. (2016) Mathematical models for immunology: current state of the art and future research directions. Bull. Math. Biol. 78, 2091–2134.
Bocharov G., Volpert V., Ludewig B., Meyerhans A. (2018) Mathematical immunology of virus infections. Cham: Springer International Publishing.
Bocharov G., Meyerhans A., Bessonov N., Trofimchuk S., Volpert V. (2016) Spatiotemporal dynamics of virus infection spreading in tissues. PLoS One. 11, e0168576.
Novkovic M., Onder L., Cheng H-W., Bocharov G., Ludewig B. (2018) Integrative computational modeling of the lymph node stromal cell landscape. Front. Immunol. 9, 2428.
Prokopiou S., Barbarroux L., Bernard S., Mafille J., Leverrier Y., Arpin C., Marvel J., Gandrillon O., Crauste F. (2014) Multiscale modeling of the early CD8 T-cell immune response in lymph nodes: an integrative study. Computation. 2, 159–181.
Gao X., Arpin C., Marvel J., Prokopiou S.A., Gandrillon O., Crauste F. (2016) IL-2 sensitivity and exogenous IL-2 concentration gradient tune the productive contact duration of CD8+ T cell-APC: a multiscale modeling study. BMC Systems Biol. 10, 77.
Girel S., Arpin C., Marvel J., Gandrillon O., Crauste F. (2019) Model-based assessment of the role of uneven partitioning of molecular content on heterogeneity and regulation of differentiation in CD8 T-cell immune responses. Front. Immunol. 10, 230.
Bouchnita A., Bocharov G., Meyerhans A., Volpert V. (2017) Hybrid approach to model the spatial regulation of T cell responses. BMC Immunology. 18, 29.
Bouchnita A., Bocharov G., Meyerhans A., Volpert V. (2017) Towards a multiscale model of acute HIV infection. Computation. 5, 6.
Grossman Z.S, Paul W.E. (2015) Dynamic tuning of lymphocytes: physiological basis, mechanisms, and function. Annu. Rev. Immunol. 33, 677–713.
Zinkernagel R.M., Hengartner H., Stitz L. (1985) On the role of viruses in the evolution of immune responses. Br. Med. Bull. 41, 92–97.
Zinkernagel R.M. (2004) On immunity against infections and vaccines: credo 2004. Scand. J. Immunol. 60, 9–13.
Oyler-Yaniv A., Oyler-Yaniv J., Whitlock B.M., Liu Z., Germain R.N., Huse M., Altan-Bonnet G., Krichevsky O. (2017) A tunable diffusion-consumption mechanism of cytokine propagation enables plasticity in cell-to-cell communication in the immune system. Immunity. 46, 609–620.
Gottschalk R.A., Martins A.J., Angermann B.R., Dutta B., Ng C.E., Uderhardt S., Tsang J.S., Fraser I.D., Meier-Schellersheim M., Germain R.N. (2016) Distinct NF-κB and MAPK activation thresholds uncouple steady-state microbe sensing from anti-pathogen inflammatory responses. Cell Systems. 2, 378–390.
Jafarnejad M., Zawieja D.C., Brook B.S., Nibbs R.J.B., Moore J.E. (2017) A novel computational model predicts key regulators of chemokine gradient formation in lymph nodes and site-specific roles for CCL19 and ACKR4. J. Immunol. 199, 2291–2304.
Bocharov G., Danilov A., Vassilevski Yu., Marchuk G.I., Chereshnev V.A., Ludewig B. (2011) Reaction-diffusion modelling of interferon distribution in secondary lymphoid organs. Math. Model. Nat. Phenom. 6, 13–26.
Fallahi-Sichani M., El-Kebir M., Marino S., Kirschner D.E., Linderman J.J. (2011) Multiscale computational modeling reveals a critical role for TNF-receptor 1 dynamics in tuberculosis granuloma formation. J. Immunol. 186, 3472–3483.
Cilfone N.A., Perry C.R., Kirschner D.E., Linderman J.J. (2013) Multi-scale modeling predicts a balance of tumor necrosis factor-α and interleukin-10 controls the granuloma environment during Mycobacterium tuberculosis infection. PLoS One. 8, e68680.
Pienaar E., Matern W.M., Linderman J.J., Bader J.S., Kirschner D.E. (2016) Multiscale model of mycobacterium tuberculosis infection maps metabolite and gene perturbations to granuloma sterilization predictions. Infect. Immun. 84, 1650–1669.
Castiglione F., Tieri P., Palma A., Jarrah A.S. (2016) Statistical ensemble of gene regulatory networks of macrophage differentiation. BMC Bioinformatics. 17, 506.
Palma A., Jarrah A.S., Tieri P., Cesareni G., Castiglione F. (2018) Gene regulatory network modeling of macrophage differentiation corroborates the continuum hypothesis of polarization states. Front. Physiol. 9, 1659.
Takaoka A., Yanai H. (2006) Interferon signalling network in innate defence. Cell. Microbiol. 8, 907–922.
Schoggins J.W., Wilson S.J., Panis M., Murphy M.Y., Jones C.T., Bieniasz P., Rice C.M. (2011) A diverse range of gene products are effectors of the type I interferon antiviral response. Nature. 472, 481–485.
Marsili G., Remoli A.L., Sgarbanti M., Perrotti E., Fragale A., Battistini A. (2012) HIV-1, interferon and the interferon regulatory factor system: an interplay between induction, antiviral responses and viral evasion. Cytokine Growth Factor Rev. 23, 255–270.
Doyle T., Goujon C., Malim M.H. (2015) HIV-1 and interferons: who’s interfering with whom? Nat. Rev. Microbiol. 13, 403–413.
Rand U., Rinas M., Schwerk J., Nöhren G., Linnes M., Kröger A., Flossdorf M., Kály-Kullai K., Hauser H., Höfer T., et al. (2012) Multi-layered stochasticity and paracrine signal propagation shape the type-I interferon response. Mol. Syst. Biol. 8, 584.
Sharpe A.H., Pauken K.E. (2017) The diverse functions of the PD1 inhibitory pathway. Nat. Rev. Immunol. 18, 153–167.
Havel J.J., Chowell D., Chan T.A. (2019) The evolving landscape of biomarkers for checkpoint inhibitor immunotherapy. Nat. Rev. Cancer. 19, 133–150.
Peligero C., Argilaguet J., Güerri-Fernandez R., Torres B., Ligero C., Colomer P., Plana M., Knobel H., García F., Meyerhans A. (2015) PD-L1 Blockade differentially impacts regulatory T cells from HIV-infected individuals depending on plasma viremia. PLoS Pathogens. 11, e1005270.
Gonzalez-Cao M., Martinez-Picado J., Karachaliou N., Rosell R., Meyerhans A. (2018) Cancer immunotherapy of patients with HIV infection. Clin. Transl. Oncol. 1‒8.
Nikolaev E.V., Zloza A., Sontag E.D. (2019) Immunobiochemical reconstruction of influenza lung infection—melanoma skin cancer interactions. Front. Immunol. 10, 4.
Wong H.S., Germain R.N. (2018) Robust control of the adaptive immune system. Semin. Immunol. 36, 17–27.
Lin F., Butcher E.C. (2008) Modeling the role of homologous receptor desensitization in cell gradient sensing. J. Immunol. 181, 8335–8343.
Nandagopal S., Wu D., Lin F. (2011) Combinatorial guidance by CCR7 ligands for T lymphocytes migration in co-existing chemokine fields. PLoS One. 6, e18183.
Chang S.L., Cavnar S.P., Takayama S., Luker G.D., Linderman J.J. (2015) Cell, isoform, and environment factors shape gradients and modulate chemotaxis. PLoS One. 10, e0123450.
Chan C., Billard M., Ramirez S.A., Schmidl H., Monson E., Kepler T.B. (2013) A model for migratory B cell oscillations from receptor down-regulation induced by external chemokine fields. Bul. Math. Biol. 75, 185–205.
Wu D., Lin F. (2011) Modeling cell gradient sensing and migration in competing chemoattractant fields. PLoS One. 6, e18805.
Eftimie R., Gillard J.J., Cantrell D.A. (2016) Mathematical models for immunology: current state of the art and future research directions. Bull. Math. Biol. 78, 2091–2134.
Chakraborty A.K. (2017) A perspective on the role of computational models in immunology. Annu. Rev. Immunol. 35, 403–439.
Ali Al-Radhawi M., Del Vecchio D., Sontag E.D. (2019) Multi-modality in gene regulatory networks with slow promoter kinetics. PLoS Comp. Biol. 15, e1006784.
Ferrell J.E. (2013) Feedback loops and reciprocal regulation: recurring motifs in the systems biology of the cell cycle. Curr. Opin. Cell Biol. 25, 676–686.
Alon U. (2007) Network motifs: theory and experimental approaches. Nat. Rev. Gen. 8, 450–461.
Albeck J.G., Pargett M., Davies A.E. (2018) Experimental and engineering approaches to intracellular communication. Essays Biochem. 62, 515–524.
Gordley R.M., Bugaj L.J., Lim W.A. (2016) Modular engineering of cellular signaling proteins and networks. Curr. Opin. Struct. Biol. 39, 106–114.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология