

Молекулярная биология, 2019, T. 53, № 5, стр. 849-859
Свой эпитоп среди чужих – Т-клетки и аутоантигены
a Кафедра иммунологии биологического факультета Московского государственного университета
им. М.В. Ломоносова
119234 Москва, Россия
* E-mail: shilov_evgeny@inbox.ru
Поступила в редакцию 30.04.2019
После доработки 30.04.2019
Принята к публикации 10.05.2019
Аннотация
Т-лимфоциты играют ключевую роль в реакциях адаптивного иммунитета, распознавая антигены при помощи вариабельных T-клеточных рецепторов (TCR). Функциональные гены субъединиц TCR образуются в результате соматической реаранжировки, при этом часть получаемых TCR реагирует на аутоантигены – собственные молекулы организма. Такие аутореактивные Т-клетки несут опасность развития иммунного ответа против собственного организма. В процессе онтогенеза иммунной системы часть аутореактивных Т-лимфоцитов удаляется путем апоптоза, часть дифференцируется в иммуносупрессорные регуляторные Т-клетки, которые поддерживают толерантность иммунитета к аутоантигенам, часть переходит в нефункциональное состояние анергии. Регуляторные Т-клетки подавляют эффекторные с помощью иммуносупрессорных цитокинов и костимуляторных молекул, деплеции стимулирующего интерлейкина-2 (IL-2), удаления аутореактивных пептидов вместе с молекулами главного комплекса гистосовместимости (MHC) и других способов. В случае нарушения толерантности возникают аутоиммунные заболевания. Однако при опухолевых заболеваниях именно нарушение иммунологической толерантности лежит в основе терапевтической стратегии, позволяющей проводить иммунотерапию и использовать потенциал эффекторных аутореактивных Т-клеток. Эффективная иммунотерапия опухолей часто сопровождается побочными аутоиммунными реакциями, что в настоящее время считается неизбежной издержкой такого подхода.
ВВЕДЕНИЕ
Т-лимфоциты узнают антигены с помощью димерных Т-клеточных рецепторов (TCR), при этом гены их полипептидных цепей возникают в результате соматической перестройки (реаранжировки). Этот процесс основан на случайном (или близком к случайному) выборе двух (α- и γ‑цепи, V- и J-сегменты) или трех (β- и δ-цепи, V-, D- и J-сегменты) фрагментов, кодирующих вариабельный домен. Состоящие из α- и β-цепей TCR узнают в качестве антигенов короткие пептиды (эпитопы), связанные с молекулами главного комплекса гистосовместимости (MHC), γδ TCR распознают белковые, липидные и углеводные антигены. Молекулы MHC делятся на два класса: MHC I, представляющие пептиды, входящие в состав внутреннего протеома клетки, и MHC II, представляющие пептиды экзогенных белков фагоцитированных частиц. Впрочем, эндогенный пептид может оказаться в комплексе с MHC II после фагоцитоза апоптотических телец, а экзогенный – в комплексе с MHC I в результате процесса кросс-презентации антигена. Распознавание антигена с помощью γδ TCR происходит либо напрямую, без специфической презентации, либо в комплексе с MHC-подобными молекулами, такими как CD1d ‒ ее лигандом могут быть различные липиды, включая церамиды и липид А липополисахарида, ‒ или бутирофилинами BTN3A, связывающими фосфорилированные метаболиты пути биосинтеза изопреноидов [1, 2]. Многие из антигенов, узнаваемых отдельными вариантами αβ и γδ TCR, представлены собственными молекулами организма, что создает опасность развития против них иммунного ответа. Однако в норме такого иммунного ответа не происходит, так как аутореактивные эффекторные клетки контролируются иммунной системой организма с помощью механизмов центральной и периферической толерантности.
СУБПОПУЛЯЦИИ АУТОРЕАКТИВНЫХ Т-КЛЕТОК
В постнатальный период γδ Т-клетки образуются в основном из нормальных тимоцитов, однако на бестимусных мышах было показано, что эти клетки могут образовываться в эмбриональной печени и кишечнике и дифференцироваться без участия тимуса [3]. Их разнообразие в организме человека относительно невелико по сравнению с αβ Т-клетками – 7 вариабельных сегментов γ-цепи и 8 вариабельных сегментов δ-цепи (из них только 3 основных) позволяют реализовать небольшое число сочетаний; при этом различные клоны γδ Т-лимфоцитов узнают множество собственных молекул организма. К числу таких аутоантигенов относятся кардиолипин, MHC I-подобные молекулы, такие как CD1a-d, MICA и MICB, ULBP, HLA-E, T10/22, бутирофилины, а также белки теплового шока, аполипопротеин A-I, ATP-синтетаза и многие другие белки и липиды [1, 2, 4–9]. В целом, γδ Т-клетки можно охарактеризовать как популяцию лимфоцитов, направленную на выявление молекулярных маркеров клеточного и тканевого стресса.
Активация γδ Т-клеток может приводить как к секреции цитокинов, так и к контактному киллингу клеток-мишеней. В связи с этим их роль в противоопухолевых и аутоиммунных реакциях велика и разнообразна; при этом в зависимости от использованной модели аутоиммунного заболевания γδ Т-лимфоциты могут как усиливать тяжесть заболевания, выделяя провоспалительные цитокины [10, 11], так и снижать ее, синтезируя противовоспалительные [12, 13]. Нужно отметить, что γδ Т-клетки ‒ высоко гетерогенная популяция, которая в тех или иных аутоиммунных условиях выделяет при активации интерлейкин (IL) 1β, IL-10, IL-17, IL-12, IL-22, интерферон-γ (IFNγ), трансформирующий фактор роста β (TGFβ), фактор некроза опухолей (TNF), CCL3, CCL4, CCL5 и многие другие регуляторные молекулы, что не позволяет классифицировать их функцию ни как патогенную, ни как протективную [14]. В отношении противоопухолевого иммунитета известно, что γδ Т-лимфоциты распознают молекулы-маркеры клеточного стресса и некоторые опухолеассоциированные антигены (например, F1-субъединицу АТР-синтетазы в плазматической мембране), хотя аффинность таких взаимодействий невелика – Kd порядка 10–4–10–6 M [15]. Для преодоления этой проблемы и использования цитотоксического потенциала γδ Т-клеток в настоящее время применяют либо опсонизацию опухоли направленными на нее антителами и последующую антителопосредованную цитотоксичность (ADCC) [16], либо химерные рецепторы CAR – также на основе антител [17]. На данный момент роль γδ Т-лимфоцитов в патогенезе аутоиммунных заболеваний представляется противоречивой и неоднозначной – одни и те же клонотипы γδ Т-клеток выступают в одних аутоиммунных моделях как протективные, а в других – как патогенные [14]. По-видимому, их функция состоит в основном в усилении иммунного ответа, исходно запускаемого другими, более специфическими лимфоцитами. Несмотря на то, что их роль сопоставима с ролью клеток врожденного иммунитета, она, как правило, вторична. По этой причине в последующей части обзора речь пойдет исключительно об аутореактивных αβ Т‑лимфоцитах. От γδ Т-клеток αβ Т-лимфоциты отличаются облигатным созреванием в тимусе, переходом в эффекторное состояние только после встречи с антигеном и специфичностью к пептидным эпитопам, презентируемым в комплексе с классическими молекулами MHC класса I (CD8+ Т-лимфоциты) на поверхности любых клеток либо MHC класса II (CD4+ Т-лимфоциты) на поверхности антигепрезентирующих клеток (АПК).
СЕЛЕКЦИЯ Т-ЛИМФОЦИТОВ В ТИМУСЕ
Представление о том, что иммунологическая толерантность к собственным антигенам возникает в результате удаления в тимусе возникших в нем же аутореактивных Т-лимфоцитов появилось в конце 1980-х годов [18]. Роль эпителиальных клеток тимуса в этом процессе была показана в экспериментах по индукции у мышей толерантности к аллотрансплантатам. Пересадка эмбриональных эпителиальных клеток мышей линии C3H бестимусным мышам BALB/c nude восстановила функции тимуса и позволила им отторгать кожные лоскуты C57BL/6; при этом сингенные трансплантаты C3H и BALB/c не отторгались [19]. Во время развития тимоциты проходят две стадии, на которых могут погибнуть путем апоптоза: 1) стадию положительной селекции, когда будущая Т-клетка проверяется на способность узнавать своими TCR молекулы MHC собственного организма, при этом не прошедший проверку тимоцит элиминируется как бесполезный для иммунной системы; 2) стадию отрицательной селекции, когда погибнуть может потенциально опасный тимоцит со слишком высокой аффинностью к MHC с аутоантигенным пептидом [20]. Процесс созревания тимоцита оказался связан с анатомическим разделением тимуса на кору (кортекс) и мозговое вещество (медулла), а именно, с различиями между функциями их эпителиальных клеток (thymus epithelial cells, TEC). Кортикальные клетки (cTEC) имеют критически важную для положительной селекции тимоцитов уникальную субъединицу протеасомы β5t, которая отличается от конститутивной β5 и иммунопротеасомной β5i субъединиц [21]. Нокаут гена Psmb11, кодирующего β5t, приводит к уменьшению количества созревающих CD8+ Т-клеток, что может служить указанием на необходимость каких-то определенных молекул MHC класса I – презентируемых аутоэпитопов для прохождения положительной селекции [22, 23]. Клетки медуллы (mTEC) считаются необходимыми для прохождения тимоцитами отрицательной селекции; причем для этого процесса они экспрессируют большое количество генов, в обычных условиях характерных для других органов и тканей. Для тканеспецифических антигенов, кодируемых такими генами, используют аббревиатуру TRA (tissue-restricted antigens), для самого феномена массовой эктопической экспрессии генов в тимусе – PGE (promiscuous gene expression). Заметим, что именно отрицательная селекция принципиально важна для установления соответствия между TCR и узнаваемыми аллелями MHC конкретного организма, так как узнавание консервативных остатков аминокислот, кодируемых многими аллелями MHC, будет приводить к слишком сильному сигналу и апоптозу [24]. Кроме того, отрицательная селекция позволяет получать молекулы TCR, сочетающие специфичность и кросс-реактивность к гомологичным эпитопам, – благодаря умеренной энергии связывания с определенным антигеном [25].
На основании анализа гибридизационных чипов считалось, что в mTEC экспрессируются гены приблизительно 3 тыс. TRA, что трактовали как ключевое отличие этих клеток от cTEC [26, 27]. Позднее показано, что различия не столь радикальны. Среди генов, ответственных за экспрессию TRA, особенно выделяют роль гена AIRE, кодирующего ремодулирующий хроматин белок. Исходно этот ген был открыт в результате скрининга мутаций у пациентов, больных аутоиммунной полиэндокринопатией-кандидозом-эпидермальной дистрофией (APECED) [28]. Это заболевание характеризуется инфильтрацией лимфоцитов в пораженные органы, высоким титром аутореактивных антител и нарушением регуляции иммунной системы, повышающим вероятность развития кандидоза. Белок AIRE содержит участки HSR, SAND (домены для связывания с другими белками) и два PHD-домена (цинковые пальцы) и участвует в сборке ремоделирующего хроматин комплекса, активирующего гены-мишени, по-видимому, в случайных участках генома [29]. Данные по массовому высокопроизводительному секвенированию отдельных популяций и субпопуляций клеток эпителия тимуса показали, что в реальности экспрессия генов TRA характерна и для mTEC, и для cTEC: при пороговом уровне 0.13 миллионных долей, нормированных на килобазу длины гена (FPKM), детектировали транскрипты 19 293 генов в mTEC и 15 198 генов в cTEC [30]. Важно, что на уровне траснскриптома отдельных эпителиальных клеток наблюдали экспрессию лишь небольшого числа дополнительных генов (не более нескольких десятков), что свидетельствует о высокой степени их функциональной гетерогенности с точки зрения отрицательной селекции тимоцитов. Особый интерес с точки зрения изучения такой гетрогенности представляют современные исследования транскриптомов единичных клеток, что позволяет выявлять в тимусе клеточные кластеры, соответствующие стадиям созревания и выполняемым функциям [31]. Высокая экспрессия Aire характерна не только для mTEC, но выявлена в семенниках, надпочечниках и поджелудочной железе [27]. Нокаут мышиного Aire приводил к супрессии примерно 2300–3300 генов в mTEC [30, 32]; при этом число экспрессируемых генов оставалось высоким, из чего можно сделать вывод о существовании альтернативных механизмов, обеспечивающих PGE. В качестве второго специфического активатора PGE у мыши на данный момент рассматривают ген Fezf2, кодирующий содержащий цинковые пальцы белок Fezf2. При кондиционном нокауте этого гена наблюдали повышение концентрации аутореактивных антител и инфильтрацию лимфоцитов в органы, а также снижение уровня экспрессии некоторых TRA в тимусе [33]. Для полноты картины нужно отметить, что Fezf2 экспрессируется преимущественно в переднем мозге, а полный нокаут этого гена у мышат летален в возрасте четырех недель по причине нарушения пищевого поведения и развития гиперактивности [34].
ПОТЕНЦИАЛЬНЫЕ СПОСОБЫ УХОДА АУТОРЕАКТИВНЫХ КЛЕТОК ОТ ОТРИЦАТЕЛЬНОЙ СЕЛЕКЦИИ
Синтез в эпителии тимуса мРНК, кодирующей белок с тканеспецифическим эпитопом, еще не гарантирует встречи между соответствующей АПК и аутореактивным тимоцитом. Во-первых, наборы дополнительно экспрессируемых генов для каждой эпителиальной клетки невелики; доля mTEC, имеющих какой-то конкретный дополнительный белок, варьирует от 0.001 до 0.01. В то же время процесс селекции ограничен по времени: для человека примерно 3‒4 суток на положительную селекцию в кортексе и 5‒6 суток на отрицательную селекцию в медулле, что по расчетам соответствует диапазону от нескольких сотен до тысяч встреченных mTEC [22, 35]. Такое количество mTEC, учитывая неоднородный характер распределения экспрессии генов при PGE, позволяет тимоциту встретить большинство основных потенциально аутоантигенных белков, но далеко не все минорные и тканеспецифические. Во-вторых, PGE какого-либо гена не означает автоматического появления в клетке mTEC всех аутореактивных эпитопов кодируемого им белка – около 95% генов человека подвергается альтернативному сплайсингу, при этом около 85% имеет редко встречающиеся минорные изоформы [36]. Хотя паттерн сплайсинга в эпителии тимуса значительно более разнообразен, чем в других типах эпителия, он покрывает отнюдь не все разнообразие сплайсоформ [37]. Минорные сплайсоформы белков иногда содержат сильные аутоэпитопы, которые могут спровоцировать аутоиммунные заболевания. Так, например, аутоиммунный диабет типа I у части пациентов вызван Т-клетками, узнающими аутореактивные эпитопы белка IA-2, у которого есть полноразмерная сплайсоформа в β‑клетках островков Лангерганса и укороченная сплайсоформа без 13 экзона в тимусе и селезенке [38]. Аналогичная ситуация характерна для целого ряда других альтернативно сплайсируемых генов [39, 40]. Нужно отметить, что альтернативный сплайсинг мРНК не обязательно приводит к дефициту в тимусе более редкой сплайсоформы. Также в силу относительно небольшого числа эффективных Т-клеточных эпитопов две различающиеся сплайсоформы белка могут оказаться идентичными по набору эпитопов. В целом, долю альтернативно сплайсируемых генов, для которых аутореактивные Т-клетки могут избежать отрицательной селекции, можно оценить в 10–15% от числа недостаточно представленных в тимусе генов [41]. Известно, что у здоровых взрослых людей доля CD8+ Т-лимфоцитов, распознающих определенный аутологичный эпитоп, как правило, находится в интервале 1 × 104–106 клеток, что соответствует доле клеток, распознающих чужеродные эпитопы (например, вирусные антигены) [42]. Такое количество аутореактивных эффекторных клеток, тем не менее, не приводит к развитию аутоиммунных заболеваний благодаря постоянно работающим механизмам периферической толерантности.
Т-лимфоцит во время созревания в тимусе может встретиться и с периферическими по происхождению антигенами, которые приносятся туда миграторными дендритными клетками CD8-SIRPα+, плазмоцитоидными дендритными клетками и В-лимфоцитами [43‒45]. Вопрос о том, какие именно АПК, mTEC или дендритные клетки, вносят основной вклад в отрицательную селекцию, на данный момент относится к дискуссионным – для одних моделей показана зависимость удаления аутореактивных лимфоцитов строго от дендритных клеток [46, 47], для других – от mTEC [48, 49]. Интересно, что агрегация молекул MHC класса II в контактных синапсах с тимоцитами характерна для cTEC, но не для mTEC, для которых характерен равномерный паттерн распределения MHC [50]. По-видимому, в случае mTEC в кластеризации MHC нет необходимости, так как процесс PGE обеспечивает довольно высокое содержание антигена на поверхности клетки, хотя вероятность экспрессии конкретного антигена отдельно взятой эпителиальной клеткой не слишком велика. В целом, относительная непродолжительность отрицательной селекции, недостаточная полнота паттерна экспрессии генов mTEC и ограниченный характер сплайсинга в тимусе могут приводить к тому, что на периферию выйдет наивная эффекторная Т-клетка, узнающая какой-то определенный аутоантиген, но не встречавшая его в тимусе.
РЕГУЛЯТОРНЫЕ Т-КЛЕТКИ СПЕЦИФИЧНЫ К АУТОАНТИГЕНАМ
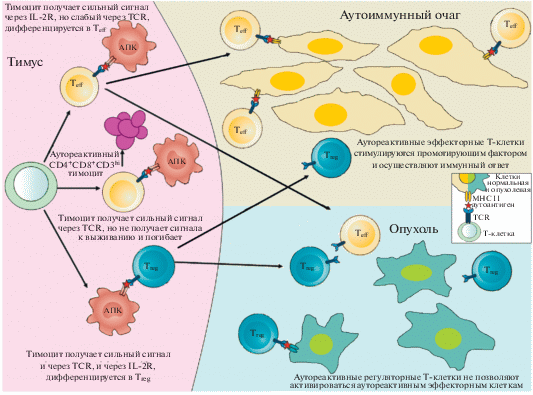
Несмотря на существование отрицательной селекции, распознавание аутологичного эпитопа не обязательно приводит к гибели Т-лимфоцита. Многие такие лимфоциты выходят на периферию и функционируют в качестве естественных регуляторных Т-клеток (Treg). В отношении механизма выбора между клеточной гибелью и дифференцировкой аутореактивного тимоцита в Treg существуют две точки зрения: модель аффинности и модель авидности. В соответствии с первой моделью, регуляторными клетками становятся лимфоциты с промежуточной аффинностью к аутоантигену в составе MHC, а высокоаффинные клетки погибают. Модель авидности предполагает, что главную роль играет авидность и плотность конкретного антигена на MHC в ходе селекции, поэтому высоко представленные антигены могут вызывать гибель среднеаффинных клеток и направлять в Treg-дифференцировку низкоаффинные лимфоциты [43, 51]. Отличительной особенностью Treg считается экспрессия транскрипционного фактора FOXP3, который критически важен для толерантности организма к аутоантигенам. Мутации потери функции в гене этого белка обнаружены у людей, больных аутоиммунным синдромом IPEX (Х-сцепленные утрата иммунорегуляции, полиэндокринопатия и энтеропатия), а также у мышей линии Scurfy, страдающих от анемии, экземы, диареи и погибающих от цитокинового шторма [52]. Иммуносупрессорный эффект Treg обусловлен секрецией противовоспалительных цитокинов IL-10, IL-35, TGFβ и иммуносупрессорного аденозина, конкуренцией с другими лимфоцитами за IL-2, а также контактным сигналингом с АПК при помощи негативных костимуляторных молекул CTLA-4 и TIGIT [53, 54]. Учитывая конформационную динамику молекул MHC и довольно высокую вероятность ухода исходно презентируемого пептида с последующей заменой его на другой [55], можно предположить, что клетки Treg могут каким-либо образом дестабилизировать или модифицировать MHC с аутологичными пептидами, узнаваемыми ими на поверхности АПК. В 2019 году показано, что клетки Treg могут отрывать от дендритных клеток и присваивать себе значительные участки плазматической мембраны, содержащие молекулы MHC класса II с антигеном, к которому специфичен данный тип Treg [56]. Такой механизм позволяет снизить количество антигена, доступного для эффекторного лимфоцита на поверхности АПК, а также установить прямое взаимодействие между двумя аутореактивными клетками – как между регуляторной и эффекторной, так и между двумя регуляторными – за счет того, что клетка Treg после этого может сама презентировать “краденый” аутоантиген.
Помимо образующихся в тимусе естественных клеток Treg, иммунный ответ также ограничивают индуцированные Treg, происходящие из числа периферических эффекторных CD4+-лимфоцитов. Таким образом, различают центральную толерантность к аутоантигенам, обусловленную удалением аутореактивных тимоцитов и образованием естественных Treg в тимусе, и периферическую толерантность, вызываемую подавлением активности эффекторных Т-клеток и иммуносупрессией со стороны индуцированных Treg. Выбор пути дифференцировки тимоцита в эффекторную или регуляторную клетку зависит от интенсивности сигнала, приходящего через TCR, и концентрации доступного IL-2. Если аутореактивный тимоцит при взаимодействии с АПК одновременно получает и достаточно сильный сигнал через TCR, и необходимый уровень IL-2, то он дифференцируется в Treg. Если тимоцит получает необходимое количество IL-2, но интенсивность сигнала от TCR невелика, то он становится обычной CD4+ эффекторной клеткой (Т-хелпером). Если сильный сигнал TCR не подкрепляется сигналом от IL-2, то срабатывает отрицательная селекция и тимоцит уходит в апоптоз [43]. Источником IL-2 может быть сама АПК, а также другие тимоциты, за исключением самих клеток Treg, которые с помощью CD25, специфической высокоаффинной α-cубъединицы рецептора IL-2R, удаляют доступный цитокин из своего микроокружения. В этом случае реализуется своеобразный принцип очереди: первый связавшийся с АПК аутореактивный тимоцит получает оба критических сигнала, дифференцируется в Treg и после этого некоторое время конкурирует с другими тимоцитами той же специфичности, не давая им, возможно, тоже стать регуляторными лимфоцитами. В качестве лимитирующего созревание Treg источника IL-2 рассматривают как дендритные клетки [57], так и сами Т-клетки [58]. В случае дефицита IL-2 роль эрзац-сигнала может выполнять IL-15, также использующий CD132 ‒ общую γ-цепь цитокиновых рецепторов IL-2R, IL-4R, IL-7R, IL-9R, IL-15R и IL-21R; при этом интенсивность сигнала TCR от MHC с аутологичным пептидом лимитируется костимуляторными молекулами CD28/CD80/CD86, в отсутствие которых число Treg значительно снижается [59]. Другим критически важным параметром для созревания Treg служит плотность MHC с антигенным пептидом на поверхности АПК. В то время как высокоаффинные тимоциты имеют слабую зависимость силы сигнала от плотности релевантных MHC, для клеток с низкой аффинностью рецептора критически значима высокая плотность антигена-лиганда [60].
Таким образом, в норме толерантность к собственным антигенам обеспечивается балансом между аутореактивными регуляторными и эффекторными Т-клетками. Образование индуцированных Treg на периферии обусловлено высоким уровнем противовоспалительного TGFβ и ретиноевой кислоты, основным источником которых служат АПК [61, 62]. Кроме того, ограничение активации аутореактивных Т-клеток на периферии может быть связано с эффектом “чувства кворума” у лимфоцитов, при котором после достижения критической плотности клеток с одинаковой специфичностью иммунный ответ запускается гораздо легче за счет локального паракринного усиления активирующего сигнала [63]. Так, недавно показано, что эффект “чувства кворума” работает при дифференцировке CD4+ T-лимфоцитов в клетки памяти [64], нельзя исключить его действие и как супрессора активации аутореактивных Т-лимфоцитов.
АУТОРЕАКТИВНЫЕ Т-ЛИМФОЦИТЫ В НОРМЕ И ПРИ ПАТОЛОГИЯХ
Механизмы периферической толерантности в норме не позволяют аутореактивным эффекторным Т-клеткам запускать иммунный ответ против распознаваемых антигенов собственных тканей [65], а также в случае совместимых аллографтов при трансплантации органов [66]. В случае нарушения такой толерантности становится возможным развитие аутоиммунных заболеваний, причем их следует отличать от аутовоспалительных заболеваний, вызываемых хронической активацией врожденного иммунитета и не зависящих от аутореактивных Т-клеток. К аутовоспалительным заболеваниям относят подагру, спондилоартрит, болезнь Крона и другие воспалительные заболевания кишечника; наследственные заболевания, связанные с мутациями некоторых генов рецепторов цитокинов и паттернов патогенности, такие как семейный псориаз (мутации гена CARD14) или синдром Блау (мутации гена NOD2, симптомы: артрит, дерматит, увеит) [67]. К аутоиммунным заболеваниям относятся рассеянный склероз, системная красная волчанка, синдром Гийена–Барре, аутоиммунные диабет, гепатит, тиреоидит и ряд других.
Весьма часто триггером к развитию аутоиммунных реакций становится инфекционное заболевание, вызванное патогеном, белковые антигены которого схожи по последовательности с определенными белками хозяина (молекулярная мимикрия). Такими инфекционными агентами могут быть вирус Эпштейна–Барр (последовательность эпитопа вирусного белка EBVNA1 сходна с основным белком миелина, MBP), Yersinia enterocolitica (белки Yomp, Ysp и SpyA показывают иммунную кросс-реактивность с тиреотропным рецептором, TSH-R), цитомегаловирус (ДНК-связывающий белок 1 вируса воспроизводит эпитопы декарбоксилазы глутаминовой кислоты, GAD65) и многие другие [68]. На вероятность развития аутоиммунного заболевания также влияет генотип MHC и соответственно спектр презентируемых пептидов [69]; при этом важны наборы аллелей MHC обоих классов, в том числе минорных и вспомогательных генов, таких как HLA-DMA и HLA-DMB [70]. Далеко не всегда для развития аутоиммунного заболевания достаточно промотирующего агента. Так, кардиомиопатия при болезни Шагаса развивается в случае неэффективности работы механизма центральной толерантности. Инфильтрация Trypanosoma cruzi в миокард формирует требуемый для заболевания цитокиновый фон, но далее в патогенезе участвуют CD4+ T-клетки, аутореактивные к α-миозину кардиомиоцитов, но не к антигенам трипаносом [71]. В целом, механизм патогенеза аутоиммунного заболевания следующий: клон аутореактивных Т-лифоцитов проходит через фильтры центральной толерантности (отрицательной селекции), после чего нарушаются механизмы периферической толерантности и смещается баланс между регуляторными и эффекторными клетками в сторону последних [72]. Это чрезвычайно важный баланс – его смещение в обратную сторону способствует выживанию опухолевых клеток.
Многие опухоли отличаются тем, что инфильтрированы множеством аутореактивных лимфоцитов, которые, тем не менее, бездействуют за счет толерогенного микроокружения и периферической толерантности [73]. В норме этот механизм реализуется при контроле иммунного ответа и защите собственных АПК, однако используется и опухолями – для ускользания от иммунного надзора. Для снятия этого эффекта можно использовать моноклональные антитела, которые подавляют передачу иммуносупрессорного сигнала от опухолевых клеток к иммунным и называются “блокаторами иммунологических контрольных точек”. Для такой иммунотерапии в настоящее время применяются антитела, ингибирующие белки CD4+ лимфоцитов (как регуляторных, так и эффекторных) и их опухолевых партнеров. Это CTLA-4 (ипилимумаб), PD-1 (ниволумаб, пембролизумаб), PD-L1 (атезолизумаб, авелумаб, дурвалумаб). Некоторые антитела находятся на стадии клинических испытаний (тислелизумаб), но есть и не прошедшие их “горнило” (тремелимумаб). Также получены антитела (урелумаб и утомилумаб), направленные на прямую активацию аутореактивных CD8+ Т-лимфоцитов, партнер которых CD137 (более известный как 4-1BB) [74]. Еще один подход, который пытались применить для иммунотерапии, состоял в использовании суперантигенов – специальных бактериальных белков, активирующих комплекс TCR/MHC напрямую, без специфического распознавания презентируемого антигена. Но пока успех на этом пути не достигнут, что связано с высоким уровнем неспецифической активации Т-клеток и иммунотоксичностью [75]. В качестве возможного решения этой проблемы предлагается разделить полипептид на две части и осуществить их целенаправленную доставку в опухоль с последующей сборкой in situ функционального иммуноактивирующего суперантигена [76]. Более подробно особенности иммунотерапии опухолей рассмотрены в ряде обзоров, специально посвященных этой теме [77–79]. Отдельного рассмотрения в контексте иммунотерапии опухолей заслуживают неоантигены – новые эпитопы, возникающие вследствие соматических мутаций во время прогрессии опухоли. Для более подробного знакомства с ними можно порекомендовать свежий обзор Schumacher и др. [80], в то же время необходимо отметить, что неоантигены нельзя отождествлять с аутоантигенами в строгом смысле слова, так как они не закодированы в генах зародышевой линии и не могут быть представлены в тимусе.
К сожалению, побочным эффектом иммунотерапии опухолей может стать развитие аутоиммунного заболевания: гепатита, колита, пульмонита, гипофизита, дисфункции почек и щитовидной железы, ревматоидного артрита или миастении гравис [81–83]. Время манифестации аутоиммунного заболевания, его характер и тяжесть зависят от множества трудно прогнозируемых факторов, включая режим терапии и генотип пациента. Так, вероятность развития аутоиммунного колита варьирует от 1.5% для авелумаба до 20% для атезолизумаба, а среднее время проявления симптомов – от 1 месяца после начала терапии ипилимумабом до 5 месяцев при применении ниволумаба, с высоким разбросом этих показателей среди пациентов [84]. С учетом корреляции между эффективностью противоопухолевой терапии и ее побочными эффектами иммунотерапия рассматривается как “наименьшее из зол” в данной ситуации, хотя подбор наиболее эффективного блокатора иммунологических контрольных точек включает в себя и задачу минимизации аутоиммунных последствий. В целом представления о роли аутореактивных Т-лимфоцитов в патогенезе различных заболеваний резюмированы на рис. 1.
БОРЬБА С АУТОРЕАКТИВНОСТЬЮ КАК ЭВОЛЮЦИОННЫЙ ТРЕНД
Возникновение адаптивного иммунитета открыло для позвоночных широкие перспективы для борьбы с распространенными патогенами, однако вместе с тем создало проблему аутоиммунитета. Снижение остроты этой проблемы – эволюционный тренд, благодаря которому, например, произошел переход от кластерной организации перестраиваемых генов иммунных рецепторов к транслоконной. Локус транслоконового типа содержит отдельные кассеты V-, D- и J-сегментов, при этом участие в реаранжировке одного определенного сегмента какого-либо типа не позволяет параллельно использовать другой сегмент того же типа. Более архаичная кластерная организация включает повторяющиеся прототипы генов со всеми сегментами сразу, реаранжировка происходит между этими генами и может идти в нескольких парах одновременно. В этом случае может возникнуть полифункциональный лимфоцит, содержащий несколько перестроенных генов в одном локусе и несколько комбинаторных сочетаний полипептидных цепей, что повышает вероятность аутореактивности получающихся рецепторов [85]. Среди современных животных кластерная организация характерна для локусов, кодирующих антитела рыб, но не характерна для локусов, кодирующих TCR во всех известных классах позвоночных [86]. Единственное исключение – кластерный локус NAR-TRD акул, который кодирует как δ-цепь TCR, так и специфические однодоменные антитела. Однако в данном случае однодоменное устройство антител и TCR исключает риск создания химерного рецептора за счет комбинации полипептидных цепей. Во всех прочих случаях запрет на кластерную организацию обычных локусов TCR соблюдается.
По-видимому, присутствие в организме аутореактивных Т-лимфоцитов гораздо более опасно, чем аутоиммунных B-лимфоцитов (наличие 30% аутореактивных В-лимфоцитов от общего числа В-лимфоцитов не приводит к патологическим процессам) [87, 88]. Как показано при изучении репертуара антител больных с IPEX-синдромом, контроль над аутореактивными В-клетками осуществляют клетки Тreg, без нормального функционирования которых доля потомков аутореактивных клонов на периферии значительно превышает долю наивных В-лимфоцитов в костном мозге [89]. Еще один механизм страхования от появления высокоаффинных аутореактивных Т-лимфоцитов обусловлен тем, что они, в отличие от В-лимфоцитов, не способны к соматическому гипермутагенезу, а значит, не могут изменить исходную специфичность [90]. Единственным исключением из этого “правила” оказались опять акулы – недавно у них обнаружен соматический гипермутагенез генов α-, γ- и δ-цепей TCR [91].
ЗАКЛЮЧЕНИЕ
В целом, наличие нормально работающих механизмов дифференцировки аутореактивных Treg и отрицательной селекции аутореактивных тимоцитов позволяет человеку и другим млекопитающим держать под контролем весь адаптивный иммунитет. Аутоиммунные заболевания в таких условиях возможны либо вследствие глобальной поломки какого-либо ключевого компонента этой системы, например, генов AIRE в случае APECED и FOXP3 в случае IPEX, либо при “неудачном стечении обстоятельств” – сочетании сразу нескольких факторов: неблагоприятного генотипа по генам MHC, промотирующего инфекционного агента, случайного ухода аутореактивных Т-клеток от отрицательной селекции и др. Кроме того, иммунотерапия опухолей, создающих толерогенное микроокружение, сопровождается нарушением системы периферической толерантности и активацией аутореактивных эффекторных клеток, что может приводить к усилению аутоиммунных процессов. В эволюционном же контексте иммунная система человека выглядит гораздо более защищенной от аутореактивных Т-лимфоцитов по сравнению с иммунной системой хрящевых рыб.
Работа поддержана грантом РФФИ № 16-04-01188.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Автор заявляет об отсутствии конфликта интересов.
Список литературы
Adams E.J., Gu S., Luoma A.M. (2015) Human gamma delta T cells: evolution and ligand recognition. Cell. Immunol. 296(1), 31–40.
Champagne E. (2011) γδ T cell receptor ligands and modes of antigen recognition. Arch. Immunol. Ther. Exp. (Warsz.). 59(2), 117–137.
Carding S.R., Kyes S., Jenkinson E.J., Kingston R., Bottomly K., Owen J.J., Hayday A.C. (1990) Developmentally regulated fetal thymic and extrathymic T-cell receptor gamma delta gene expression. Genes Dev. 4, 1304–1315.
Groh V., Steinle A., Bauer S., Spies T. (1998) Recognition of stress-induced MHC molecules by intestinal epithelial gamma delta T cells. Science. 279(5357), 1737–1740.
Scotet E., Martinez L.O., Grant E., Barbaras R., Jenö P., Guiraud M., Monsarrat B., Saulquin X., Maillet S., Estève J.P., Lopez F., Perret B., Collet X., Bonneville M., Champagne E. (2005) Tumor recognition following Vgamma9Vdelta2 T cell receptor interactions with a surface F1-ATPase-related structure and apolipoprotein A-I. Immunity. 22(1), 71–80.
Zhang L., Jin N., Nakayama M., O’Brien R.L., Eisenbarth G.S., Born W.K. (2010) Gamma delta T cell receptors confer autonomous responsiveness to the insulin-peptide B:9-23. J. Autoimmun. 34(4), 478–484.
O’Brien R.L., Fu Y.X., Cranfill R., Dallas A., Ellis C., Reardon C., Lang J., Carding S.R., Kubo R., Born W. (1992) Heat shock protein Hsp60-reactive gamma delta cells: a large, diversified T-lymphocyte subset with highly focused specificity. Proc. Natl. Acad. Sci. USA. 89(10), 4348–4352
Born W.K., Vollmer M., Reardon C., Matsuura E., Voelker D.R., Giclas P.C., O’Brien R.L. (2003) Hybridomas expressing gamma delta T-cell receptors respond to cardiolipin and beta2-glycoprotein 1 (apolipoprotein H). Scand. J. Immunol. 58(3), 374–381.
Kong Y., Cao W., Xi X., Ma C., Cui L., He W. (2009) The NKG2D ligand ULBP4 binds to TCRgamma9/ delta2 and induces cytotoxicity to tumor cells through both TCR gamma delta and NKG2D. Blood. 114(2), 310–317.
Roark C.L., French J.D., Taylor M.A., Bendele A.M., Born W.K., O’Brien R.L. (2007) Exacerbation of collagen-induced arthritis by oligoclonal, IL-17-producing gamma delta T cells. J. Immunol. 179, 5576–5583.
Markle J.G., Mortin-Toth S., Wong A.S., Geng L., Hayday A., Danska J. S. (2013) T cells are essential effectors of type 1 diabetes in the nonobese diabetic mouse model. J. Immunol. 190, 5392–5401.
Harrison LC, Dempsey-Collier M, Kramer DR, Takahashi K. (1996) Aerosol insulin induces regulatory CD8 gamma delta T cells that prevent murine insulin-dependent diabetes. J. Exp. Med. 184(6), 2167–2174.
Ponomarev E.D., Dittel B.N. (2005) Gamma delta T cells regulate the extent and duration of inflammation in the central nervous system by a Fas ligand-dependent mechanism. J. Immunol. 174, 4678–4687.
Paul S., Shilpi, Lal G. Role of gamma-delta (γδ) T cells in autoimmunity. (2015) J. Leukoc. Biol. 97(2), 259–271.
Legut M., Cole D.K., Sewell A.K. (2015) The promise of γδ T cells and the γδ T cell receptor for cancer immunotherapy. Cell. Mol. Immunol. 12(6), 656–668.
Oberg H.H., Peipp M., Kellner C., Sebens S., Krause S., Petrick D., Adam-Klages S., Röcken C., Becker T., Vogel I., Weisner D., Freitag-Wolf S., Gramatzki M., Kabelitz D., Wesch D. (2014) Novel bispecific antibodies increase γδ T-cell cytotoxicity against pancreatic cancer cells. Cancer Res. 74(5), 1349–1360.
Deniger D.C., Switzer K., Mi T., Maiti S., Hurton L., Singh H., Huls H., Olivares S., Lee D.A., Champlin R.E., Cooper L.J. (2013) Bispecific T-cells expressing polyclonal repertoire of endogenous γδ T-cell receptors and introduced CD19-specific chimeric antigen receptor. Mol. Ther. 21(3), 638–647.
Kappler JW, Roehm N, Marrack P. (1987) T cell tole-rance by clonal elimination in the thymus. Cell. 49(2), 273–280.
Salaün J., Bandeira A., Khazaal I., Calman F., Coltey M., Coutinho A., Le Douarin N.M. (1990) Thymic epithelium tolerizes for histocompatibility antigens. Science. 247(4949 Pt 1), 1471–1474.
Surh C.D., Sprent J. (1994) T-cell apoptosis detected in situ during positive and negative selection in the thymus. Nature. 372(6501), 100–103.
Murata S., Sasaki K., Kishimoto T., Niwa S., Hayashi H., Takahama Y., Tanaka K. (2007) Regulation of CD8+ T cell development by thymus-specific proteasomes. Science. 316(5829), 1349–1353.
Klein L., Kyewski B., Allen P.M., Hogquist K.A. (2014) Positive and negative selection of the T cell repertoire: what thymocytes see (and don’t see). Nat. Rev. Immunol. 14, 377–391.
Kincaid E.Z., Murata S., Tanaka K., Rock K.L. (2016) Specialized proteasome subunits have an essential role in the thymic selection of CD8(+) T cells. Nat. Immunol. 17(8), 938–945. https://doi.org/10.1038/ni.3480
Huseby E.S., Crawford F., White J., Marrack P., Kappler J.W. (2006) Interface-disrupting amino acids establish specificity between T cell receptors and complexes of major histocompatibility complex and peptide. Nat. Immunol. 7(11), 1191–1199.
Kosmrlj A., Jha A.K., Huseby E.S., Kardar M., Chakraborty A.K. (2008) How the thymus designs antigen-specific and self-tolerant T cell receptor sequences. Proc. Natl. Acad. Sci. USA. 105(43), 16671–16676.
Kyewski B., Derbinski J. (2004) Self-representation in the thymus: an extended view. Nat. Rev. Immunol. 4(9), 688–698.
Derbinski J., Gabler J., Brors B., Tierling S., Jonnakuty S., Hergenhahn M., Peltonen L., Walter J., Kyewski B. (2005) Promiscuous gene expression in thymic epithelial cells is regulated at multiple levels. J. Exp. Med. 202, 33–45.
Bjorses P., Aaltonen J., Horelli-Kuitunen N., Yaspo M.L., Peltonen L. (1998) Gene defect behind APECED: a new clue to autoimmunity. Hum. Mol. Genet. 7(10), 1547–1553.
Passos G.A., Speck-Hernandez C.A., Assis A.F., Mendes-da-Cruz D.A. (2018) Update on Aire and thymic negative selection. Immunology. 153(1), 10–20.
Sansom S.N., Shikama-Dorn N., Zhanybekova S., Nusspaumer G., Macaulay I.C., Deadman M.E., Heger A., Ponting C.P., Hollander G.A. (2014) Population and single-cell genomics reveal the Aire dependency, relief from Polycomb silencing, and distribution of self-antigen expression in thymic epithelia. Genome Res. 24(12), 1918–1931.
Kernfeld E.M., Genga R.M.J., Neherin K., Magaletta M.E., Xu P., Maehr R. (2018) A single-cell transcriptomic atlas of thymus organogenesis resolves cell types and developmental maturation. Immunity. 48(6), 1258–1270.
St-Pierre C., Trofimov A., Brochu S., Lemieux S., Perreault C. (2015) Differential features of AIRE-induced and AIRE-independent promiscuous gene expression in thymic epithelial cells. J. Immunol. 195, 498–506.
Takaba H., Morishita Y., Tomofuji Y., Danks L., Nitta T., Komatsu N., Kodama T., Takayanagi H. (2015) Fezf2 orchestrates a thymic program of self-antigen expression for immune tolerance. Cell. 163(4), 975–987.
Hirata T., Suda Y., Nakao K., Narimatsu M., Hirano T., Hibi M. (2004) Zinc finger gene fez-like functions in the formation of subplate neurons and thalamocortical axons. Dev. Dyn. 230(3), 546–556.
Le Borgne M., Ladi E., Dzhagalov I., Herzmark P., Liao Y.F., Chakraborty A.K., Robey E.A. (2009) The impact of negative selection on thymocyte migration in the medulla. Nat. Immunol. 10(8), 823–830.
Burge C.B. (2008) Alternative isoform regulation in human tissue transcriptomes. Nature. 456(7221), 470–476.
Keane P., Ceredig R., Seoighe C. (2015) Promiscuous mRNA splicing under the control of AIRE in medullary thymic epithelial cells. Bioinformatics. 31(7), 986–990.
Diez J., Park Y., Zeller M., Brown D., Garza D., Ricordi C., Hutton J., Eisenbarth G.S., Pugliese A. (2001) Differential splicing of the IA-2 mRNA in pancreas and lymphoid organs as a permissive genetic mechanism for autoimmunity against the IA-2 type 1 diabetes autoantigen. Diabetes. 50(4), 895–900.
Dogra R.S., Vaidyanathan P., Prabakar K.R., Marshall K.E., Hutton J.C., Pugliese A. (2006) Alternative splicing of G6PC2, the gene coding for the islet-specific glucose-6-phosphatase catalytic subunit-related protein (IGRP), results in differential expression in human thymus and spleen compared with pancreas. Diabetologia. 49(5), 953–957.
Ng B., Yang F., Huston D.P., Yan Y., Yang Y., Xiong Z., Peterson L.E., Wang H., Yang X.F. (2004) Increased noncanonical splicing of autoantigen transcripts provides the structural basis for expression of untolerized epitopes. J. Allergy Clin. Immunol. 114(6), 1463–1470.
Шилов Е.С., Горшкова Е.А., Миннегалиева А.Р., Поташникова Д.М. (2019) Паттерн сплайсинга мРНК в клетках эпителия тимуса ограничивает транскриптом, доступный для отрицательной селекции аутореактивных Т-лимфоцитов. Молекуляр. биология. 53, 109–119.
Yu W., Jiang N., Ebert P.J., Kidd B.A., Müller S., Lund P.J., Juang J., Adachi K., Tse T., Birnbaum M.E., Newell E.W., Wilson D.M., Grotenbreg G.M., Valitutti S., Quake S.R., Davis M.M. (2015) Clonal deletion prunes but does not eliminate self-specific αβ CD8(+) T lymphocytes. Immunity. 42(5), 929–941.
Klein L., Robey E.A., Hsieh C.S. (2019) Central CD4+ T cell tolerance: deletion versus regulatory T cell diffe-rentiation. Nat. Rev. Immunol. 19(1), 7–18.
Bonasio R., Scimone M.L., Schaerli P., Grabie N., Lichtman A.H., von Andrian U.H. (2006) Clonal deletion of thymocytes by circulating dendritic cells homing to the thymus. Nat. Immunol. 7(10), 1092–1100.
Oh J., Shin J.S. (2015) The role of dendritic cells in central tolerance. Immune Netw. 15(3), 111–120.
Brocker T. (1999) The role of dendritic cells in T cell selection and survival. J. Leukoc. Biol. 66(2), 331–335.
Proietto A.I., van Dommelen S., Zhou P., Rizzitelli A., D’Amico A., Steptoe R.J., Naik S.H., Lahoud M.H., Liu Y., Zheng P., Shortman K., Wu L. (2008) Dendritic cells in the thymus contribute to T-regulatory cell induction. Proc. Natl. Acad. Sci. USA. 105(50), 19869–19874.
Hinterberger M., Aichinger M., Prazeres da Costa O., Voehringer D., Hoffmann R., Klein L. (2010) Autonomous role of medullary thymic epithelial cells in central CD4(+) T cell tolerance. Nat. Immunol. 11(6), 512–519.
Aschenbrenner K., D’Cruz L.M., Vollmann E.H., Hinterberger M., Emmerich J., Swee L.K., Rolink A., Klein L. (2007) Selection of Foxp3+ regulatory T cells specific for self antigen expressed and presented by Aire+ medullary thymic epithelial cells. Nat. Immunol. 8(4), 351–358.
Yang S.J., Ahn S., Park C.S., Holmes K.L., Westrup J., Chang C.H., Kim M.G. (2006) The quantitative assessment of MHC II on thymic epithelium: implications in cortical thymocyte development. Int. Immunol. 18(5), 729–739.
Relland L.M., Mishra M.K., Haribhai D., Edwards B., Ziegelbauer J., Williams C.B. (2009) Affinity-based selection of regulatory T cells occurs independent of agonist-mediated induction of Foxp3 expression. J. Immunol. 182(3), 1341–350.
Patel D.D. (2001) Escape from tolerance in the human X-linked autoimmunity-allergic disregulation syndrome and the Scurfy mouse. J. Clin. Invest. 107(2), 155–157.
Wing K., Onishi Y., Prieto-Martin P., Yamaguchi T., Miyara M., Fehervari Z., Nomura T., Sakaguchi S. (2008) CTLA-4 control over Foxp3+ regulatory T cell function. Science. 322(5899), 271–275.
Joller N., Hafler J.P., Brynedal B., Kassam N., Spoerl S., Levin S.D., Sharpe A.H., Kuchroo V.K. (2011) Cutting edge: TIGIT has T cell-intrinsic inhibitory functions. J. Immunol. 186(3), 1338–1342.
Wieczorek M., Abualrous E.T., Sticht J., Álvaro-Benito M., Stolzenberg S., Noé F., Freund C. (2017) Major histocompatibility complex (MHC) class I and MHC class II proteins: conformational plasticity in antigen presentation. Front. Immunol. 8, 292.
Akkaya B., Oya Y., Akkaya M., Al Souz J., Holstein A.H., Kamenyeva O., Kabat J., Matsumura R., Dorward D.W., Glass D.D., Shevach E.M. (2019) Regulatory T cells mediate specific suppression by depleting peptide-MHC class II from dendritic cells. Nat. Immunol. 20(2), 218–231.
Weist B.M., Kurd N., Boussier J., Chan S.W., Robey E.A. (2015) Thymic regulatory T cell niche size is dictated by limiting IL-2 from antigen-bearing dendritic cells and feedback competition. Nat. Immunol. 16(6), 635–641.
Owen D.L., Mahmud S.A., Vang K.B., Kelly R.M., Blazar B.R., Smith K.A., Farrar M.A. (2018) Identification of cellular sources of IL-2 needed for regulatory T cell development and homeostasis. J. Immunol. 200(12), 3926–3933.
Salomon B., Lenschow D.J., Rhee L., Ashourian N., Singh B., Sharpe A., Bluestone J.A. (2000) B7/CD28 costimulation is essential for the homeostasis of the CD4+CD25+ immunoregulatory T cells that control autoimmune diabetes. Immunity. 12(4), 431–440.
Martinic M.M., Rocha B., McCoy K.D., Hengartner H., Zinkernagel R.M. (2004) Role of TCR-restricting MHC density and thymic environment on selection and survival of cells expressing low-affinity T cell receptors. Eur. J. Immunol. 34(4), 1041–1049.
Luo X., Tarbell K.V., Yang H., Pothoven K., Bailey S.L., Ding R., Steinman R.M., Suthanthiran M. (2007) Dendritic cells with TGF-β1 differentiate naive CD4+CD25− T cells into islet-protective Foxp3+ regulatory T cells. Proc. Natl. Acad. Sci. USA. 104, 2821–2826.
Yamazaki S., Dudziak D., Heidkamp G.F., Fiorese C., Bonito A.J., Inaba K., Nussenzweig M.C., Steinman R.M. (2008) CD8+CD205+ splenic dendritic cells are specialized to induce Foxp3+ regulatory T cells. J. Immunol. 181, 6923–6933.
Butler T.C., Kardar M., Chakraborty A.K. (2013) Quorum sensing allows T cells to discriminate between self and nonself. Proc. Natl. Acad. Sci. USA. 110(29), 11833–11838.
Polonsky M., Rimer J., Kern-Perets A., Zaretsky I., Miller S., Bornstein C., David E., Kopelman N.M., Stelzer G., Porat Z., Chain B., Friedman N. (2018) Induction of CD4 T cell memory by local cellular collectivity. Science. 360(6394). pii: eaaj1853. https://doi.org/10.1126/science.aaj1853
Sakaguchi S., Yamaguchi T., Nomura T., Ono M. (2008) Regulatory T cells and immune tolerance. Cell. 133(5), 775–787.
Walsh P.T., Taylor D.K., Turka L.A. (2004) Tregs and transplantation tolerance. J. Clin. Invest. 114(10), 1398–1403.
Park H., Bourla A.B., Kastner D.L., Colbert R.A., Siegel R.M. (2012) Lighting the fires within: the cell bio-logy of autoinflammatory diseases. Nat. Rev. Immunol. 12(8), 570–580.
Rojas M., Restrepo-Jiménez P., Monsalve D.M., Pacheco Y., Acosta-Ampudia Y., Ramírez-Santana C., Leung P.S.C., Ansari A.A., Gershwin M.E., Anaya J.M. (2018) Molecular mimicry and autoimmunity. J. Autoimmun. 95, 100–123.
Trowsdale J. (2011) The MHC, disease and selection. Immunol. Lett. 137(1‒2), 1–8.
Alvaro-Benito M., Morrison E., Wieczorek M., Sticht J., Freund C. (2016) Human leukocyte antigen-DM polymorphisms in autoimmune diseases. Open Biol. 6(8), pii: 160165. https://doi.org/10.1098/rsob.160165
Lv H., Lipes M.A. (2012) Role of impaired central tolerance to α-myosin in inflammatory heart disease. Trends Cardiovasc. Med. 22(5), 113–117.
Kitz A., Singer E., Hafler D. (2018) Regulatory T cells: from discovery to autoimmunity. Cold Spring Harb. Perspect. Med. 8(12). pii: a029041. https://doi.org/10.1101/cshperspect.a029041
Bogolyubova A.V., Belousov P.V. (2016) Inflammatory immune infiltration in human tumors: role in pathogenesis and prognostic and diagnostic value. Biochemistry (Mosc.). 81(11), 1261–1273.
Chester C., Sanmamed M.F., Wang J., Melero I. (2018) Immunotherapy targeting 4-1BB: mechanistic rationale, clinical results, and future strategies. Blood. 131(1), 49–57.
Brodin T.N., Persson R., Soegaard M., Ohlsson L., D’Argy R., Olsson J., Molander A., Antonsson P., Gunnarsson P O., Kallan T., Dohlsten M. (1998) Man-made superantigens: tumor-selective agents for T-cell-based therapy. Adv. Drug Deliv. 31, 131–142.
Golob-Urbanc A., Rajcevic U., Strmsek Z., Jerala R. (2019) Design of split superantigen fusion proteins for cancer immunotherapy. J. Biol. Chem. pii: jbc.RA118.006742. https://doi.org/10.1074/jbc.RA118.006742
Sharma P., Allison J.P. (2015) Immune checkpoint targeting in cancer therapy: towards combination strategies with curative potential. Cell. 161(2), 205–214.
Bogolyubova A.V., Efimov G.A., Drutskaya M.S., Nedospasov S.A. (2015) Cancer immunotherapy based on the blockade of immune chekpoints. Med. Immunol. (Russia). 17(5), 395–406.
Khair D.O., Bax H.J., Mele S., Crescioli S., Pellizzari G., Khiabany A., Nakamura M., Harris R.J., French E., Hoffmann R.M., Williams I.P., Cheung A., Thair B., et al. (2019) Combining immune checkpoint inhibitors: established and emerging targets and strategies to improve outcomes in melanoma. Front. Immunol. 10, e453. https://doi.org/10.3389/fimmu.2019.00453
Schumacher T.N., Scheper W., Kvistborg P. (2019) Cancer neoantigens. Annu. Rev. Immunol. 37, 173–200.
Makarious D., Horwood K., Coward J.I.G. (2017) Myasthenia gravis: an emerging toxicity of immune checkpoint inhibitors. Eur. J. Cancer. 82, 128–136.
Lidar M., Giat E., Garelick D., Horowitz Y., Amital H., Steinberg-Silman Y., Schachter J., Shapira-Frommer R., Markel G. (2018) Rheumatic manifestations among cancer patients treated with immune checkpoint inhibitors. Autoimmun. Rev. 17(3), 284–289.
Cukier P., Santini F.C., Scaranti M., Hoff A.O. (2017) Endocrine side effects of cancer immunotherapy. Endocr. Relat. Cancer. 24(12), 331–347.
Sriratana P., Norton J. (2018) New immunotherapies in oncology treatment and their side effect profiles. J. Am. Board Fam. Med. 31(4), 620–627.
Shilov E.S., Kuprash D.V. (2016) Genetic mechanisms of adaptive immunity emergence in vertebrates. Russ. J. Genetics. 52(7), 664–675.
Flajnik M.F., Kasahara M. (2010) Origin and evolution of the adaptive immune system: genetic events and selective pressures, Nat. Rev. Genet. 11(1), 47–59.
Wardemann H., Yurasov S., Schaefer A., Young J.W., Meffre E., Nussenzweig M.C. (2003) Predominant autoantibody production by early human B cell precursors. Science. 301(5638), 1374–1377.
Zikherman J., Parameswaran R., Weiss A. (2012) Endogenous antigen tunes the responsiveness of naive B cells but not T cells. Nature. 489(7414), 160–164.
Kinnunen T., Chamberlain N., Morbach H., Choi J., Kim S., Craft J., Mayer L., Cancrini C., Passerini L., Bacchetta R., Ochs H.D., Torgerson T.R., Meffre E. (2013) Accumulation of peripheral autoreactive B cells in the absence of functional human regulatory T cells. Blood. 121(9), 1595–1603.
Bachl J., Wabl M. (1995) Hypermutation in T cells questioned. Nature. 375(6529), 285–286.
Ott J.A., Castro C.D., Deiss T.C., Ohta Y., Flajnik M.F., Criscitiello M.F. (2018) Somatic hypermutation of T cell receptor α chain contributes to selection in nurse shark thymus. eLife. 7, e28477. https://doi.org/10.7554/eLife.28477
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология