

Молекулярная биология, 2019, T. 53, № 6, стр. 954-967
Мутантная РНК: роль в патогенезе болезни Гентингтона и других полиглутаминовых заболеваний
А. Н. Богомазова a, *, А. В. Еремеев a, Г. Е. Позмогова a, М. А. Лагарькова a, **
a Федеральный научный клинический центр физико-химической медицины
Федерального медико-биологического агентства России
119435 Москва, Россия
* E-mail: abogomazova@rcpcm.org
** E-mail: lagar@rcpcm.org
Поступила в редакцию 04.06.2019
После доработки 04.06.2019
Принята к публикации 18.06.2019
Аннотация
Полиглутаминовые заболевания ‒ редкие наследственные нейродегенеративные патологии, обусловленные экспансией тринуклеотидных CAG-повторов в кодирующей части определенных генов. Экспансия CAG-повторов приводит к появлению в клетках мРНК с аномально протяженными участками CAG-триплетов (mCAG-РНК) и белков со сверхдлинными полиглутаминовыми (PolyQ) участками, по которым эти патологии были названы полиглутаминовыми заболеваниями, или PolyQ болезнями. Всего описано девять PolyQ заболеваний: болезнь (хорея) Гентингтона (HD), дентаторубро-паллидолюисовая атрофия (DRPA), спино-бульбарная мышечная атрофия (SBMA), а также шесть различных типов спиноцеребеллярной атаксии (SCA 1, 2, 3, 6, 7 и 17). PolyQ болезни приводят к серьезнейшим, неуклонно прогрессирующим нарушениям в нервной и/или мышечной системах, а подходы к их успешной терапии на сегодняшний день не разработаны. За последние годы убедительно показано, что в патологическом процессе при PolyQ заболеваниях может активно участвовать mCAG-РНК. Мутантная РНК вовлечена в различные молекулярные механизмы, приводящие к нарушению транскрипции, сплайсинга, трансляции, структуры цитозоля, транспорта РНК из ядра в цитоплазму и, в конечном итоге, к нейродегенерации. В этом обзоре рассмотрено участие мутантной mCAG-РНК в процессах нейродегенерации при PolyQ заболеваниях.
ВВЕДЕНИЕ
Полиглутаминовым (PolyQ) заболеваниям присущи такие общие признаки, как гибель нейронов, обратная корреляция между числом CAG-повторов и возрастом проявления симптомов, нестабильность числа CAG-повторов в поколениях и склонность белковых продуктов к образованию больших внутриклеточных агрегатов. Все PolyQ заболевания ‒ это аутосомно-доминантные патологии, за исключением Х-сцепленной спино-бульбарной мышечной атрофии (SBMA), которая поражает исключительно взрослых мужчин. Следует отметить, что хотя PolyQ болезни имеют сходные характеристики, однако их неврологические проявления различны, так как при каждой из них преимущественной дегенерации подвергаются разные типы нейронов (табл. 1).
Таблица 1.
Основные характеристики PolyQ болезней
| Название, #OMIM |
Ген. Число CAG-повторов в норме (wt) и патологии (m) | Клетки ЦНС, подверженные дегенерации, и патоморфологические проявления | Другие соматические патологические проявления |
|---|---|---|---|
| Болезнь
Гентингтона* #143100 |
HTT wtHTT: CAG11–34 mHTT: CAG36–121 |
Утрата нейронов в полосатом теле, коре головного мозга, таламусе и ядрах гипоталамуса, утрата до 95% ГАМКергических нейронов в чечевичном теле и черной субстанции | Сердечно-сосудистая недостаточность; остеопороз и скелетно-мышечная атрофия; нарушение толерантности к глюкозе, гипотиреоз; снижение уровня тестостерона у мужчин и тестикулярная атрофия; снижение массы тела |
| DRPA** #125370 |
ATN1 wtATN1: CAG7–25 mATN1: CAG≥48 |
Атрофические изменения клеток мозжечка, ствола мозга и больших полушарий, очаги демиелинизации в белом веществе перивентрикулярной области и овального центра больших полушарий | Очень редкая форма. Данное заболевание является популяционно-специфичным и встречается почти исключительно в Японии (в Европе и Северной Америке описаны лишь единичные случаи DRPA) |
| SBMA* #313200 |
AR wtAR: CAG11–31 mAR: CAG40–62 |
Утрата нижних моторных нейронов в переднем роге спинного мозга, а также в моторных ядрах ствола мозга и спинальных ганглиях | Мышечная дистрофия, связанная с нарушением формирования волокон, дистрофические поражения мужских половых желез, почек |
| SCA1* #164400 |
ATXN1 wtATXN1: CAG6–39 mATNX1: CAG41–81 |
Дегенерация коры мозжечка, в первую очередь, за счет утраты клеток Пуркинье, демиелинизация белого вещества мозжечка, дегенерация нижних олив, ядер и поперечных волокон моста мозга, ядер каудального ствола. В процесс могут вовлекаться также проводники спинного мозга, зрительный нерв | Миодистрофия скелетных мышц и сердца; тазовые расстройства |
| SCA2* #183090 |
ATXN2 wtATXN2: CAG13–31 mATXN2: CAG32–79 |
Утрата клеток Пуркинье, клеток нижних олив, ядер моста и черной субстанции, дегенерация волокон моста, задних столбов и, в меньшей степени, спиноцеребеллярных участков; в ряде случаев изменения затрагивают клетки передних рогов спинного мозга, спинальные корешки, лобную и височную кору | Миодистрофия скелетных, глазодвигательных мышц, мочеполовой системы (недержание мочи) |
| Болезнь Мачадо‒Джозефа* (SCA3) #109150 |
ATXN3 wtATXN3: CAG12–44 m ATXN3: CAG45–86 |
Дегенерация нейронов в зубчатых ядрах, верхней и средней ножках мозжечка, черной субстанции, красных и субталамических ядрах, ядрах черепных нервов, передних рогов и спиноцеребеллярных участков спинного мозга | Дисфункции тазовых органов, миодистрофия скелетных, глазодвигательных мышц |
| SCA6* #183086 |
CACNA1A wtCACNA1A: CAG4–18 mCACNA1A:CAG19–33 |
Утрата клеток Пуркинье в мозжечке, особенно в верхних и задних отделах червя; характерна относительная сохранность нижних олив, вовлекаемых в процесс вторично | Миодистрофия скелетных мышц и сердца, языкоглоточного аппарата, желудочно-кишечного тракта |
| SCA7** #164500 |
ATXN7 wtATXN7: CAG<35 mATXN7: CAG37–300 |
Утрата клеток Пуркинье в мозжечке, утрата мотонейронов ядер моста, зубчатых ядер и ядер нижней оливы, атрофия спиноцеребральных и пирамидных участков, атрофия миелина в белом веществе мозжечка, зрительного нерва | Миодистрофия скелетных мышц, конусных фоторецепторов, макулодистрофия (сосудистого и пигментного слоев), поражения мембраны Бранча, поражения сфинктеров, гепатомегалия и сердечная недостаточность в детском возрасте и при тяжелом течении |
| SCA17** #607136 |
TBP wtTBP: CAG25–42 mTBP: CAG48–66 |
Утрата клеток Пуркинье в мозжечке; атрофия мозжечка и менее выраженная атрофия больших полушарий мозга; гибель нейронов и глиоз в области хвостатого ядра, скорлупы, таламуса, нижних олив, лобной и височной коры | Сопутствующие патологии органов сходны с болезнью Гентингтона |
* По данным монографии Nóbrega C., Almeida L Polyglutamine Disorders. Изд-во: “Springer”, 2018, c. 458. ** По данным web-сайта https://medicalplanet.su/neurology/ataksii.html.
До сих пор нет полного понимания молекулярных механизмов патогенеза PolyQ заболеваний. Следует отметить, что наибольший интерес исследователей всегда вызывали токсические эффекты PolyQ белков, особенно их внутриклеточных агрегатов. Например, известно, что при болезни Гентингтона мутантный белок HTT (mHTT) нарушает аутофагию, везикулярный транспорт, передачу нейротрансмиттеров и функции митохондрий. Однако в последнее время стало ясно, что в патологическом процессе может активно участвовать mCAG-РНК (рис. 1 ). Вторичные структуры, образуемые избыточными повторами в РНК, влияют на транскрипцию и сплайсинг, вызывая глобальные изменения транскриптома клеток. Они способны нарушать трансляцию, что приводит к появлению нефункциональных или токсичных белковых продуктов. Вторичные структуры и длинные повторы провоцируют также образование внутриядерных фокальных скоплений аномальной РНК.
Рис. 1.
Молекулярные механизмы возможного участия мутантной РНК в патогенезе полиглутаминовых заболеваний.
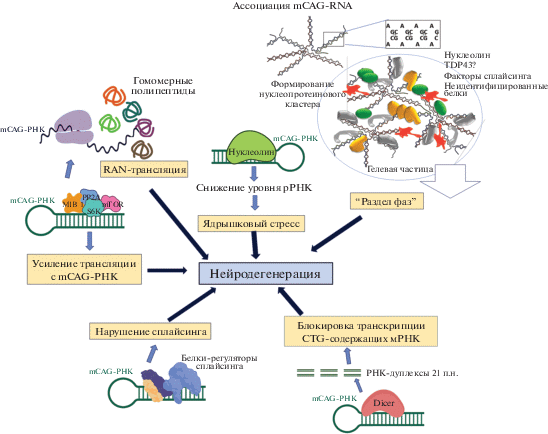
В этом обзоре рассмотрены вопросы участия mCAG-РНК в процессах нейродегенерации при PolyQ заболеваниях. Интерес к mCAG-РНК вызван не только ее возможным участием в патогенезе, но и тем, что одним из наиболее перспективных подходов к терапии PolyQ заболеваний считается применение антисмысловых олигонуклеотидов (ASO), комплементарных mCAG-РНК [1‒3]. Более подробно будет рассмотрена болезнь Гентингтона ‒ наиболее изученное PolyQ заболевание.
ГЕН HTT ПРИ БОЛЕЗНИ ГЕНТИНГТОНА И ОСОБЕННОСТИ ЕГО ТРАНСКРИПЦИИ
Болезнь Гентингтона вызвана экспансией тринуклеотидных CAG-повторов в первом экзоне гена HTT. Этот ген находится на коротком плече хромосомы 4 (4p16.3), содержит 67 экзонов и кодирует белок размером примерно 350 кДа. Ген HTT экспрессируется во всех типах клеток на примерно одинаковом уровне [4], хотя клетки головного мозга содержат несколько больше НТТ. Экспансия CAG-триплетов в первом экзоне гена HTT приводит к дегенерации полосатого тела головного мозга и проявляет себя как аутосомно-доминантная мутация с приобретением новых патологических функций (gain-of-function mutation). Если число CAG-повторов в гене НТТ превышает 40, то эта мутация обладает 100%-ной пенетрантностью. Чем больше протяженность участка CAG-повторов, тем раньше появляются признаки болезни. Если число повторов незначительно превышает 40, то первые клинические симптомы диагностируют, как правило, после 40‒50 лет. При числе повторов более 70 развивается ювенильная форма болезни Гентингтона с первыми симптомами, диагностируемыми в детском возрасте [5]. Полная делеция гена HTT приводит к гибели плода на ранней стадии эмбрионального развития [6], при гемизиготности аномальный фенотип не формируется, проявления гомозиготной и гетерозиготной экспансии CAG-триплетов не отличаются [7, 8].
При терминальной форме болезни Гентингтона образцы ткани лобной доли головного мозга содержат примерно на четверть меньше транскриптов, генерируемых мутантным аллелем гена HTT, чем нормальным. Меньшее количество мутантного транскрипта обнаружено также в фибробластах при ювенильной форме болезни Гентингтона [9]. Различия в экспрессии нормального и мутантного аллелей гена HTT с умеренным числом CAG-повторов не выявлены ни в культивируемых лимфоцитах, ни в фибробластах [10].
В норме ген HTT транскрибируется с образованием двух альтернативных транскриптов (10.3 и 13.7 т.н.), различающихся длиной 3'-нетранслируемой области (3'-UTR). В дополнение к двум полноразмерным транскриптам экспрессия мутантного гена HTT может приводить к образованию укороченного полиаденилированного транскрипта, содержащего первый экзон и часть первого интрона [11]. Этот транскрипт длиной около 7.9 т.н. может транслироваться с образованием укороченного высокотоксичного PolyQ-белка. Аберрантный транскрипт формируется в результате полиаденилирования по слабому полиA-сигналу в первом интроне, когда не происходит сплайсинг между первым и вторым экзонами. Этот сигнал срабатывает, если первый экзон содержит удлиненный участок CAG-повторов, причем существует положительная корреляция между длиной этого участка и уровнем аномального полиаденилирования. Предполагается, что подавление сплайсинга между экзонами 1 и 2 обусловлено абсорбцией факторов сплайсинга на CAG-повторах, в частности фактора SRSF6 [12]. Кроме того, доступность слабого полиА-сигнала в первом интроне для полиаденилирования может повышаться из-за того, что CAG-повторы снижают скорость РНК-полимеразы II в ходе транскрипции [13].
Транскрипты HTT различаются стабильностью, длиной полиА-последовательности, а также локализацией в теле или в отростках нейронов. Они имеют разные сайты для РНК-связывающих белков и микроРНК [14, 15]. Короткий транскрипт содержит полиА-последовательность длиной около 50 н., в то время как у средней и длинной мРНК HTT длина полиА-последовательности составляет всего 5 и 10 н. соответственно. Короткий транскрипт имеет самый большой период полужизни, самой нестабильной является длинная HTT-мРНК. Средний и длинный транскрипты имеют больше сайтов связывания микроРНК, чем короткий, причем микроРНК-221 обладает селективностью в отношении длинной и средней мРНК. Выявлены различия в клеточной локализации средней и длинной HTT-мРНК, а также в эффективности их трансляции [16]. Хотя сравнительную эффективность трансляции короткой HTT-мРНК не определяли, однако можно ожидать, что в силу большей стабильности и меньшего количества регуляторных сайтов короткая мРНК будет транслироваться более эффективно, чем нормальные полноразмерные транскрипты [13].
При некоторых наследственных болезнях, обусловленных экспансией микросателлитных повторов, антисмысловые транскрипты, включающие зону экспансии, могут быть функциональными или оказывать дополнительное патогенное действие [17, 18]. Ген HTT занимает протяженный участок на хромосоме 4, поэтому неудивительно, что выявлены транскрипты, читающиеся с его антисмысловой цепи. Некоторые транскрипты, а именно, альтернативные транскрипты гена HTT-AS, включают зону экспансии, соответственно, такие транскрипты несут учаcток CUG-повторов. Изучение этих транскриптов показало, что они экспрессируются на невысоком уровне практически во всех типах клеток. Промотор гена HTT-AS из образцов мозга пациентов с болезнью Гентингтона гораздо менее активен, чем промотор гена HTT, а длина участка тринуклеотидных повторов обратно коррелирует с уровнем экспрессии таких транскриптов [19]. Таким образом, сомнительно, что антисмысловые транскрипты гена HTT играют заметную роль в патогенезе болезни Гентингтона. Не исключено, однако, что антисмысловые транскрипты соответствующих генов вносят некий вклад в развитие других PolyQ болезней. В частности, антисмысловые транскрипты с CUG-повторами обнаружены при SCA2. Ген, кодирующий эти транскрипты, назван ATXN2-AS, его экспрессия обнаружена в образцах мозга пациентов со SCA2, а также в различных типах клеток, полученных от таких пациентов, включая индуцированные плюрипотентные стволовые клетки (ИПСК). На клеточных моделях показана токсичность мРНК, которая транскрибируется с гена ATXN2-AS, содержащего увеличенное количество CTG-повторов [20].
Итак, при изучении возможного патогенного действия мРНК при PolyQ болезнях важно охарактеризовать все альтернативные транскрипты генов с экспансией CAG-повторов, включая незаконные транскрипты, полученные с участием скрытых промоторов и скрытых сайтов полиаденилирования. Кроме того, необходимо оценить возможный вклад транскрипции с антисмысловой цепи.
ЯДЕРНЫЕ ФОКУСЫ мРНК С ДЛИННЫМИ УЧАСТКАМИ ТРИНУКЛЕОТИДНЫХ ПОВТОРОВ
Область CAG-повторов в мРНК, транскрибирующейся с нормального или мутантного гена HTT, способна к формированию шпильки, стабильность которой поддерживается за счет прилежащих небольших участков из CCG-триплетов. Единственное отличие mCAG-РНК от нормальной мРНК состоит в увеличенной длине стебля шпильки, состоящей исключительно из CAG-повторов. Тем не менее, этого достаточно, чтобы mCAG-РНК накапливалась в ядре с образованием фокусов, визуализируемых при помощи флуоресцентной гибридизации in situ (РНК-FISH) [21, 22]. При болезни Гентингтона такие РНК-фокусы наблюдали в различных клетках, включая фибробласты, лимфоциты и нейрональные предшественники [23].
Нужно отметить, что феномен ядерных РНК-фокусов наиболее хорошо изучен при миотонической дистрофии типа 1 (DM1), еще одной болезни, связанной с экспансией числа тринуклеотидных (CTG) повторов в 3'-UTR гена DMPK. Первые описания ядерных фокусов в клетках и тканях пациентов с DM1 относятся к 1995 году [24]. Многие работы, посвященные ядерным РНК-фокусам при болезни Гентингтона, инспирированы более ранними исследованиями DM, поэтому мы рассмотрим основные их результаты.
Как для DM1, так и для PolyQ болезней характерен патогенный порог числа CTG-повторов (>50), а также обратная зависимость между числом триплетных повторов в зоне экспансии и возрастом, в котором появляются первые клинические симптомы [25]. Однако в отличие от PolyQ болезней, при которых CAG-экспансия приводит к одновременному появлению и аномальной мРНК, и аномального белка, патогенез DM1 никак не связан с белком, кодируемым мутантным геном DMPK. Считается, что развитие DM1 индуцируется токсичностью мРНК, содержащей большое число CUG-повторов (mCUG-РНК). CUG-повторы образуют шпильку, где в антипараллельных РНК-цепях напротив GC-пар стоят комплементарные GC-пары (GC-CG), а напротив урацилов находятся урацилы (U-U). При этом CUG-шпилька представляет собой РНК-спираль с конформацией, очень близкой к А-форме. В малой бороздке этой РНК-спирали чередуются положительные и отрицательные электростатические заряды, что формирует характерный паттерн связывания некоторых РНК-связывающих белков [26]. Токсический эффект mCUG-РНК реализуется за счет того, что шпилька из CUG-повторов провоцирует образование внутриядерных агрегатов (фокусов) mCUG-РНК с захватом РНК-связывающих белков, в частности MBNL1 и других белков семейства MBNL, которые регулируют сплайсинг.
Известно, что белок MBNL1 образует кольцеобразную структуру, которая связывается с РНК-спиралью из CUG-повторов. При этом с РНК связывается N-концевой участок белка MBNL1, а С-концевой участок вовлечен в гомотипические взаимодействия, которые могут стабилизировать внутри- и/или межкольцевые контакты [27]. Захват MBNL в ядерных РНК-фокусах и обусловленный этим их дефицит приводят к нарушению сплайсинга, недостатку необходимых транскриптов с одновременным появлением альтернативных фетальных транскриптов, не соответствующих состоянию терминальной дифференцировки мышечных клеток [28]. Мишени сплайсинга с участием белка MBNL включают такие важные для мышечных клеток гены, как ген тропонина, ген рецептора инсулина и ген, кодирующий один из белков хлоридного канала [29, 30]. Таким образом, ведущим патогенным фактором при DM1 является мутантная РНК, а РНК-связывающие белки играют роль промежуточных звеньев.
Динамические наблюдения показали, что ядерные фокусы mCUG-РНК при DM1 представляют собой лабильные структуры, которые постоянно дезагрегируют и вновь образуются стохастически. Ядерные фокусы mCUG-РНК могут распадаться на части, вновь объединяться и сливаться с соседними фокусами, напоминая динамикой скорее вязкую жидкость, чем твердое тело [31]. Количество ядерных фокусов mCUG-РНК в клетках больных DM1 можно уменьшить, понизив уровень MBNL1, что указывает на этот белок как на один из ключевых участников формирования РНК-фокусов [32]. Образование токсичных РНК-фокусов можно подавить, уменьшив количество mCUG-РНК при помощи РНК-интерференции [33] или ASO [34]. В качестве альтернативного подхода к подавлению образования РНК-фокусов рассматривается воздействие малых молекул на вторичную структуру mCUG-РНК, формируемую зоной CUG-экспансии [35, 36]. Подавление образования ядерных фокусов mCUG-РНК нормализует клеточный транскриптом, снижая число событий аномального сплайсинга.
Как уже сказано, CAG-повторы также образуют шпильку в РНК. При этом белок MBNL1 обладает почти одинаковым сродством к CUG- и CAG-шпилькам, поскольку он взаимодействует в основном с GC-элементом повторяющейся последовательности [37]. Однако существует ряд тонких молекулярных различий между CUG- и CAG-повторами. Так, CAG-шпилька образует дцРНК-спираль с параметрами, промежуточными между А- и B-конформациями [38]. Термодинамическая стабильность шпильки, формируемой CAG-триплетами, повторенными 20 раз, примерно в 1.5 раза ниже, чем у аналогичного CUG-олигонуклеотида. В отличие от CUG-, CAG-олигонуклеотиды крайне неустойчивы к действию РНКаз из-за большого числа фосфодиэфирных связей CрA [39]. Помимо небольших молекулярных различий во вторичной структуре РНК, формируемых CUG- и GAC-повторами, следует отметить и существенное различие в числе повторов. При болезни Гентингтона число повторов редко превышает 100, в то время как пациенты с классической DM1 обычно наследуют аллели с несколькими сотнями повторов CTG. При DM1 экспансия CTG-повторов продолжается и в соматических клетках, достигая нескольких тысяч повторов в мышечной и сердечной ткани. Ядерные РНК-фокусы, формируемые mCAG-РНК, существенно хуже визуализируются при помощи FISH [23]. CAG-фокусы несколько отличаются морфологически от РНК-фокусов при DM1: они более рыхлые и занимают большую площадь [40]. Интересно также, что экзогенно экспрессируемая в клетках HeLa нетранслируемая mCUG-РНК формирует больше фокусов, чем mCAG-РНК с тем же числом повторов [41].
Показано, что морфология и количество РНК-фокусов в фибробластах больных с различными PolyQ патологиями (HD, DRPA, SCA1, SCA3, SCA7) довольно сходны, хотя наибольший размер и наибольшая площадь РНК-фокусов выявлены в клетках от больных DRPA. Количество РНК-фокусов зависело от длины CAG-повтора, но не от уровня экспрессии mCAG-РНК. В PolyQ фибробластах не обнаружено фокальных скоплений транскриптов, читающихся с CTG-повторов антисмысловой цепи. В отличие от РНК-фокусов при DM1, РНК-фокусы в ядрах фибробластов, полученных от пациентов с PolyQ заболеваниями, лишь частично локализовались вместе с белком MBNL1. При этом во всех проанализированных PolyQ фибробластах РНК-фокусы локализовались вместе со спекл-маркером сплайсинга SC35 [39]. Это также отличает PolyQ заболевания от DM1, где колокализация РНК-фокусов и спекл-маркера SC35 носила случайный характер [24].
Таким образом, существует ряд различий между РНК-фокусами при DM1 и при PolyQ заболеваниях, т.е. роль ядерного фокального накопления mCAG-РНК в патогенезе PolyQ заболеваний еще предстоит определить. Кроме того, следует отметить, что присутствие выраженных и многочисленных РНК-фокусов в ядре не всегда ведет к клеточной гибели. По крайней мере, об этом свидетельствуют результаты изучения РНК-токсичности при боковом амиотрофическом склерозе с лобно-височной деменцией, где патологический процесс вызван увеличением числа повторов GGGGCC в интроне гена C9orf72. Так, в частности, показано, что способность РНК с GGGGCC-повторами агрегировать в ядерные фокусы в нейральных клетках не коррелировала с повышенной нейродегенерацией [42].
ВЛИЯНИЕ mCAG-РНК НА ТРАНСКРИПТОМ
На ранних стадиях болезни Гентингтона наблюдается нарушение экспрессии множества генов в нейронах, локализованных в областях мозга, наиболее подверженных дегенерации [43]. Некоторые исследователи связывают эти нарушения со способностью mCAG-РНК запускать незаконную РНК-интерференцию, влияющую на множество клеточных транскриптов. Показано, что mCAG-РНК, содержащая длинные CAG-шпильки, служит мишенью рибонуклеазы Dicer, основного компонента РНК-интерференции [44]. В принципе, все типы CNG-шпилек достаточной длины (>17 н.) могут быть субстратами Dicer. При этом Dicer взаимодействует со стеблем РНК-шпильки, поскольку эта двухцепочечная структура, несмотря на неполную комплементарность цепей, сохраняет сходство с совершенным РНК-дуплексом. В экспериментах in vitro показано, что Dicer разрезает CAG-шпильку с образованием коротких РНК-дуплексов (sCAG) длиной 21 п.н. Скорее всего, такой процесс может также протекать in vivo, так как фрагменты sCAG аналогичной длины обнаружены в постмортальных образцах мозга пациентов с болезнью Гентингтона. Полагают, что с активностью рибонуклеазы Dicer может быть связан упомянутый феномен сниженного количества мутантных транскриптов HTT по сравнению с нормальной HTT-мРНК [44]. После того, как CAG-шпилька разрезается, одна цепь sCAG объединяется с комплексом RISC, который расщепляет целевую РНК с использованием нуклеазной активности белка Argonaute (Ago1). Целевой мРНК для комплекса RISC могут оказаться транскрипты с CTG-повторами в кодирующих областях или в 3'-UTR. Однако следует отметить, что в экспериментах in vitro, выполненных на клетках нейробластомы SH-SY5Y, обнаружено лишь незначительное (около 10%) снижение экспрессии подобных транскриптов после трансфекции клеток mHTT-РНК, содержащей 80 CAG-повторов [45]. sCAG не выявлены также в линии Drosophila, модельной для PolyQ заболеваний [46]. Иными словами, феномен sCAG является спорным, и вклад sCAG в патогенез PolyQ заболеваний требует дальнейшего изучения.
Интересные результаты, не связанные с вкладом sCAG в нейродегенерацию, получены недавно Murmann и соавт. [47], которые утверждают, что sCAG обладают высокой токсичностью для раковых клеток in vitro и in vivo, не вызывая гибель нетрансформированных клеток. По мнению авторов данной работы, это может объяснять давно отмеченный факт пониженной частоты онкологических заболеваний при болезни Гентингтона [48].
ВЛИЯНИЕ mCAG-РНК НА СПЛАЙСИНГ
Как уже сказано, фактор сплайсинга MBNL1 связывается с mCAG-РНК. Как следствие, при болезни Гентингтона в клетках изменяются паттерны сплайсинга известных мРНК-мишеней MBNL1. Это подтверждено также на клеточных моделях болезни Гентингтона и SCA3 [41].
Наиболее подробно роль мутантной РНК в повреждении механизмов сплайсинга изучена J. Schilling и соавт. [49]. Проведя масс-спектрометрический анализ белков, которые преимущественно связывают мутантную HTT-РНК, они показали, что это в основном белки сплайсосомы, в том числе PRPF8, SF3B2, SNRNP40 и SON. Белки PRPF8 и SNRNP40 являются компонентами малой субъединицы сплайсосомы U5, а белок SF3B3 входит в состав другой малой субъединицы сплайсосомы – U2. Белок SON, вероятно, облегчает взаимодействие между сплайсосомным белком SRSF2 и РНК-полимеразой II. Такое взаимодействие необходимо для образования самого раннего АТР-зависимого комплекса сплайсинга и для взаимодействия субъединиц U1 и U2 сплайсосомы с пре-мРНК.
Показано, что экспрессия mCAG-РНК вызывает неправильный сплайсинг в клеточной модели на основе клеток нейробластомы SHSY5Y, а сверхэкспрессия гомолога фактора сплайсинга PRPF8 − Prp8 − ослабляет фенотип болезни Гентингтона у Drosophila melanogaster [49]. Наконец, чтобы проверить возможность неправильного сплайсинга в клетках мозга при болезни Гентингтона, методом количественной ОТ-ПЦР сравнили транскрипты CREB1 в образцах мозга больных и в контрольной группе. Выявлено значительное увеличение удержания интрона CREB1 в РНК из образцов, полученных от больных [49].
Следует отметить, что участие mCAG-РНК в связывании белков сплайсинга и в нарушениях сплайсинга показано только при болезни Гентингтона и SCA3, но пока не выявлено при других заболеваниях.
ДИСФУНКЦИЯ ЯДРЫШКА ПРИ PolyQ ЗАБОЛЕВАНИЯХ
В нейронах модельных мышей и в клетках пациентов с болезнью Гентингтона иногда обнаруживают существенное снижение экспрессии рРНК, что может приводить к ядрышковому стрессу с последующим апоптозом [50]. Ученые из Китайского университета Гонгконга смогли проследить цепь событий, которая связывает mCAG-РНК с апоптозом, индуцированным ядрышковым стрессом [51]. Показано, что в клетке mCAG-РНК ассоциирована с нуклеолином ‒ многофункциональным белком, который играет критическую роль в транскрипции пре-рРНК, процессинге и сборке пре-рибосом. Взаимодействие с mCAG-РНК препятствует связыванию нуклеолина и регуляторного элемента рРНК. Это, в свою очередь, вызывает гиперметилирование регуляторного элемента и снижение транскрипции рРНК. Уменьшение количества рРНК приводит к появлению свободных рибосомных белков (RpL5, RpL11 и RpL23), которые объединяются с E3-убиквитинлигазой MDM2, одна из клеточных функций которой ‒ опосредованное участие в протеасомной деградации р53. Взаимодействие рибосомных белков с MDM2 нарушает деградацию p53, из-за чего в клетках, экспрессирующих mCAG-РНК, повышается уровень p53, который перемещается в митохондрии и активирует апоптоз [51].
Те же ученые из Китайского университета Гонконга позднее показали, что при помощи смоделированного пептида BIND (англ. beta-structured inhibitor for neurodegenerative diseases) можно блокировать связывание mCAG-РНК с нуклеолином [52]. Обеспечив доставку этого пептида в клетку за счет конъюгации с транслокационным TAT-пептидом, показали, что пептид TAT-BIND значительно снижает гибель клеток HEK293, экспрессирующих экзогенную mCAG-РНК. При этом применение TAT-BIND уменьшило проявление структурных аномалий ядрышка, характерных для клеток, испытывающих ядрышковый стресс. Нормализовался также такой характерный для ядрышкового стресса показатель, как сниженный уровень экспрессии 45S рРНК. Эксперименты in vivo, проведенные на модельных мышах и дрозофиле, подтвердили результаты, полученные на клеточных моделях. Кроме того, они показали специфичность корректирующего действия пептида TAT-BIND при PolyQ заболеваниях.
Пока не ясно, какое место занимает ядрышковый стресс в цепи патогенетических событий при PolyQ болезнях. Тем не менее установлено, что аномалии ядрышка, вызванные взаимодействием с мутантной мРНК, могут служить перспективной мишенью для разработки лекарственных средств.
НАРУШЕНИЯ ТРАНСЛЯЦИИ mCAG-РНК ПРИ PolyQ ЗАБОЛЕВАНИЯХ
mCAG-РНК способна вызывать не только нарушения процессов, происходящих в ядре, но также процессов, не связанных с ядром, прежде всего трансляции. Одно из таких нарушений ‒ нетипичная трансляция, ассоциированная с повторами (RAN ‒ repeat associated non-ATG), которая приводит к появлению в клетке нефункциональных пептидов и белков, иногда обладающих токсичными свойствами. RAN-трансляция возникает вследствие того, что шпилька, формируемая в мРНК зоной экспансии CAG-повторов, может инициировать старт трансляции в отсутствие канонического триплета ATG. При этом RAN-трансляция может начинаться как с неканонического сайта инициации, так и непосредственно с участка повторов. В 2015 году впервые показали, что при болезни Гентингтона в мозге накапливаются четыре гомополимерных белка (полиаланин, полисерин, полилейцин, полицистеин) [53]. Это свидетельствует о RAN-трансляции при болезни Гентингтона, которая происходит со смысловой и с антисмысловой цепей. Наибольшие количества этих белков обнаружены в областях мозга с потерей нейронов, активацией микроглии и апоптозом. Эти области включают хвостатое ядро, белое вещество, а в случае ювенильной формы − мозжечок. Гомотипичные белки, возникающие в результате RAN-трансляции, обнаружены также в мышиной модели болезни Гентингтона [53]. Таким образом, при болезни Гентингтона в клетках в дополнение к mНТТ генерируются нефункциональные и потенциально токсичные белки, появление которых вызвано аномальной вторичной структурой mCAG-РНК. Более подробное рассмотрение феномена RAN-трансляции при PolyQ заболеваниях можно найти в нашем обзоре [54].
Некоторые исследователи также отмечают, что mCAG-РНК транслируется более эффективно, чем нормальная мРНК. Один из механизмов, который приводит к увеличению трансляции, запускается связыванием удлиненного CAG-повтора с РНК-связывающим белком MID1. MID1, в свою очередь, рекрутирует в этот комплекс протеинфосфатазу 2A (PP2A) и 40S рибосомную киназу S6K. MID1 ‒ это E3-убиквитинлигаза, S6K является регулятором трансляции, а фосфатаза PP2A подавляет фосфозависимую активность S6K. После связывания с PP2A белок MID1 катализирует убиквитинзависимую деградацию PP2A. Таким образом, MID1 действует как негативный регулятор активности этой фосфатазы. S6K, рекрутируемая к mCAG-РНК через MID1, в отсутствие фосфатазной активности PP2A индуцирует повышение трансляции мутантной РНК [55].
Позднее показали, что связывание MID1 с mCAG-РНК, кодируемой генами ATXN2, ATXN3 и ATXN7, индуцирует повышение трансляции мутантного белка в культурах нейронов трансгенных мышей, а также в фибробластах пациентов с соответствующими PolyQ заболеваниями [56]. Таким образом, подтверждено, что белок MID1 может быть мишенью для разработки терапии PolyQ заболеваний, направленной на снижение количества мутантных белков в нейронах.
ПОЧЕМУ ДЛЯ РАЗВИТИЯ ЗАБОЛЕВАНИЙ НЕОБХОДИМО ПОРОГОВОЕ ЗНАЧЕНИЕ ДЛИНЫ ПОВТОРОВ?
При всех выявленных к настоящему времени патологиях, связанных с экспансией коротких нуклеотидных повторов, заболевание развивается после того, как число повторов в РНК достигнет критического числа, т.е. РНК приобретает патологические свойства при превышении определенной пороговой длины [57, 58]. Обсуждаются различные гипотезы, объясняющие этот феномен (см., например, обзор [59]). Наиболее убедительное, на наш взгляд, предположение состоит в том, что для запуска патологических процессов мутантная мРНК должна быть способна формировать дополнительные или более стабильные вторичные структуры (в основном, шпильки) в отличие от мРНК с нормальным числом повторов. Эти вторичные структуры могут вступать в незаконные взаимодействия с РНК-связывающими белками, такими как белки сплайсинга, рибонуклеаза Dicer или иными белками метаболизма РНК, нарушая тем самым их функцию.
Другая гипотеза, объясняющая пороговый эффект повторов, подразумевает, что патология возникает из-за структурных внутриклеточных нарушений, вызванных присутствием мутантной РНК. Такие нарушения могут быть вызваны способностью протяженных повторов в мРНК к межмолекулярной самоассоциации, ведущей к процессу разделения фаз или, иными словами, структурной сегрегации молекул внутри клетки. Так, межмолекулярное взаимодействие мутантной мРНК может приводить к образованию сложных разветвленных надмолекулярных РНК-комплексов, разрастание которых может быть причиной формирования коллоидных частиц, способных сливаться в гидрогели. Со временем аморфные частицы могут приобрести упорядоченную, термодинамически наиболее выгодную, твердую структуру. В пользу “структурной” гипотезы свидетельствуют результаты опубликованного в 2017 году исследования, в котором показано, что раствор (CNG)n-РНК подвергается фазовому переходу in vitro [60], что выражается в капельной агрегации (CNG)n-РНК. Этот процесс зависит от количества повторов в РНК и их последовательности. Так, кластеры РНК, содержащих (CAG)n и (CUG)n, регистрировали при n ≥ 31, тогда как в случае РНК с (CCGGG)n − при n > 5. При этом именно число повторов определяет способность РНК к межмолекулярной ассоциации за счет комплементарных и/или Хугстиновских взаимодействий гетероциклов. Формированию гелевых частиц РНК микронного размера способствуют известные факторы компактизации полинуклеотидов, такие как ионы двухвалентных металлов, концентрация РНК и молекулярное сгущение раствора (molecular crowding).
Следует отметить, что РНК может способствовать разделу фаз не только посредством взаимодействия РНК-РНК, но и РНК-белок. Некоторые исследователи считают, что абсорбция специфических РНК-связывающих белков на РНК-повторах может способствовать разделу фаз, проявляясь в виде ядерных фокусов РНК и внося свой вклад в структурирование нуклеоплазмы [61].
Как известно, раздел фаз вовлечен в формирование безмембранных органелл клетки, таких как ядрышко, тельца Кахаля, стресс-гранулы и другие [62]. Кроме того, за последние годы показано, что структурная внутриклеточная сегрегация за счет раздела фаз происходит при взаимодействии молекулярных комплексов на суперэнхансерах [63], при формировании гетерохроматина [64], репарации [65], ответе на окислительный стресс [66]. Возможно, подобные клеточные структуры и процессы могут быть нарушены в присутствии mCAG-РНК, например, из-за конкуренции за факторы компактизации.
Итак, патологические эффекты мутантной мРНК с пороговым числом CAG-повторов могут быть связаны с их вмешательством в клеточные процессы или структуры. Очевидно, что структурные и функциональные аномалии не являются взаимоисключающими, однако их количественный и качественный вклад в формирование патологического фенотипа при PolyQ заболеваниях еще предстоит оценить.
МУТАНТНАЯ мРНК КАК МИШЕНЬ ДЛЯ ТЕРАПЕВТИЧЕСКОГО ВОЗДЕЙСТВИЯ
Перспективность использования mCAG-РНК в качестве мишени для терапевтического воздействия была показана еще в 2005 году, когда снижение уровня mCAG-РНК при помощи РНК-интерференции привело к снижению патологических проявлений у мышей линии HD-N171-82Q, служащей моделью болезни Гентингтона [67]. Конструкцию на основе аденоассоциированного вируса, экспрессирующую короткую шпилечную РНК, инъецировали в различные участки мозга и показали безопасность этого подхода.
Несколько компаний проводят клинические испытания препаратов, действие которых направлено на блокирование экспрессии mCAG-РНК при болезни Гентингтона.
Компании “Voyager Therapeutics” и “uniQure” разрабатывают терапию на основе микроРНК. Эти компании сконструировали модифицированные вирусные векторы, которые доставляют непосредственно в мозг. В частности, “uniQure” использует вектор на основе аденоассоциированного вируса (AAV5) AMT-130, который постоянно экспрессирует микроРНК, избирательно блокирующую mCAG-РНК. С помощью инъекции вектор АМТ-130, экспрессирующий микроРНК, вводят непосредственно в пораженные ткани мозга, что вызывает селективный нокдаун мутантного гена. В апреле 2018 года “uniQure” представила обзор доклинических данных, доказывающих эффективность концепции AMT-130. Результаты доклинических испытаний на животных показывают, что однократное введение АМТ-130 привело к дозозависимому и устойчивому снижению содержания mHTT как в глубоких структурах мозга, так и в коре. Исследование, проведенное на приматах, показало, что через 6 месяцев после введения АМТ-130 содержание mHTT было снижено на 68% в стриатуме и на 47% во фронтальной коре. В доклинических исследованиях на грызунах снижение содержания mHTT привело к значительному улучшению нейрональной функции полосатого тела и моторной координации, снижению потери массы тела и увеличению медианы выживаемости на 24% по сравнению с контрольной группой. В апреле 2019 FDA разрешило “uniQure” проведение клинических испытаний фазы I/II АМТ-130 [68].
Компания “Ionis Pharmaceuticals” использует другой подход, в котором подавление экспрессии mCAG-РНК достигается при помощи ASO, доставляемых интратекально. Разработанный этой компанией препарат RG6042 представляет собой синтетический олигонуклеотид, фосфатный остов которого содержит фосфоротиатные связи вместо фосфодиэфирных. На концах этого ASO cодержатся нуклеотиды с 2'-O-метоксиэтильными группами. Такая комбинация модификаций влияет на распределение ASO в тканях мозга, период полураспада, клеточную ассимиляцию и активность РНКаз [69]. Предполагалось, что RG6042 может снижать уровень не только mHTT, но и белка дикого типа. Эксперименты in vitro, проведенные на фибробластах, полученных от пациентов с болезнью Гентингтона, показали, что терапевтический ASO приводит к снижению уровня мРНК НТТ ‒ мутантной на 83%, нормальной ‒ на 43%.
Обширные доклинические исследования, проведенные на мышах линий YAC128 и BACHD, моделирующих медленно прогрессирующие формы болезни Гентингтона, и на линии R6/2 ‒ модели быстро прогрессирующего заболевания, показали безопасность и эффективность препарата RG6042. Препарат уже прошел несколько этапов клинических испытаний. В 2015–2017 г.г. было проведено клиническое испытание (фазы I/II) безопасности и переносимости препарата RG6042 (NCT02519036), включавшее 46 взрослых пациентов с ранней стадией болезни Гентингтона [70]. Оказалось, что 4-кратное интеркатальное введение RG6042 с интервалом 4 недели снизило уровень mHTT в спинномозговой жидкости до 60%. Это соответствовало уменьшению содержания mHTT в коре мозга на 55‒85% и в спинном мозге на 20‒50%. Препарат при этом распределялся по всем тканям мозга – нейронам и глии. Положительные результаты отмечены также при проведении когнитивных тестов и энцефалографических исследований.
В декабре 2018 года стартовала третья фаза мультицентровых клинических исследований безопасности и эффективности введения препарата RO7234292 (RG6042). Это исследование компания “Ionis Pharmaceuticals” проводит в партнерстве с гигантом фарминдустрии “Roche”, в нем будут участвовать 46 центров и 660 пациентов с манифестацией хореи. Окончание исследования ожидается в 2022 году. Планируется испытать два режима: препарат будут вводить каждые 4 или 8 недель интратекально (под оболочки мозга − субарахноидально или эпидурально интралюмбально путем инъекции на уровне L4–L5 поясничных позвонков). Основная цель исследования ‒ доказать эффективность препарата по изменению в динамике двигательных нарушений. Эффективность препарата оценивали также по 17 неврологическим показателям (двигательные, когнитивные и психомоторные функции) и биомаркерам, характеризующим применение тестируемого препарата, в частности, по концентрации мутантного белка.
Терапевтический подход, основанный на применении ASO, использует не только “Ionis Pharmaceuticals”, но также компания “Wave Life Sciences”. В настоящее время эта компания испытывает два олигонуклеотидных препарата (WVE-120101 и WVE-120102), которые блокируют экспрессию mCAG-РНК. Мутантный аллель идентифицируют по наличию сцепленного однонуклеотидного полиморфизма (SNP). WVE-120101 предназначен для пациентов с одним SNP, а WVE-120102 с другим. “Wave Life Sciences” проводит фазу I/II клинического испытания (NCT03225833 и NCT03225846) для оценки безопасности и переносимости этих препаратов у пациентов с ранними проявлениями заболевания. Препарат будут вводить в позвоночный канал.
Разрабатываются и другие подходы к снижению уровня mHTT путем воздействия на мРНК-мишень. Так, с 2014 года изучают возможность использования малых молекул, обладающих высокой биодоступностью при оральном применении [71, 72].
ЗАКЛЮЧЕНИЕ
Исследование механизмов и поиск новых способов компенсации повреждений и защиты нейронов при хронических нейродегенеративных заболеваниях остается актуальной проблемой современной медицины. Выяснение причин нейродегенерации и исследование изменений, происходящих при развитии PolyQ патологий, важно как в связи с особой тяжестью таких заболеваний, так и в связи с отсутствием лекарственных препаратов, эффективных при такой нозологии. Функции как мутантных, так и нормальных генов при большинстве PolyQ заболеваний остаются слабо изученными, не установлен максимально токсичный агент – мутантный белок или мутантная РНК. Непонятно, почему PolyQ болезни избирательно повреждают нервную систему, хотя мутантные гены и кодируемые ими белки широко представлены во всех тканях. Неясно, какие клеточные органеллы и сигнальные пути наиболее уязвимы для токсического повреждения; какие пусковые механизмы являются общими для болезней этого типа, а какие зависят от конкретного мутантного гена?
В последние годы широко обсуждается роль именно мутантной РНК в патогенезе PolyQ болезней. В ряде работ убедительно показано, что при болезни Гентингтона mCAG-РНК вовлечена в аномальные проявления транскрипции, сплайсинга, трансляции, структуры цитозоля, транспорта РНК из ядра в цитоплазму. Однако вполне вероятно, что вклад механизмов, связанных с PolyQ белками или mCAG-РНК, различен при каждом PolyQ заболевании. Например, недавно выявили ключевое токсичное действие комплекса мутантного атаксина-1 с белком-партнером CIC при SCA1. При внесении мутации, блокирующей связывание атаксина-1 c CIC, у мышей не наблюдается дегенерации нейронов Пуркинье, хотя у них экспрессируется mCAG-мРНК, имеющая длинный участок CAG-повторов [73].
До недавнего времени изучение PolyQ заболеваний было ограничено постмортальными образцами нейронов человека, трансформированными клеточными линиями либо трансгенными животными. Не столь давно разработанная технология получения ИПСК [74] и их дифференцировки в нейральные клетки позволяет расширить спектр моделей нейродегенеративных заболеваний. За последние годы показана перспективность использования моделей на основе ИПСК для изучения роли РНК в патогенезе нейродегенеративных заболеваний [76 ] . Интересным кажется также сравнительное изучение PolyQ болезней, направленное на выявление общих и частных механизмов этих заболеваний.
Работа поддержана Российским научным фондом (№ 19-15-00425).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Aronin N., DiFiglia M. (2014) Huntingtin-lowering strategies in Huntington’s disease: antisense oligonucleotides, small RNAs, and gene editing. Movement Disorders. 29, 1455‒1461.
Hu J., Matsui M., Gagnon K.T., Schwartz J.C., Gabillet S., Arar K., Wu J., Bezprozvanny I., Corey D.R. (2009) Allele-specific silencing of mutant huntingtin and ataxin-3 genes by targeting expanded CAG repeats in mRNAs. Nat. Biotechnol. 27, 478.
Hu J., Liu J., Corey D.R. (2010). Allele-selective inhibition of huntingtin expression by switching to an miRNA-like RNAi mechanism. Chem. Biol. 17, 1183‒1188.
Sharp A.H., Loev S.J., Schilling G., Li S.H., Li X.J., Bao J., Wagster M.V., Kotzuk J.A., Steiner J.P., Lo A., Hedreen J. (1995) Widespread expression of Huntington’s disease gene (IT15) protein product. Neuron. 14, 1065‒1074.
Иллариошкин С.Н., Клюшников С.А., Селивёрстов Ю.А. (2018) Болезнь Гентингтона. М.: Атмосфера.
Nasir J., Floresco S.B., O’Kusky J.R., Diewert V.M., Richman J.M., Zeisler J., Borowski A., Marth J.D., Phillips A.G., Hayden M.R. (1995) Targeted disruption of the Huntington’s disease gene results in embryonic lethality and behavioral and morphological changes in heterozygotes. Cell. 81, 811‒823.
Wexler N.S., Young A.B., Tanzi R.E., Travers H., Starosta-Rubinstein S., Penney J.B., Snodgrass S.R., Shoulson I., Gomez F., Ramos Arroyo M.A., Penchaszadeh G.K. (1987) Homozygotes for Huntington’s disease. Nature. 326, 194.
Myers R.H., Leavitt J.L., Farrer L.A., Jagadeesh J., McFarlane H., Mastromauro C.A., Mark R.J., Gu-sella J.F. (1989) Homozygote for Huntington disease. Am. J. Hum. Genet. 45, 615.
Evers M.M., Schut M.H., Pepers B.A., Atalar M., van Belzen M.J., Faull R.L., Roos R.A., van Roon-Mom W.M. (2015) Making (anti-) sense out of huntingtin levels in Huntington disease. Mol. Neurodegener. 10, 21.
Shin A., Shin B., Shin J.W., Kim K.H., Atwal R.S., Hope J.M., Gillis T., Leszyk J.D., Shaffer S.A., Lee R., Kwak S., MacDonald M.E., Gusella J.F., Seong I.S., Lee J.M. (2017) Novel allele-specific quantification methods reveal no effects of adult onset CAG repeats on HTT mRNA and protein levels. Hum. Mol. Genet. 26, 1258‒1267.
Neueder A., Landles C., Ghosh R., Howland D., Myers R.H., Faull R.L., Tabrizi S.J., Bates G.P. (2017) The pathogenic exon 1 HTT protein is produced by incomplete splicing in Huntington’s disease patients. Sci. Rept. 7, 1307.
Sathasivam K., Neueder A., Gipson T.A., Landles C., Benjamin A.C., Bondulich M.K., Smith D.L., Faull R.L., Roos R.A., Howland D., Detloff P.J., Housman D.E., Bates G.P. (2013) Aberrant splicing of HTT generates the pathogenic exon 1 protein in Huntington disease. Proc. Natl. Acad. Sci. USA. 110, 2366‒2370.
Neueder A., Dumas A.A., Benjamin A.C., Bates G.P. (2018) Regulatory mechanisms of incomplete huntingtin mRNA splicing. Nat. Commun. 9, 3955.
Romo L., Ashar-Patel A., Pfister E., Aronin N. (2017) Alterations in mRNA 3′ UTR isoform abundance accompany gene expression changes in human Huntington’s disease brains. Cell Rept. 20, 3057‒3070.
Romo L., Mohn E.S., Aronin N. (2018) A fresh look at huntingtin mRNA processing in Huntington’s disease. J. Huntington’s Dis. 7, 101‒108.
Xu H., An J.J., Xu B. (2017) Distinct cellular toxicity of two mutant huntingtin mRNA variants due to translation regulation. PLoS One. 12, e0177610.
Khalil A.M., Faghihi M.A., Modarresi F., Brothers S.P., Wahlestedt C. (2008) A novel RNA transcript with antiapoptotic function is silenced in fragile X syndrome. PLoS One. 3, e1486.
Daughters R.S., Tuttle D.L., Gao W., Ikeda Y., Moseley M.L., Ebner T.J., Swanson M.S., Ranum L.P. (2009) RNA gain-of-function in spinocerebellar ataxia type 8. PLoS Genetics. 5, e1000600.
Chung D.W., Rudnicki D.D., Yu L., Margolis R.L. (2011) A natural antisense transcript at the Huntington’s disease repeat locus regulates HTT expression. Hum. Mol. Genet. 20, 3467‒3477.
Li P.P., Sun X., Xia G., Arbez N., Paul S., Zhu S., Peng H.B., Ross C.A., Koeppen A.H., Margolis R.L., Pulst S.M., Ashizawa T., Rudnicki D.D. (2016) ATXN2-AS, a gene antisense to ATXN2, is associated with spinocerebellar ataxia type 2 and amyotrophic lateral sclerosis. Ann. Neurol. 80, 600‒615.
de Mezer M., Wojciechowska M., Napierala M., Sobczak K., Krzyzosiak W.J. (2011) Mutant CAG repeats of Huntingtin transcript fold into hairpins, form nuclear foci and are targets for RNA interference. Nucl. Acids Res. 39, 3852‒3863.
Wojciechowska M., Krzyzosiak W.J. (2011) Cellular toxicity of expanded RNA repeats: focus on RNA foci. Hum. Mol. Genet. 20, 3811‒3821.
Urbanek M.O., Krzyzosiak W.J. (2016) RNA FISH for detecting expanded repeats in human diseases. Methods. 98, 115‒123.
Taneja K.L., McCurrach M., Schalling M., Housman D., Singer R.H. (1995) Foci of trinucleotide repeat transcripts in nuclei of myotonic dystrophy cells and tissues. J. Cell. Biol. 128, 995‒1002.
Wheeler T.M., Thornton C.A. (2007) Myotonic dystrophy: RNA-mediated muscle disease. Curr. Opin. Neurol. 20, 572‒576.
Mooers B.H., Logue J.S., Berglund J.A. (2005) The structural basis of myotonic dystrophy from the crystal structure of CUG repeats. Proc. Natl. Acad. Sci. USA. 102, 16626‒16631.
Yuan Y., Compton S.A., Sobczak K., Stenberg M.G., Thornton C.A., Griffith J.D., Swanson M.S. (2007) Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucl. Acids Res. 35, 5474‒5486.
Nakamori M., Sobczak K., Puwanant A., Welle S., Eichinger K., Pandya S., Dekdebrun J., Heatwole C.R., McDermott M.P., Chen T., Cline M., Tawil R., Osborne R.J., Wheeler T.M., Swanson M.S., Moxley R.T. 3rd, Thornton C.A. (2013) Splicing biomarkers of disease severity in myotonic dystrophy. Ann. Neurol. 74, 862‒872.
Botta A., Vallo L., Rinaldi F., Bonifazi E., Amati F., Biancolella M., Gambardella S., Mancinelli E., Angelini C., Meola G., Novelli G. (2006) Gene expression analysis in myotonic dystrophy: indications for a common molecular pathogenic pathway in DM1 and DM2. Gene expression. J. Liver Res. 13, 339‒351.
Salvatori S., Furlan S., Fanin M., Picard A., Pastorello E., Romeo V., Trevisan C.P., Angelini C. (2009) Compa-rative transcriptional and biochemical studies in muscle of myotonic dystrophies (DM1 and DM2). Neurol. Sci. 30, 185‒192.
Querido E., Gallardo F., Beaudoin M., Ménard C., Chartrand P. (2011) Stochastic and reversible aggregation of mRNA with expanded CUG-triplet repeats. J. Cell. Sci. 124, 1703‒1714.
Konieczny P., Stepniak-Konieczna E., Sobczak K. (2014) MBNL proteins and their target RNAs, interaction and splicing regulation. Nucl. Acids Res. 42, 10873‒10887.
Sobczak K., Wheeler T.M., Wang W., Thornton C.A. (2013) RNA interference targeting CUG repeats in a mouse model of myotonic dystrophy. Mol. Therapy. 21, 380‒387.
Jauvin D., Chrétien J., Pandey S. K., Martineau L., Revillod L., Bassez G., Thornton C.A. (2017) Targeting DMPK with antisense oligonucleotide improves muscle strength in myotonic dystrophy type 1 mice. Mol. Therapy-Nucl. Acids. 7, 465‒474.
Rzuczek S.G., Colgan L.A., Nakai Y., Cameron M.D., Furling D., Yasuda R., Disney M.D. (2017) Precise small-molecule recognition of a toxic CUG RNA repeat expansion. Nat. Chem. Biol. 13, 188.
Angelbello A.J., Rzuczek S.G., Mckee K.K., Chen J.L., Olafson H., Cameron M.D., Moss W.N., Wang E.T., Disney M.D. (2019) Precise small-molecule cleavage of an r (CUG) repeat expansion in a myotonic dystrophy mouse model. Proc. Natl. Acad. Sci. USA. 116, 7799–7804.
Yuan Y., Compton S.A., Sobczak K., Stenberg M.G., Thornton C.A., Griffith J.D., Swanson M.S. (2007) Muscleblind-like 1 interacts with RNA hairpins in splicing target and pathogenic RNAs. Nucl. Acids Res. 35, 5474‒5486.
Tawani A., Kumar A. (2015) Structural insights reveal the dynamics of the repeating r (CAG) transcript found in Huntington’s disease (HD) and spinocerebellar ataxias (SCAs). PLoS One. 10, e0131788.
Sobczak K., Michlewski G., de Mezer M., Kierzek E., Krol J., Olejniczak M., Kierzek R., Krzyzosiak W.J. (2010) Structural diversity of triplet repeat RNAs. J. Biol. Chem. 285, 12755‒12764.
Urbanek M.O., Jazurek M., Switonski P.M., Figura G., Krzyzosiak W.J. (2016) Nuclear speckles are detention centers for transcripts containing expanded CAG repeats. Biochim. Biophys. Acta (BBA)–Mol. Basis Disease. 1862, 1513‒1520.
Mykowska A., Sobczak K., Wojciechowska M., Kozlowski P., Krzyzosiak W.J. (2011) CAG repeats mimic CUG repeats in the misregulation of alternative splicing. Nucl. Acids Res. 39, 8938‒8951.
Mizielinska S., Grönke S., Niccoli T., Ridler C.E., Clayton E.L., Devoy A., Moens T., Norona F.E., Woollacott I.O.C., Pietrzyk J., Cleverley K., Nicoll A.J., Pickering-Brown S., Dols J., Cabecinha M., Hendrich O., Fratta P., Fisher E.M.C., Partridge L., Isaacs A.M. (2014) C9orf72 repeat expansions cause neurodegeneration in Drosophila through arginine-rich proteins. Science. 345, 1192‒1194.
Hodges A., Strand A.D., Aragaki A.K., Kuhn A., Sengstag T., Hughes G., Elliston L.A., Hartog C., Goldstein D.R., Thu D., Hollingsworth Z.R., Collin F., Synek B., Holmans P.A., Young A.B., Wexler N.S., Delorenzi M., Kooperberg C., Augood S.J., Faull R.L., Olson J.M., Jones L., Luthi-Carter R. (2006) Regional and cellular gene expression changes in human Huntington’s disease brain. Hum. Mol. Genet. 15, 965‒977.
Krol J., Fiszer A., Mykowska A., Sobczak K., de Mezer M., Krzyzosiak W.J. (2007) Ribonuclease dicer cleaves triplet repeat hairpins into shorter repeats that silence specific targets. Mol. Cell. 25, 575‒586.
Bañez-Coronel M., Porta S., Kagerbauer B., Mateu-Huertas E., Pantano L., Ferrer I., Guzmán M., Estivill X., Martí E. (2012) A pathogenic mechanism in Huntington’s disease involves small CAG-repeated RNAs with neurotoxic activity. PLoS Genet. 8, e1002481.
Reinhardt A., Feuillette S., Cassar M., Callens C., Thomassin-Bourrel H., Birman S., Lecourtois M., Antoniewski C., Tricoire H. (2012) Lack of miRNA misregulation at early pathological stages in Drosophila neurodegenerative disease models. Front. Genet. 3, 226.
Murmann A.E., Gao Q.Q., Putzbach W.E., Patel M., Bartom E.T., Law C.Y., Bridgeman B., Chen S., McMahon K.M., Thaxton C.S., Peter M.E. (2018) Small interfering RNAs based on huntingtin trinucleotide repeats are highly toxic to cancer cells. EMBO Repts. 19, e45336.
Sørensen S.A., Fenger K., Olsen J.H. (1999) Significantly lower incidence of cancer among patients with Huntington disease: an apoptotic effect of an expanded polyglutamine tract? Cancer. 86, 1342‒1346.
Schilling J., Broemer M., Atanassov I., Duernberger Y., Vorberg I., Dieterich C., Dagane A., Dittmar G., Wanker E., van Roon-Mom W., Winter J., Krauß S. (2019) Deregulated splicing is a major mechanism of RNA-induced toxicity in Huntington’s disease. J. Mol. Biol. 19, 1869‒1877.
Lee J., Hwang Y.J., Boo J.H., Han D., Kwon O.K., Todorova K., Kowall N.W., Kim Y., Ryu H. (2011) Dysregulation of upstream binding factor-1 acetylation at K352 is linked to impaired ribosomal DNA transcription in Huntington’s disease. Cell Death Differ. 18, 1726.
Tsoi H., Lau T.C.K., Tsang S.Y., Lau K.F., Chan H.Y.E. (2012) CAG expansion induces nucleolar stress in polyglutamine diseases. Proc. Natl. Acad. Sci. USA. 109, 13428‒13433.
Zhang Q., Chen Z.S., An Y., Liu H., Hou Y., Li W., Lau K.F., Koon A.C., Ngo J.C.K., Chan H. Y.E. (2018) A peptidylic inhibitor for neutralizing expanded CAG RNA-induced nucleolar stress in polyglutamine diseases. RNA. 24, 486‒498.
Bañez-Coronel M., Ayhan F., Tarabochia A.D., Zu T., Perez B.A., Tusi S.K., Pletnikova O., Borchelt D.R., Ross C.A., Margolis R.L., Yachnis A.T., Troncoso J.C., Ranum L.P. (2015) RAN translation in Huntington disease. Neuron. 88, 667‒677.
Воловиков Е.А., Давиденко А.В., Лагарькова М.А. (2019) Молекулярные механизмы атаксии первого типа. Генетика. 55, в печати.
Krauss S., Griesche N., Jastrzebska E., Chen C., Rutschow D., Achmüller C., Dorn S., Boesch S.M., Lalowski M., Wanker E., Schneider R., Schweiger S. (2013) Translation of HTT mRNA with expanded CAG repeats is regulated by the MID1-PP2A protein complex. Nat. Commun. 4, 1511.
Griesche N., Schilling J., Weber S., Rohm M., Pesch V., Matthes F., Auburger G., Krauss S. (2016) Regulation of mRNA translation by MID1: a common mechanism of expanded CAG repeat RNAs. Front. Cell Neurosci. 7, 226.
La Spada A.R., Taylor J.P. (2010) Repeat expansion disease: progress and puzzles in disease pathogenesis. Nat. Rev. Genet. 11, 247.
Babić Leko M., Župunski V., Kirincich J., Smilović D., Hortobágyi T., Hof P.R., Šimić G. (2019) Molecular mechanisms of neurodegeneration related to C9orf72 hexanucleotide repeat expansion. Behav. Neurol. 2019, 2909168.
Lee D.Y., McMurray C.T. (2014) Trinucleotide expansion in disease: why is there a length threshold? Curr. Opin. Genet. Dev. 26, 131‒140.
Jain A., Vale R.D. (2017) RNA phase transitions in repeat expansion disorders. Nature. 546, 243.
Fay M.M., Anderson P.J. (2018) The role of RNA in biological phase separations. J. Mol. Biol. 430, 4685‒4701.
Boeynaems S., Alberti S., Fawzi N.L., Mittag T., Polymenidou M., Rousseau F., Schymkowitz J., Shorter J., Wolozin B., Van Den Bosch L., Tompa P., Fuxreiter M. (2018) Protein phase separation: a new phase in cell biology. Trends Cell Biol. 28, 420‒435.
Sabari B.R., Dall’Agnese A., Boija A., Klein I.A., Coffey E.L., Shrinivas K., Abraham B.J., Hannett N.M., Zamudio A.V., Manteiga J.C., Li C.H., Guo Y.E., Day D.S., Schuijers J., Vasile E., Malik S., Hnisz D., Lee T.I., Cisse I.I., Roeder R.G., Sharp P.A., Chakra-borty A.K., Young R.A. (2018) Coactivator condensation at super-enhancers links phase separation and gene control. Science. 361, eaar3958.
Larson A.G., Narlikar G.J. (2018) The role of phase separation in heterochromatin formation, function, and regulation. Biochemistry. 57, 2540‒2548.
Singatulina A.S., Hamon L., Sukhanova M.V., Desforges B., Joshi V., Bouhss A., Lavrik O.I., Pastré D. (2019) PARP-1 activation directs FUS to DNA damage sites to form PARG-reversible compartments enriched in damaged DNA. Cell Rept. 27, 1809‒1821.
Kato M., Yang Y.S., Sutter B.M., Wang Y., McKnight S.L., Tu B.P. (2019) Redox state controls phase separation of the yeast ataxin-2 protein via reversible oxidation of its methionine-rich low-complexity domain. Cell. 177, 711‒721.
Harper S.Q., Staber P.D., He X., Eliason S.L., Martins I.H., Mao Q., Yang L, Kotin R.M., Paulson H.L., Davidson B. L. (2005) RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc. Natl. Acad. Sci. USA. 102, 5820‒5825.
http://www.uniqure.com/gene-therapy/huntingtons-disease.php.
Bennett C.F., Swayze E.E. (2010) RNA targeting therapeutics: molecular mechanisms of antisense oligonucleotides as a therapeutic platform. Annu. Rev. Pharmacol. Toxicol. 50, 259‒293.
https://clinicaltrials.gov/ct2/show/NCT02519036.
Naryshkin N.A., Weetall M., Dakka A., Narasimhan J., Zhao X., Feng Z., Ling K.K., Karp G.M., Qi H., Woll M.G., Chen G., Zhang N., Gabbeta V., Vazirani P., Bhattacharyya A., Furia B., Risher N., Sheedy J., Kong R., Ma J., Turpoff A., Lee C.S., Zhang X., Moon Y.C., Trifillis P., Welch E.M., Colacino J.M., Babiak J., Almstead N.G., Peltz S.W., Eng L.A., Chen K.S., Mull J.L., Lynes M.S., Rubin L.L., Fontoura P., Santarelli L., Haehnke D., McCarthy K.D., Schmucki R., Ebeling M., Sivaramakrishnan M., Ko C.P., Paushkin S.V., Ratni H., Gerlach I., Ghosh A., Metzger F. (2014) SMN2 splicing modifiers improve motor function and longevity in mice with spinal muscular atrophy. Science. 345, 688‒693.
Doherty E.M. (2017) Screening approaches to identify small-molecule modulators of huntingtin protein levels. CHDI Foundation Annual Therapeutics Conference; Malta. 2017. (abstr).
Rousseaux M.W., Tschumperlin T., Lu H.C., Lackey E.P., Bondar V.V., Wan Y.W., Tan Q., Adamski C.J., Friedrich J., Twaroski K., Chen W., Tolar J., Henzler C., Sharma A., Bajić A., Lin T., Duvick L., Liu Z., Sillitoe R.V., Zoghbi H.Y., Orr H.T., Chen W. (2018) ATXN1-CIC complex is the primary driver of cerebellar pathology in spinocerebellar ataxia type 1 through a gain-of-function mechanism. Neuron. 97, 1235‒1243.
Takahashi K., Yamanaka S. (2006) Induction of pluripotent stem cells from mouse embryonic and adult fibroblast cultures by defined factors. Cell. 126, 663‒676.
Nekrasov E.D., Vigont V.A., Klyushnikov S.A., Lebedeva O.S., Vassina E.M., Bogomazova A.N., Chestkov I.V., Semashko T.A., Kiseleva E., Suldina L.A., Bobrovsky P.A., Zimina O.A., Ryazantseva M.A., Skopin A.Y., Illarioshkin S.N., Kaznacheyeva E.V., Lagarkova M.A., Kiselev S.L. (2016) Manifestation of Huntington’s disease pathology in human induced pluripotent stem cell-derived neurons. Mol. Neurodege-neration. 11, 27.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология