

Молекулярная биология, 2019, T. 53, № 6, стр. 982-997
Методы оценки эффективности работы систем CRISPR/Cas при геномном редактировании
Н. А. Ломов a, b, *, В. С. Вьюшков a, b, **, А. П. Петренко a, М. С. Сыркина a, b, М. А. Рубцов a, b, c, ***
a Биологический факультет Московского государственного университета им. М.В. Ломоносова
119991 Москва, Россия
b Международная ассоциированная лаборатория LIA LFR2O “Laboratoire franco-russe de recherches en oncologie”, Московский государственный университет им. М.В. Ломоносова
119991 Москва, Россия
c Первый Московский государственный медицинский университет им. И.М. Сеченова Минздрава России
(Сеченовский университет)
119991 Москва, Россия
* E-mail: lomov13@gmail.com
** E-mail: vyushkov22@gmail.com
*** E-mail: ma_rubtsov@mail.ru
Поступила в редакцию 16.05.2019
После доработки 16.05.2019
Принята к публикации 04.07.2019
Аннотация
На сегодняшний день для редактирования геномов широко используется система CRISPR/Cas. В процессе редактирования генома клеток необходимо, во-первых, подобрать наиболее эффективный РНК-гид и, во-вторых, провести анализ полученных клонов на наличие искомой мутации. В представленном обзоре рассмотрены методы, позволяющие оценить эффективность работы систем CRISPR/Cas и подтвердить внесение мутации в целевой локус, а также отмечены их сильные и слабые стороны. Данный обзор призван помочь исследователям выбрать оптимальный метод в зависимости от конкретной задачи и доступных ресурсов.
ВВЕДЕНИЕ
Редактирование генома широко применяют в исследовательских целях: точечных мутантов используют для изучения функций генов и белков. Также редактирование генома позволяет создавать клеточные модели заболеваний, вызванных мутациями. На таких моделях изучают механизмы развития заболеваний и используют их для разработки новых терапевтических подходов. Наконец, редактирование генома само по себе может стать доступным и эффективным подходом к лечению наследственных заболеваний и опухолей.
В последнее десятилетие появилось несколько инструментов, благодаря которым редактирование генома перестало быть трудоемким процессом, во многом зависящим от удачи. Эти инструменты основаны на трех системах узнавания и разрезания ДНК: ZFN (zinc-finger nuclease), TALEN (transcription activator-like effector nuclease) и CRISPR/Cas (clustered regularly interspaced short palindromic repeats/CRISPR-associated protein). Перечисленные системы позволяют при внедрении в клетку направленно вносить двухцепочечный разрыв (ДЦР) в заданную последовательность ДНК, что служит основой соответствующих технологий редактирования генома [1]. Первые две системы предполагают предварительное конструирование белковых доменов, узнающих целевую последовательность ДНК, тогда как система CRISPR/Cas ‒ конструирование короткого РНК-гида (gРНК), который и будет направлять нуклеазу в место внесения разрыва в ДНК. Узнающая часть gРНК составляет всего 20 нуклеотидов, поэтому модификация системы CRISPR/Cas для узнавания новой целевой последовательности крайне проста. Применение системы CRISPR/Cas стало настоящим прорывом в области редактирования генома ‒ достаточно подобрать последовательность gРНК и ввести в клетки эту РНК и редактирующую нуклеазу (например, Cas9), чтобы получить хотя бы в некоторых клетках искомую мутацию [2]. Именно поэтому система CRISPR/Cas получила самое широкое распространение, и в обзоре мы сосредоточимся на практических аспектах работы именно с ней.
СИСТЕМА CRISPR/Cas КАК ИНСТРУМЕНТ РЕДАКТИРОВАНИЯ ГЕНОМА
В природе система CRISPR/Cas представляет собой прокариотический аналог адаптивного иммунитета, который направлен на борьбу с бактериофагами и другими инородными молекулами ДНК [3]. Существует несколько типов CRISPR/Cas-систем (всего на конец 2018 года известно 6 типов [4]). Для редактирования генома обычно используют систему II типа. Системы других типов применяют значительно реже, поэтому их описание выходит за рамки этого обзора.
Эффекторный элемент системы CRISPR/Cas II типа ‒ нуклеаза Cas9. Этот фермент образует комплекс с gРНК, которая по принципу комплементарности узнает ДНК-мишень, что необходимо для внесения ДЦР. Помимо gРНК в эффекторный комплекс входит и трансактивирующая РНК, которая комплементарно взаимодействует с gРНК. Соединение gРНК и трансактивирующей РНК в одну химерную молекулу, sgРНК, не влияет на эффективность внесения разрывов, но в большинстве случаев удобнее для генной инженерии (рис. 1) [5]. Далее по тексту мы не будем делать различий между терминами “РНК-гид” и “химерный РНК-гид”. Нуклеаза Cas9 может внести разрыв в ДНК только рядом с последовательностью, которая называется мотивом, примыкающим к протоспейсеру (protospacer adjacent motif, PAM). Для Сas9 из Streptococcus pyogenes последовательность PAM – это NGG, где N – любой нуклеотид. PAM примыкает к 3'‑концу последовательности, распознаваемой gРНК. Нуклеаза Cas9 вносит ДЦР на расстоянии 3‒4 нуклеотидов от PAM в направлении 5'‑конца цепи ДНК (рис. 1) [6].
Рис. 1.
Принцип работы нуклеазы Cas9. Показана нуклеаза Cas9 в комплексе с ДНК-мишенью. Для узнавания последовательности ДНК нуклеазе Cas9 необходима молекула РНК-гида (gРНК), состоящая из 20-нуклеотидной последовательности (показана оранжевым цветом), комплементарной целевому участку на ДНК, и константного каркасного участка (показан зеленым цветом). Cas9 вносит двухцепочечный разрыв в ДНК, отступив 3 нуклеотида от последовательности PAM (NGG для Сas9 из Streptococcus pyogenes). Для редактирования генома в клетку вводят плазмидные конструкции, содержащие ген Cas9 и ген gРНК, причем за выбор точки редактирования отвечает лишь небольшой участок длиной 20 нуклеотидов в гене gРНК.
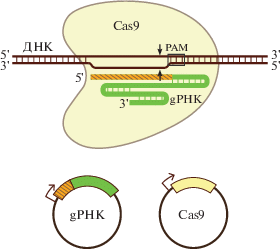
Репарация внесенного нуклеазой Cas9 разрыва по пути негомологичного соединения концов (non-homologous end joining, NHEJ) часто приводит к инсерциям или делециям. При внесении разрыва в кодирующую последовательность можно добиться сдвига рамки считывания, что используется для генного нокаута. А если вводить в клетку матрицу для гомологичной рекомбинации одновременно с внесением разрыва, то можно направленно измененять кодирующую последовательность генов или интегрировать в геном новые конструкции [3].
Методика кажется простой, но в ней скрыто множество технических деталей. Получение клеток с отредактированным геномом подразумевает несколько основных стадий. Во-первых, это трансфекция исходной культуры клеток компонентами системы: плазмидами, содержащими гены нуклеазы Cas9 и gРНК. Во-вторых, это разделение культуры клеток после трансфекции и выращивание клонов ‒ колоний из одиночных клеток. Клоны проверяют на наличие искомой мутации, и из клонов с нужной мутацией получают линию клеток с отредактированным геномом.
Получение клонов и последующий анализ их генома ‒ самая затратная по времени и средствам часть работы. И чем ниже вероятность редактирования, тем больше приходится выращивать и анализировать клонов для обнаружения клеток с нужной мутацией. Известно, что не все gРНК эффективно выполняют свою функцию, даже если они были подобраны с помощью компьютерных алгоритмов [7]. С целью упростить задачу проводят предварительную проверку или даже количественную оценку эффективности этих РНК. Такая проверка позволяет заранее отбросить неудачные варианты и повысить вероятность редактирования. Первой группой методов, которые мы рассмотрим в обзоре, будут методы проверки эффективности gРНК.
Для анализа полученных клонов на предмет редактирования ДНК (наличия необходимой мутации) с ДНК каждого клона проводят амплификацию области редактирования. Секвенирование дает полную информацию о последовательности нуклеотидов в районе редактирования, но секвенирование ДНК каждого из многочисленных клонов затратно и занимает несколько дней. Разработаны методы, которые позволяют обойтись без секвенирования всех клонов. Требуемым результатом редактирования может быть: 1) любой сдвиг рамки считывания в случае генного нокаута, 2) конкретные нуклеотидные замены или 3) вставка/делеция по матрице для гомологичной рекомбинации. В зависимости от типа требуемой мутации выбирают тот или иной подход для ее детектирования. При выборе важным фактором становится доступность метода в условиях конкретной лаборатории, что включает стоимость реагентов и наличие необходимого оборудования.
Уже есть обзоры по методам, предназначенным для детектирования событий редактирования генома. Germini с соавт. [8] рассмотрели методики, пригодные для оценки эффективности нуклеаз, в то время как Zischewski с соавт. [9] сосредоточили внимание на методах выявления событий редактирования в клонах. Здесь мы обсуждаем и те, и другие методы, в том числе появившиеся в последнее время, и акцентируем внимание на их различиях. В результате читатель сможет легко сориентироваться и выбрать подходящий для решения его задачи метод с учетом возможностей лаборатории.
Редактирование генома в терапевтических целях пока не распространено из-за внесения системой CRISPR/Cas разрывов также и в нецелевые локусы ‒ так называемые офтаргеты (off-target), ‒ что ведет к риску возникновения побочных эффектов. Для применения в терапии необходимо, во-первых, использовать системы редактирования с высочайшей специфичностью и, во-вторых, проверять нуклеазы не только на эффективность внесения разрывов в целевые локусы, но также и на минимализацию офтаргетов. Методы обнаружения офтаргетов трудозатратны, так как требуется проверка всех возможных вариантов или хотя бы нескольких наиболее вероятных, предсказанных с помощью компьютерных алгоритмов. Именно поэтому такую проверку часто опускают, когда редактирование генома проводится исключительно для исследовательских целей. Для ознакомления с методами проверки gРНК на предмет офтаргетов предлагаем обратиться к обзору Zischewski и др. [9].
МЕТОДЫ ОЦЕНКИ ЭФФЕКТИВНОСТИ РНК-ГИДА
Как правило, оценку эффективности редактирования проводят по следующей схеме: культуру клеток трансфицируют генами gРНК и нуклеазы Cas9 или в клетки вводят очищенный фермент и gРНК. Затем всю популяцию анализируют на предмет наличия клеток, в которых произошла какая-либо мутация. Оценив долю мутантных аллелей относительно немутантных (wt, дикий тип), делают вывод об эффективности гида. Среди отредактированных молекул ДНК встречаются как нуклеотидные замены, так и инсерции или делеции, причем различной длины [10]. В связи с этим методы оценки эффективности редактирования должны быть не специфичны к типам мутаций, которые возникают при внесении ДЦР. Наиболее релевантным методом считается высокопроизводительное секвенирование (next generation sequencing, NGS), однако существуют и другие методы ‒ более дешевые, хотя и менее точные: T7E1-тест, SURVEYOR, RFLP (полиморфизм длин рестрикционных фрагментов), TIDE (метод отслеживания инделей через декомпозицию), IDAA (анализ ампликонов с инделями), pDR/FACS (плазмида/активируемая флуоресценцией сортировка клеток), ENIT (индуцированные нуклеазами транслокации), qEva-CRISPR. Методы отличаются по чувствительности ‒ порогу детекции клеток с мутацией (в процентах от общего числа клеток). Отметим, что чувствительность метода зависит и от типа мутаций. При оценке эффективности особенно важно ставить контроль с клетками, не трансфицированными gРНК, так как исходные клетки могут быть гетерозиготными по редактируемому локусу, что даст ложноположительный результат. Также стоит отметить, что большинство описываемых ниже методов включает в себя этап амплификации ДНК. В связи с этим при расчетах эффективности редактирования следует учитывать, что различные последовательности ДНК могут амплифицироваться с разной эффективностью.
NGS ‒ высокопроизводительное секвенирование
Технология NGS считается наиболее полным и достоверным подходом для анализа эффективности редактирования генома. С помощью NGS исследователь получает четкое представление о результатах редактирования: типах мутаций в точке редактирования (инсерции/делеции, нуклеотидные замены, транслокации), частоте мутаций, размере инсерций и делеций [6–11].
Очевидный недостаток подходов, основанных на NGS, ‒ высокая стоимость, поэтому они практически не применимы для рутинного анализа gРНК. Однако только NGS позволяет одновременно проверить gРНК еще и на возможные офтаргеты. В качестве более дешевой альтернативы NGS можно использовать такой подход: продукты амплификации области редактирования клонируют в плазмиды, и несколько десятков или сотен таких плазмид секвенируют по Сэнгеру [12]. Но такой подход занимает гораздо больше времени, чем NGS.
T7E1-тест и SURVEYOR ‒ методы, основанные на разрезании ДНК по неправильно спаренным основаниям
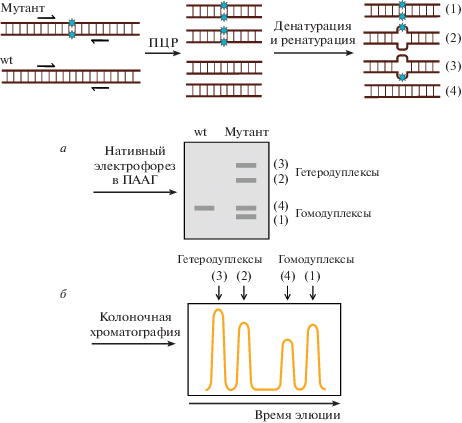
Методы, основанные на разрезании нуклеазами T7E1 и SURVEYOR гетеродуплексных фрагментов ДНК, наиболее широко используются для анализа результатов редактирования генома. Когда одноцепочечные фрагменты, содержащие мисматчи (неправильно спаренные основания) и инсерционно-делеционные мутации (индели), отжигаются на фрагменты немодифицированных молекул ДНК, получаются гетеродуплексы [13]. По точкам мисматчей в таких гетеродуплексах происходит разрезание нуклеазой. В случае T7E1-анализа используется резолваза E1 бактериофага T7 [14], а в случае SURVEYOR – нуклеазы семейства CEL из сельдерея [15].
Как в случае T7E1-анализа, так и SURVEYOR, участок ДНК в районе точки редактирования амплифицируют с помощью ПЦР. Затем ПЦР-продукты нагревают для денатурации и медленно охлаждают для ренатурации. В связи с тем, что в ПЦР-смеси присутствуют разные молекулы ДНК ‒ одни содержат индели в результате NHEJ, а другие представляют собой неотредактированные молекулы, ‒ после ренатурации формируются гетеродуплексы. Чем эффективнее Cas9-нуклеаза вносит разрыв, тем больше образуется инделей и больше гетеродуплексных молекул ДНК сформируется после ренатурации. Затем ПЦР-продукты обрабатывают нуклеазами T7E1 или SURVEYOR и продукты разрезания анализируют электрофорезом в агарозном или полиакриламидном геле (рис. 2). По отношению интенсивности полос продуктов разрезания и интактных молекул вычисляют эффективность внесения ДЦР нуклеазой Cas9 с проверяемой gРНК [6, 13]. В этом методе обязателен отрицательный контроль ‒ все процедуры повторяют с клетками, не трансфицированными gРНК. В противном случае можно получить ложноположительный результат, например, из-за возможных однонуклеотидных полиморфизмов (SNP) в амплифицируемой области в популяции исходных клеток.
Рис. 2.
Принцип методов, основанных на разрезании гетеродуплексов ДНК нуклеазами T7E1 и SURVEYOR. Долю молекул ДНК с мутациями рассчитывают по соотношению интенсивности полос продуктов разрезания и интенсивности полосы полноразмерного ПЦР-продукта на электрофореграмме (R ‒ редактируемая ДНК, wt ‒ нередактируемая ДНК). Стрелками указаны продукты разрезания ДНК нуклеазами при использовании РНК-гида низкой эффективности редактирования (1) и высокой (2).
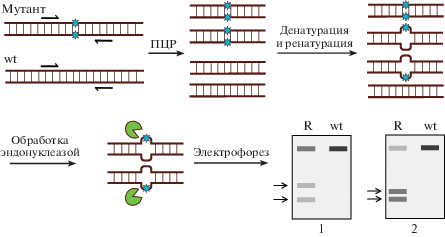
При сравнении T7E1- и SURVEYOR-нуклеаз следует отметить, что первая более чувствительна к крупным инделям (возможно детектировать молекулы с инделями, составляющие 0.5% от общего пула молекул), но малоэффективна при разрезании коротких инделей и особенно мисматчей. Эффективность T7E1-нуклеазы зависит от условий инкубации, соотношения ДНК/фермент и т.д., поэтому в каждом случае необходима оптимизация всех параметров. SURVEYOR-нуклеаза дороже, обладает меньшей чувствительностью (порог ‒ около 3% мутантных молекул), однако подходит для детекции коротких инделей и мисматчей и, что важно, имеет стандартизированный протокол [13].
Несмотря на популярность T7E1-теста, он дает лишь приблизительную оценку эффективности внесения разрывов. Так, при сравнении результатов T7E1-теста и данных NGS по популяции трансфицированных клеток первый давал заниженные результаты с большинством из исследованных gРНК. Более того, результаты T7E1-теста скорее отражали разнообразие продуктов редактирования ‒ чем больше вариантов инсерций и делеций среди продуктов редактирования с использованием данной gРНК, тем более эффективной она окажется по результатам T7E1-теста. Для тех gРНК, которые давали один преимущественный продукт редактирования (например, делецию одного нуклеотида), одинаковые мутантные молекулы отжигались друг на друга без образования гетеродуплексов с мисматчами. По этой причине результаты T7E1-анализа оказывались заниженными по сравнению с результатами NGS для тех же gРНК. Тем не менее, результаты T7E1-теста никогда не оказывались завышенными, т.е. он не дает ложноположительных результатов при анализе популяции клеток [12].
Методы T7E1 и SURVEYOR применяют также для анализа клонов на наличие мутации в целевом локусе. В этом случае к протоколу добавляется еще один шаг: перед денатурацией ДНК, выделенную из клона, смешивают с ДНК исходных немодифицированных клеток. Это необходимо, чтобы исключить получение ложноотрицательных результатов для клонов, содержащих одинаковую (биаллельную) мутацию в гомологичных хромосомах.
RFLP ‒ полиморфизм длин рестрикционных фрагментов
Полиморфизм длин рестрикционных фрагментов (restriction fragment length polymorphism, RFLP) ‒ давно разработанный метод [16], который применялся для сравнения геномов, а затем и для геномного редактирования [17]. Именно RFLP первоначально использовали для детекции инделей в точке, разрезаемой CRISPR/Cas. При использовании этого метода РНК-гиды изначально подбирают так, чтобы точка расщепления оказалась внутри сайта узнавания какой-нибудь рестриктазы. Из смешанной популяции клеток после трансфекции выделяют ДНК и амплифицируют область, включающую точку редактирования. Затем ПЦР-продукт обрабатывают рестриктазой и анализируют электрофоретически. Полосы соответствуют продуктам расщепления и нерасщепленным молекулам, которые несут мутации в сайте узнавания рестриктазой вследствие возникновения инделей. По соотношению интенсивностей полос делают вывод об эффективности внесения разрыва (рис. 3).
Рис. 3.
Принцип метода RFLP. Зеленой рамкой обозначен сайт узнавания эндонуклеазой. На электрофоретическом геле стрелками указаны продукты рестрикции. R ‒ редактируемая ДНК, wt ‒ нередактируемая ДНК. Доля молекул ДНК с мутациями вычисляется на основании отношения интенсивности полосы полноразмерного ПЦР-продукта к интенсивности полос продуктов разрезания (указаны стрелками): 1 – результат для РНК-гида с более высокой эффективностью редактирования, 2 – результат для РНК-гида с низкой эффективностью редактирования.
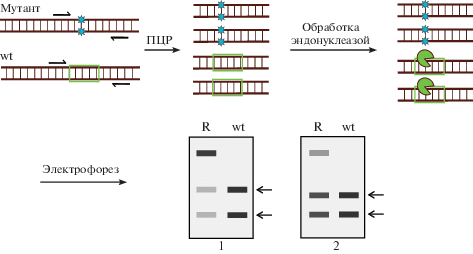
Важное преимущество метода RFLP ‒ при анализе клонов можно отличить биаллельную мутацию от дикого типа, что не получается сделать популярными методами Т7Е1 или SURVEYOR в стандартном исполнении. Метод довольно надежный, но изначально накладывает ограничения на последовательности, в которые будет вноситься разрыв. Также для него требуется наличие соответствующей рестриктазы.
Для обхода ограничения на последовательность, в которую будет вноситься разрыв, вместо эндонуклеаз рестрикции применяют фермент Cas9 в комплексе с соответствующей gРНК. Метод носит название RGEN-RFLP (RNA-guided engineered nucleases). Этот метод гораздо более точно отражает число событий редактирования, чем Т7Е1 [18]. Недостаток ‒ необходимость в выделенной рекомбинантной Cas9 и предварительно синтезированной in vitro gРНК, а также в оптимизации условий реакции для Cas9 (подбор концентрации фермента).
TIDE ‒ отслеживание инделей через декомпозицию
Метод отслеживания инделей через декомпозицию (tracking of indeles by decomposition, TIDE) основывается на биоинформатическом анализе результатов сэнгеровского секвенирования ПЦР-продуктов амплификации редактируемого локуса в культуре трансфицированных клеток [19]. Этот метод позволяет с высокой точностью предсказывать частоты различных делеций и инсерций, возникающих в редактируемом локусе (рис. 4). Необходимо заметить, что в базовом варианте учитываются только инсерции +1, как наиболее часто встречающиеся. TIDE достоверно предсказывает индели, если их частота превышает 1‒2%, что в некоторых случаях превосходит чувствительность T7E1-анализа и SURVEYOR [13, 19]. Точность предсказания по алгоритму TIDE сопоставима с результатами NGS [12]. К недостатками этого метода относится ошибочность предсказаний для часто повторяющихся последовательностей, а также требовательность к высокому качеству ПЦР-продукта и его прочтения [19].
Рис. 4.
Метод TIDE. Из популяции трансфицрованных клеток выделяют ДНК и проводят ПЦР-амплификацию редактируемого локуса; при этом получают как ПЦР-продукты с исходной последовательностью (wt), так и ПЦР-продукты с инсерциями и делециями (1‒4), возникающими при негомологичном соединении концов. При сэнгеровском секвенировании такой смеси ПЦР-продуктов после точки разрыва происходит наложение последовательностей от разных матриц. Путем декомпозиции данных секвенирования рассчитывают частоты различных делеций и однонуклеотидных инсерций.
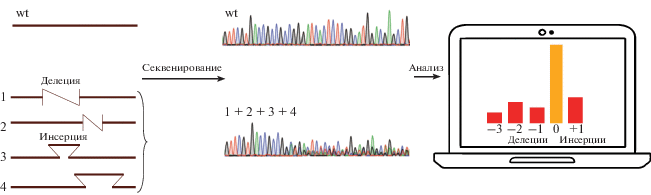
IDAA ‒ анализ ампликонов с инделями
IDAA расшифровывается как indel detection by amplicon analysis ‒ анализ ампликонов с инделями. Как и описанные ранее методы, IDAA предполагает амплификацию области редактирования с фланкирующих праймеров. В одном из праймеров содержится флуорофор. После амплификации ПЦР-продукты разделяют методом капиллярного электрофореза и детектируют по флуоресценции (рис. 5). Разрешающей способности метода достаточно для различения молекул, отличающихся по длине на один нуклеотид [20]. Таким образом, метод IDAA можно использовать для определения частот инсерций и делеций широкого диапазона (от –100 до +100) в пуле трансфицированных клеток. Чувствительность метода составляет около 1% мутантных молекул среди немодифицированных, что сопоставимо с чувствительностью NGS. Однако разрешающей способности капиллярного электрофореза бывает недостаточно для различения +1 и –1 инделей с аллелями дикого типа. А главный недостаток IDAA заключается в неспособности детектировать замены нуклеотидов и транслокации с участием локуса редактирования [12].
Рис. 5.
Принцип метода IDAA для оценки частоты инсерций и делеций в пуле трансфицированных клеток. Область ДНК в месте редактирования амплифицируется с помощью трех праймеров, один из которых универсальный и содержит флуорофор (FLU_F), а два других подобраны под конкретный редактируемый участок. ПЦР-продукты анализируют капиллярным электрофорезом. На графике справа от ПЦР продуктов дикого типа (wt) находятся молекулы ДНК с инсерциями, а слева – с делециями.
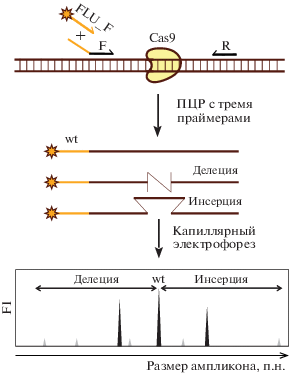
Вместо капиллярного электрофореза для анализа ампликонов можно использовать электрофорез в 15%-ном ПААГ в неденатурирующих условиях при низкой скорости [21]. В таком варианте метод доступен в лабораториях, где нет системы капиллярного электрофореза, однако требует значительных затрат времени (до нескольких часов).
Метод pDR/FACS
Метод pDR/FACS (от названия плазмиды pDR и метода FACS ‒ fluorescence-activated cell sorting) отличается от описанных ранее, так как эффективность редактирования в нем оценивают не по частоте мутаций, возникающих в ходе NHEJ, а по частоте гомологичной рекомбинации ‒ другого пути репарации ДЦР. Система на основе pDR-плазмиды была создана для оценки эффективности процесса гомологичной рекомбинации [22]. Аналогичный подход стали использовать для оценки эффективности работы нуклеаз. В этом методе клетки трансфицируют плазмидами, содержащими ген сas9 и ген проверяемой gРНК, а также плазмидой pDR. Плазмида pDR включает в себя 2 копии кодирующих последовательностей зеленого флуоресцентного белка (GFP), но обе они нефункциональны. Первая копия содержит сайт узнавания проверяемого гида, сдвигающий рамку считывания, а вторая содержит лишь центральную область гена gfp, но с правильной последовательностью. Копии разделены последовательностью длиной почти 4 тыс.п.н., что делает возможным использование одной копии в качестве матрицы для репарации другой копии после внесения в нее разрыва нуклеазой.
В случае успешной работы системы CRISPR/Cas в дефектный ген gfp будет внесен разрыв, и после гомологичной рекомбинации с участием второго гена gfp образуется полноценный ген (рис. 6). Эффективность репарации оценивают с помощью FACS-анализа клеток [23]. Однако в связи с тем, что большинство разрывов ДНК в клетках млекопитающих репарируется не гомологичной рекомбинацией, а путем NHEJ [24], оценка эффективности редактирования генома методом pDR/FACS оказывается заниженной.
Рис. 6.
Принцип использования pDR-плазмиды для оценки эффективности редактирования генома. В левую копию гена gfp вставлен участок узнавания проверяемого РНК-гида, что вызывает сдвиг рамки считывания. Правая копия гена gfp лишена промотора и терминатора. Репарация внесенного нуклеазой Cas9 двухцепочечного разрыва (ДЦР) приводит к восстановлению функционального гена gfp.
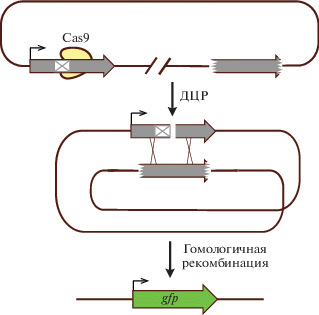
Таким образом, метод pDR/FACS подразумевает сначала получение плазмиды pDR со вставкой последовательности целевого локуса. Хотя на этот этап уходит определенное время, использование метода pDR/FACS позволяет оценить, какая из gРНК больше подходит именно для интеграции в геном конструкции по пути гомологичной рекомбинации.
ENIT ‒ индуцированные нуклеазами транслокации
Метод ENIT (engineered nuclease-induced translocations) основан на том, что при внесении нескольких разрывов в ДНК образуются хромосомные транслокации [25]. В исходном варианте метод применяли для оценки эффективности работы нуклеаз TALEN [26], а в нашей лаборатории ‒ и для системы CRISPR/Cas (неопубликованные данные). Клетки трансфицируют тремя плазмидами. Одна из них содержит ген сas9. Вторая плазмида содержит ген gРНК (к какому-либо участку “X” ДНК), для которой ранее уже была показана высокая эффективность внесения разрывов. Третья плазмида содержит ген проверяемой gРНК (к участку ДНК “Y”). Если Cas9 в комплексе с проверяемой gРНК эффективно вносит разрывы, то между локусами X и Y возникают хромосомные транслокации, которые могут быть детектированы с помощью ПЦР. Сравнение эффективности гидов может быть проведено по разнице в интенсивности полос на электрофорезе (рис. 7) или количественной ПЦР [26].
Рис. 7.
Принцип метода ENIT. Знаком “?” отмечена плазмида с проверяемым РНК-гидом к гену Y. После внесения разрывов между генами X и Y возможно образование четырех вариантов хромосомных транслокаций, которые детектируют c помощью ПЦР. X_F, X_R, Y_F, Y_R ‒ праймеры для ПЦР. Справа показан результат электрофоретического разделения ПЦР-продуктов амплификации транслокаций, индуцированных при проверке gРНК к гену Y (в данном случае ‒ ген AML1). Контроль ‒ нетрансфицированные клетки.
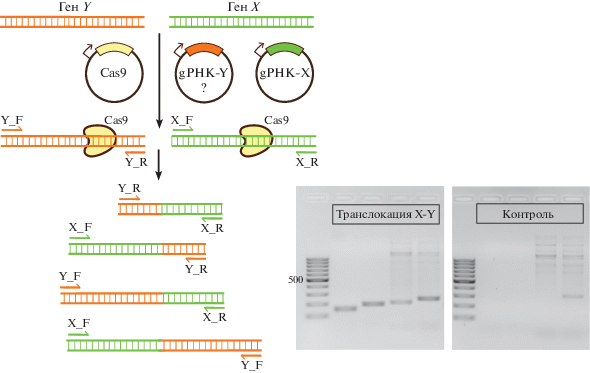
К недостаткам метода ENIT следует отнести определение только относительной эффективности редактирования. Это не позволяет узнать, какая доля клеток содержит индуцированную транслокацию. Тем не менее, метод подходит, когда подбираемую gРНК в дальнейшем планируют использовать для индукции транслокаций, например, при моделировании опухолевых заболеваний [27]. Преимущество ENIT состоит в том, что, в отличие от остальных методов, для его применения не требуется сложного оборудования или редких реактивов ‒ только стандартное оборудование и реактивы для ПЦР и электрофореза.
Цифровая капельная ПЦР
При выполнении цифровой капельной ПЦР (droplet digital PCR) 20 мкл реакционной смеси предварительно разделяют на 20 000 капель, в которых реакции ПЦР протекают независимо. Затем подсчитывают число капель, в которых прошла требуемая реакция, и делают вывод о количественном содержании амплифицируемого фрагмента ДНК в исходной смеси [28].
Для проверки эффективности gРНК с помощью цифровой капельной ПЦР используют 2 гибридизационные пробы, которые, будучи отожжены, дают флуоресцентный сигнал (например, TaqMan-пробы). Первая проба отжигается на все молекулы ДНК вне зависимости от наличия мутаций (референс). Участок отжига второй флуоресцентной пробы перекрывается с сайтом редактирования, поэтому сигнал второго цвета возникает только для молекул ДНК дикого типа. С помощью анализатора капель вычисляют долю капель, флуоресцирующих только одним цветом, и на основании этого ‒ эффективность редактирования (рис. 8).
Рис. 8.
Использование цифровой капельной ПЦР для оценки эффективности редактирования. Амплификация молекул ДНК дикого типа (wt) и молекул ДНК с мутациями проходит в каплях эмульсии. В каплях также содержатся флуоресцентные пробы, одна из которых (оранжевая*) отжигается как на продукты амплификации молекул ДНК дикого типа, так и на продукты амплификации молекул с мутациями. Вторая проба (зеленая) отжигается только на молекулы ДНК без мутаций в области редактирования. Анализ флуоресценции капель после ПЦР позволяет вычислить долю молекул ДНК с мутациями и оценить эффективность редактирования.
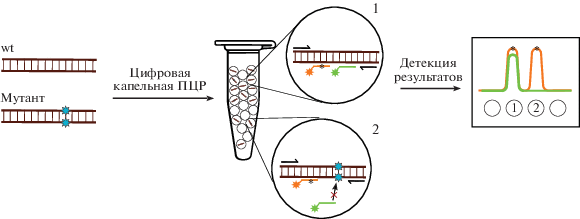
К недостаткам использования цифровой капельной ПЦР относится необходимость в дорогостоящем оборудовании, реагентах и программном обеспечении.
qEva-CRISPR
Метод qEva-CRISPR основан на лигировании двух половинок олигонуклеотидной пробы, подобранной к участку редактирования, и определении количества продуктов лигирования [29]. Проба подбирается так, что половинки отжигаются на ДНК с двух сторон от точки редактирования, и их концы оказываются рядом. ДНК клеток, трансфицированных для редактирования, смешивают с олигонуклеотидными пробами и проводят реакцию лигирования. Затем количество продуктов лигирования оценивают, амплифицируя их с помощью ПЦР и анализируя на капиллярном электрофорезе. Если происходит мутация, то конец половинки пробы не отжигается на ДНК и лигирования половинок пробы не происходит, что приводит к отсутствию ПЦР-продукта. Количество мутантной ДНК нормируется на общее количество ДНК, для чего в ту же реакцию лигирования добавляют состоящую из двух половинок пробу к интактному участку генома и праймеры для ее амплификации (рис. 9).
Рис. 9.
Метод qEva-CRISPR для оценки эффективности редактирования. ДНК клеток смешивают с олигонуклеотидными пробами, одна пара которых отжигается в области редактирования (зеленые), а другая ‒ на референсном участке, который не подвергался редактированию (оранжевая*). Затем проводят лигирование и добавляют праймеры, специфичные к олигонуклеотидным пробам. ПЦР с последующим капиллярным электрофорезом позволяют рассчитать соотношение продуктов лигирования пробы к локусу редактирования и к референсному локусу, оценив частоту событий редактирования.
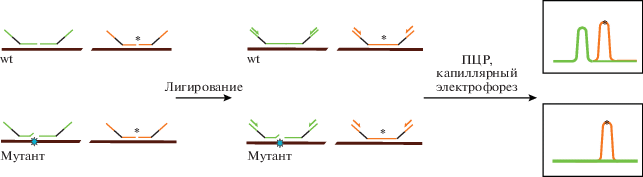
Метод qEva-CRISPR имеет ряд преимуществ. Во-первых, в одной реакции можно проводить детекцию сразу нескольких фрагментов. Во-вторых, его точность превышает таковую среди всех перечисленных методов количественной оценки частоты редактирования. Это объясняется тем, что qEva-CRISPR совершенно не чувствителен к типу мутаций, будь то нуклеотидные замены, транслокации или индели любого размера. В-третьих, метод может быть адаптирован для обнаружения нужной мутации среди клонов. В этом случае проба подбирается к отредактированной области, будь то точечная мутация или хромосомная транслокация; при этом можно детектировать как гомозиготные, так и гетерозиготные мутанты. Отметим, что вместо подсчета продукта ПЦР на капиллярном электрофорезе можно использовать количественную ПЦР. К недостаткам метода относится необходимость в длинных олигонуклеотидных пробах для каждой проверяемой gРНК, затраты времени на проведение реакции лигирования и не очень высокая чувствительность (от 5% мутантных клеток в популяции).
ПРОВЕРКА КЛОНОВ
Для проверки клонов на наличие мутации можно использовать некоторые методы из перечисленных выше: T7E1- и SURVEYOR-анализы, RFLP, IDAA, qEva-CRISPR. Их удобно применять в тех случаях, когда на стадии проверки gРНК уже отработан протокол, подобраны праймеры и условия протекания реакций. Кроме того, есть методы, которые пригодны только для проверки клонов: конкурентная ПЦР, анализ плавления высокого разрешения (HRMA), полиморфизм конформаций одноцепочечных ДНК (SSCP) и высокоэффективная хроматография в денатурирующих условиях (DHPLC). Эти методы просты в исполнении и не требуют больших финансовых затрат. После предварительного скрининга клонов с помощью перечисленных методов наличие искомой мутации подтверждают секвенированием по Сэнгеру. Для быстрого выявления клонов с конкретной мутацией используют регистрацию продуктов лигирования (ligation detection reaction, LDR).
Перечисленные методы подходят не только для скрининга клонов при редактировании генома, но и просто для поиска мутаций в интересующих локусах. Более того, большинство из этих методов были разработаны задолго до появления CRISPR/Cas и использовались как раз для изучения процессов мутагенеза или при SNP-генотипировании.
Важная характеристика многих методов, применяющихся для анализа клонов, ‒ возможность распознавания одноаллельных или биаллельных мутаций.
Конкурентная ПЦР
Для проверки клонов на наличие инделей в редактируемом локусе (например, при необходимости нокаутировать ген) разработан простой и дешевый метод, основанный на ПЦР с тремя праймерами ‒ конкурентная ПЦР (competition-based PCR, cbPCR). Два праймера фланкируют точку разрыва (внешние праймеры), а третий 3'-концом перекрывает точку разрыва (внутренний праймер). При ПЦР внешний и внутренний праймеры “конкурируют” за одну и ту же матрицу и образуют два продукта в определенном соотношении. Если же внутренний праймер отжигается на ДНК с меньшей эффективностью, чем внешний, что случается при внесении инделей, то соотношение между двумя продуктами изменяется в сторону продукта с внешних праймеров (рис. 10). Соотношение измеряется по картинке электрофореза в программе Fiji (ImageJ) [30] или других. Важно, что ПЦР-продукт, который свидетельствует о наличии инделей в точке разреза, может быть сразу клонирован в плазмиду, которой затем трансформируют компетентные бактерии. С выросших бактериальных колоний ставят ПЦР для скрининга на наличие вставки в плазмиде и этот ПЦР-продукт сразу отправляют на секвенирование для подтверждения события редактирования [31].
Рис. 10.
Метод конкурентной ПЦР. Амплификацию проводят с тремя праймерами. Участок отжига внутреннего праймера перекрывается с сайтом редактирования. При наличии мутации эффективность отжига внутреннего праймера падает, что детектируют по уменьшению интенсивности окраски короткого ПЦР-продукта на агарозном геле.
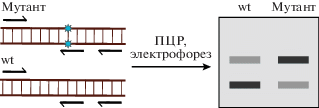
Усовершенствование описанного метода конкурентной ПЦР — использование количественной ПЦР для подсчета соотношения продуктов. После анализа кривых накопления ДНК оценивают соотношение продуктов, полученных с внешнего и внутреннего праймеров, и делают вывод о том, в каких клонах произошло редактирование.
К недостаткам метода конкурентной ПЦР относится низкая чувствительность к мутациям, которые мало влияют на отжиг внутреннего праймера. Это могут быть инсерции, не перекрываемые праймером, или мисматчи, оказывающиеся не на самом конце праймера. В связи с этим рекомендуется подбирать два комплекта праймеров ‒ чтобы внутренний праймер отжигался либо на верхнюю, либо на нижнюю цепь ДНК в точке редактирования [32].
HRMA ‒ анализ плавления высокого разрешения
Анализ плавления высокого разрешения (high resolution melt analysis, HRMA) позволяет различать продукты амплификации ДНК без мутаций (отрицательный контроль) и продукты амплификации ДНК с точечными мутациями, возникающими в результате редактирования. Праймеры подбираются так, чтобы различия в кривых плавления ампликонов, отличающихся на несколько нуклеотидов, были максимальными. Оптимальная длина ПЦР-продукта для детекции небольших инделов – 100 п.н., для различий в 1 нуклеотид оптимальный размер ампликона – 50 п.н. [33]. Обычно для подбора праймеров используют специальное программное обеспечение [34]. Также может потребоваться предварительная оптимизация протокола ПЦР.
Продукты амплификации денатурируют и ренатурируют в присутствии интеркалирующего красителя, после чего медленно нагревают, регистрируя снижение флуоресценции (рис. 11) [35]. В этом случае на кривой плавления регистрируют точки плавления гомодуплексов с мутациями, гомодуплексов дикого типа и гетеродуплексов. Существуют варианты HRMA без стадии образования гетеродуплексов ‒ в этом случае на кривой плавления регистрируют точки плавления только мутантных гомодуплексов и гомодуплексов дикого типа [36].
Рис. 11.
Принцип HRMA. После амплификации редактируемого локуса ПЦР-продукты подвергают денатурация и ренатурации и проводят анализ кривой плавления. Знаком (*) на кривой плавления отмечены температуры плавления гомо- и гетеродуплексов.
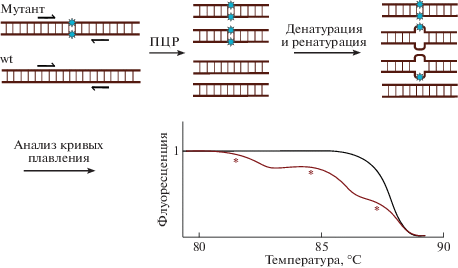
Разрешающая способность метода HRMA достаточна для обнаружения SNP [37], и иногда его используют в качестве альтернативы секвенированию для генотипирования организмов или клонов клеток. В связи с тем, что форма кривых плавления зависит от относительного содержания мутантных молекул ДНК среди всех ПЦР-продуктов, и при этом чувствительность метода составляет до 2% [35], его можно применять для количественного анализа эффективности работы gРНК. Метод HRMA легко масштабируется, и анализ множества образцов ДНК можно проводить в 96-лучночных планшетах. Удобно, что выявленные методом HRMA образцы мутантной ДНК могут быть сразу отправлены на секвенирование. К недостаткам метода относится необходимость в дорогостоящем оборудовании для анализа кривых плавления или амплификаторе для ПЦР в реальном времени с возможностью снятия кривых плавления и последующим анализом с помощью специальных программ (например, https://dna.utah.edu/uv/uanalyze.html).
SSCP ‒ полиморфизм конформаций одноцепочечных ДНК
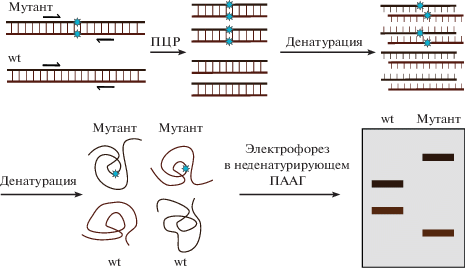
Метод SSCP (полиморфизм конформаций одноцепочечных ДНК, single-strand conformational polymorphism) основан на различной электрофоретической подвижности одноцепочечных молекул ДНК (оцДНК), содержащих различные мутации, в ПААГ или при капиллярном электрофорезе. ОцДНК в неденатурирующих условиях принимает определенную стабильную конформацию. Сначала амплифицируют участок редактирования, затем денатурируют ДНК (для разделения цепей ДНК используют формамид) и анализируют электрофорезом в ПААГ в неденатурирующих условиях ‒ когда фрагменты оцДНК принимают определенную конформацию. Наличие мутаций, таких как мисчматчи и индели, влияет на эту конформацию, что отражается на электрофоретической подвижности фрагментов оцДНК (рис. 12) [38, 39]. Для визуализации полос оцДНК можно использовать окрашивание нитратом серебра или предварительное введение флуорофоров в цепи при проведении ПЦР. Как и метод HRMA, SSCP можно рассматривать как альтернативу секвенированию для проверки наличия мутаций в редактируемом локусе [40].
Рис. 12.
Принцип SSCP-анализа. Проводят амплификацию ДНК из трансфицированных и контрольных клеток, после чего цепи ДНК разделяют с помощью формамида и анализируют электрофорезом в неденатурирующих условиях (с сохранением конформации одноцепочечных участков). Подвижность цепей ДНК с мутацией отличается от цепей ДНК дикого типа.
Анализ регибирдизованных ампликонов
Метод основан на образовании и последующей детекции гетеродуплексных молекул. Этот метод включает следующие стадии: амплификация области редактирования, денатурация ПЦР-продукта и последующая ренатурация, в ходе которой формируются гомо- и гетеродуплексы; регибридизованные ампликоны разделяют электрофорезом в неденатурирующем ПААГ или методом DHPLC.
Анализ с помощью неденатурирующего электрофореза основан на различии в подвижности гетеродуплексов и гомодуплексов (рис. 13а). Фрагменты размером 140‒170 п.н. разделяют электрофорезом в 15%-ном ПААГ. В этом случае чувствительность метода составляет до 0.5% мутантных молекул среди немутантных. К ограничениям метода относится то, что им детектируют только мисматчи и небольшие инсерции в связи с малым размером разделяемых фрагментов ДНК [41].
Рис. 13.
Методы, основанные на разделении гомо- и гетеродуплексов. После амплификации ПЦР-продукты денатурируют и ренатурируют для формирования гомо- и гетеродуплексов (если все ПЦР-продукты идентичны, то формируются только гомодуплексы). Затем такие молекулы ДНК разделяют электрофорезом в ПААГ в неденатурирующих условиях (а) или высокоразрешающей жидкостной хроматографией в денатурирующих условиях (DHPLC) при повышенной температуре (в данном случае 56°С) (б). Профиль элюции строят по поглощению при 254 нм. Для определения того, какие мутантные молекулы ДНК соответствуют пикам на профиле элюции, необходимо иметь калибровочные профили, построенные для молекул ДНК с известными мутациями.
Для обнаружения гетеродуплексных молекул ДНК можно использовать метод обращенно-фазовой хроматографии в частично денатурирующих условиях, DHPLC [42]. Как и при анализе регибридизованных молекул в нативном ПААГ, участок редактирования амплифицируют, после чего проводят денатурацию ПЦР-продукта и отжиг, при котором формируются гомо- и гетеродуплексы. В частично денатурирующих условиях (при повышенной температуре 48‒67°С) участки с мисматчами плавятся, что уменьшает взаимодействие таких молекул ДНК с носителем и приводит к снижению времени удержания их на колонке (рис. 13б). Время элюции гомодуплексов зависит также от их GC-состава.
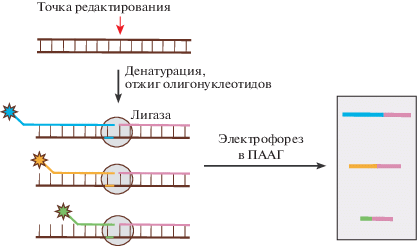
LDR ‒ регистрация продуктов лигирования
Использование вышеприведенных методов позволяет обнаружить только наличие мутации, но не идентифицировать ее. В то же время иногда требуется получение мутанта с какой-то конкретной мутацией – например, заменой одного нуклеотида на другой. В таких случаях, как правило, сначала выявляют клоны с мутациями, а затем область редактирования у таких клонов секвенируют по Сэнгеру, чтобы найти, какая именно из мутаций: инсерция, делеция или замена ‒ произошла в каждом из них.
В качестве более дешевой альтернативы такого подхода можно использовать метод регистрации продуктов лигирования (ligation detection reaction, LDR). Метод похож на вышеописанный qEva-CRISPR, но не предполагает стадии ПЦР и предназначен для работы с ДНК, выделенной из клеточных клонов, а не из смешанной популяции клеток. В методе LDR используют 2 олигонуклеотида. Первый подобран к последовательности с искомой мутацией таким образом, что его 3'-конец отжигается прямо на нуклеотиды, появившиеся в результате редактирования исходной последовательности. Второй олигонуклеотид комплементарен нижележащей последовательности и при отжиге на ДНК вплотную прилегает к первому (рис. 14). Авторы метода [43] детектировали сразу несколько разных мутаций и поэтому использовали несколько первых олигонуклеотидов, которые отличались по длине, а также несли флуорофоры разных цветов. В методе LDR оба олигонуклеотида отжигают на ДНК, выделенную из клеточного клона, после чего проводят реакцию лигирования. Если первый олигонуклеотид эффективно отжегся на ДНК (то есть его 3'-концевой нуклеотид оказался комплементарен нуклеотиду в точке редактирования), произойдет лигирование со вторым олигонуклеотидом и продукт реакции лигирования при электрофорезе в ПААГ будет давать полосу соответствующей длины. Продукты реакции можно детектировать и капиллярным электрофорезом с флуоресцентной детекцией [43].
Рис. 14.
Метод детекции продуктов лигирования. Из клонов, которые проверяют на наличие определенной мутации, выделяют ДНК и смешивают ее с олигонуклеотидами (отмечены сиреневым, а также синим, желтым и зеленым цветом), которые комплементарны участкам справа и слева от точки редактирования. После реакции лигирования продукты разделяют в ПААГ и детектируют наличие тех или иных полос.
ЗАКЛЮЧЕНИЕ
Определение эффективности работы нуклеаз – необходимый этап для успешного редактирования генома, особенно при работе с живыми организмами и в терапевтических целях. На сегодняшний день существует достаточно широкий спектр различных методик для оценки эффективности работы редактирующих нуклеаз. Эти методы различаются по стоимости, затратам времени и характеру получаемой информации. Кроме того, разные методы требуют различного оборудования – от простейших приборов для ПЦР и электрофореза до современного высокопроизводительного секвенатора.
До сих пор нет метода, который совмещает в себе все 3 параметра: высокую точность, быстроту выполнения и низкую стоимость. Подводя итог, можно отметить, что в большинстве случаев исследователи все еще используют методы T7E1 и SURVEYOR, которые, несмотря на достаточно грубую оценку степени редактирования, позволяют быстро сравнивать и выбирать более эффективную gРНК. Когда на выбор gРНК влияет желаемый спектр мутаций, предпочтение отдают подходам, позволяющим определить как частоту, так и характер мутаций среди продуктов редактирования (например, TIDE, IDAA и NGS). Самым простым и наименее затратным на сегодняшний день методом грубой оценки эффективности gРНК можно считать ENIT, в то время как для количественной оценки эффективности процесса редактирования в некоторых случаях оптимален pDR/FACS или qEva-CRISPR. При наличии соответствующего оборудования и возможностей для приобретения реактивов самым удобным может оказаться метод цифровой капельной ПЦР.
Для анализа клонов клеток в поисках искомой мутации можно использовать методы конкурентной ПЦР, HRMA, SSCP и DHPLC с последующим секвенированием по Сэнгеру, а для подтверждения наличия в геноме конкретных мутаций без стадии секвенирования подойдет метод LDR.
Работа выполнена при частичной финансовой поддержке РФФИ в рамках научных проектов № 19-04-00531_А и 19-54-16002 НЦНИЛ_а.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Gaj T., Sirk S.J., Shui S.L., Liu J. (2016) Genome-editing technologies: principles and applications. Cold Spring Harb. Perspect. Biol. 8(12), pii: a023754. https://doi.org/10.1101/cshperspect.a023754
Lomov N.A., Borunova V.V., Rubtsov M.A. (2015) CRISPR/Cas9 technology for targeted genome editing. Biopolym. Cell. 31(4), 243–248.
Sorek R., Lawrence C.M., Wiedenheft B. (2013) CRISPR-mediated adaptive immune systems in Bacteria and Archaea. Annu. Rev. Biochem. 82(1), 237–266.
Makarova K.S., Wolf Y.I., Koonin E.V. (2018) Classification and nomenclature of CRISPR-Cas systems: where from here? Cris. J. 1(5), 325–336.
Jinek M., Chylinski K., Fonfara I., Hauer M., Doudna J.A., Charpentier E. (2012) A programmable dual-RNA-guided DNA endonuclease in adaptive bacterial immunity. Science. 337(6096), 816–821.
Ran F.A., Hsu P.D., Wright J., Agarwala V., Scott D.A., Zhang F. (2013) Genome engineering using the CRIPR-Cas9 system. Nat. Protoc. 8(11), 2281‒2308.
Haeussler M., Schönig K., Eckert H., Eschstruth A., Mianné J., Renaud J.B., Schneider-Maunoury S., Shkumatava A., Teboul L., Kent J., Joly J.S., Concordet J.P. (2016) Evaluation of off-target and on-target scoring algorithms and integration into the guide RNA selection tool CRISPOR. Genome Biol. 17(1), 148.
Germini D., Tsfasman T., Zakharova V.V., Sjakste N., Lipinski M., Vassetzky Y. (2018) A comparison of techniques to evaluate the effectiveness of genome editing. Trends Biotechnol. 36(2), 147–159.
Zischewski J., Fischer R., Bortesi L. (2017) Detection of on-target and off-target mutations generated by CRISPR/Cas9 and other sequence-specific nucleases. Biotechnol. Adv. 35(1), 95–104.
Hartlerode A., Scully R. (2009) Mechanisms of double-strand break repair in somatic mammalian cells. Biochem. J. 423(2), 157–168.
Bell C.C., Magor G.W., Gillinder K.R., Perkins A.C. (2014) A high-throughput screening strategy for detecting CRISPR-Cas9 induced mutations using next-generation sequencing. BMC Genomics. 15(1), 1002.
Sentmanat M.F., Peters S.T., Florian C.P., Connelly J.P., Pruett-Miller S.M. (2018) A survey of validation strategies for CRISPR-Cas9 editing. Sci. Rep. 8(1), 888.
Vouillot L., Thélie A., Pollet N. (2015) Comparison of T7E1 and Surveyor mismatch cleavage assays to detect mutations triggered by engineered nucleases. G3 (Bethesda). 5(3), 407–415.
Mashal R.D., Koontz J., Sklar J. (1995) Detection of mutations by cleavage of DNA heteroduplexes with bacteriophage resolvases. Nat. Genet. 9(2), 177‒183.
Qiu P., Shandilya H., D’Alessio J.M., O’Connor K., Durocher J., Gerard G.F. (2004) Mutation detection using SurveyorTM nuclease. BioTechniques. 36(4), 702‒707.
Saiki R.K., Scharf S., Faloona F., Mullis K.B., Horn G.T., Erlich H.A., Arnheim N. (1985) Enzymatic amplification of β-globin genomic sequences and restriction site analysis for diagnosis of sickle cell anemia. Science. 230(4732), 1350‒1354.
Urnov F.D., Miller J.C., Lee Y.L., Beausejour C.M., Rock J.M., Augustus S., Jamieson A.C., Porteus M.H., Gregory P.D., Holmes M.C. (2005) Highly efficient endogenous human gene correction using designed zinc-finger nucleases. Nature. 435(7042), 646‒651.
Kim J.M., Kim D., Kim S., Kim J.S. (2014) Genotyping with CRISPR-Cas-derived RNA-guided endonucleases. Nat. Commun. 5, 3157.
Brinkman E.K., Chen T., Amendola M., van Steensel B. (2014) Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res. 42(22), pii: e168. https://doi.org/10.1093/nar/gku936
Yang Z., Steentoft C., Hauge C., Hansen L., Thomsen A.L., Niola F., Vester-Christensen M.B., Frödin M., Clausen H., Wandall H.H., Bennett E.P. (2015) Fast and sensitive detection of indels induced by precise gene targeting. Nucleic Acids Res. 43(9), pii: e59. https://doi.org/10.1093/nar/gkv126
VanLeuven A.J., Park S., Menke D.B, Lauderdale J.D. (2018) A PAGE screening approach for identifying CRISPR-Cas9-induced mutations in zebrafish. Biotechniques. 64(6), 275–278.
Pierce A.J., Johnson R.D., Thompson L.H., Jasin M. (1999) XRCC3 promotes homology-directed repair of DNA damage in mammalian cells. Genes Dev. 13(20), 2633–2638.
Menzorov A.G., Lukyanchikova V.A., Korablev A.N., Serova I.A., Fishman V.S. (2016) Genome editing using CRISPR/ Cas9 system: a practical guide. Russ. J. Genet.: Appl. Res. 20(6), 930–944.
Shahar O.D., Ram E.V.S.R., Shimshoni E., Hareli S., Meshorer E., Goldberg M. (2012) Live imaging of induced and controlled DNA double-strand break formation reveals extremely low repair by homologous recombination in human cells. Oncogene. 31(30), 3495–3504.
Tsai S.Q., Zheng Z., Nguyen N.T., Liebers M., Topkar V.V., Thapar V., Wyvekens N., Khayter C., Iaf-rate A.J., Le L.P., Aryee M.J., Joung J.K. (2015) GUIDE-seq enables genome-wide profiling of off-target cleavage by CRISPR-Cas nucleases. Nat. Biotechnol. 33(2), 187‒197.
Germini D., Bou Saada Y., Tsfasman T., Osina K., Robin C., Lomov N., Rubtsov M., Sjakste N., Lipinski M., Vassetzky Y. (2017) A one-step PCR-based assay to evaluate efficiency and precision of genomic DNA-editing tools. Mol. Ther. Methods Clin. Dev. 5(6), 43–50.
Torres R., Martin M.C., Garcia A., Cigudosa J.C., Ramirez J.C., Rodriguez-Perales S. (2014) Engineering human tumour-associated chromosomal translocations with the RNA-guided CRISPR-Cas9 system. Nat. Commun. 5, 1–8.
Pinheiro L.B., Coleman V.A., Hindson C.M., Herrmann J., Hindson B.J., Bhat S., Emslie K.R. (2012) Evaluation of a droplet digital polymerase chain reaction format for DNA copy number quantification. Anal. Chem. 84(2), 1003–1011.
Dabrowska M., Czubak K., Juzwa W., Krzyzosiak W.J., Olejniczak M., Kozlowski P. (2018) qEva-CRISPR: a method for quantitative evaluation of CRISPR/Cas-mediated genome editing in target and off-target sites. Nucleic Acids Res. 46(17), pii: e101. https://doi.org/10.1093/nar/gky505
Schindelin J., Arganda-Carreras I., Frise E., Kaynig V., Longair M., Pietzsch T., Preibisch S., Rueden C., Saalfeld S., Schmid B., Tinevez J.Y., White D.J., Hartenstein V., Eliceiri K., Tomancak P., Cardona A. (2012) Fiji: an open-source platform for biological-image analysis. Nat. Methods. 9(7), 676‒682.
Harayama T., Riezman H. (2017) Detection of genome-edited mutant clones by a simple competition-based PCR method. PLoS One. 12(6), 1–16.
Yu C., Zhang Y., Yao S., Wei Y. (2014) A PCR based protocol for detecting indel mutations induced by TALENs and CRISPR/Cas9 in zebrafish. PLoS One. 9(6), pii: e98282. https://doi.org/10.1371/journal.pone.0098282
Thomas H.R., Percival S.M., Yoder B.K., Parant J.M. (2014) High-throughput genome editing and phenotyping facilitated by high resolution melting curve analysis. PLoS One. 9(12), pii: e114632. https://doi.org/10.1371/journal.pone.0114632
Dwight Z., Palais R., Wittwer C.T. (2011) uMELT: Prediction of high-resolution melting curves and dynamic melting profiles of PCR products in a rich web application. Bioinformatics. 27(7), 1019‒1020.
Dahlem T.J., Hoshijima K., Jurynec M.J., Gunther D., Starker C.G., Locke A.S., Weis A.M., Voytas D.F., Grunwald D.J. (2012) Simple methods for generating and detecting locus-specific mutations induced with TALENs in the Zebrafish genome. PLoS Genet. 8(8), pii: e1002861. https://doi.org/10.1371/journal.pgen.1002861
Samarut E., Lissouba A., Drapeau P. (2016) A simplified method for identifying early CRISPR-induced indels in zebrafish embryos using high resolution melting analysis. BMC Genomics. 17, 547.
Liew M., Pryor R., Palais R., Meadows C., Erali M., Lyon E., Wittwer C. (2004) Genotyping of single-nucleotide polymorphisms by high-resolution melting of small amplicons. Clin. Chem. 50(7), 1156‒1164.
Inazuka M., Wenz H.M., Sakabe M., Tahira T., Hayashi K. (1997) A streamlined mutation detection system: multicolor post-PCR fluorescence labeling and single-strand conformational polymorphism analysis by capillary electrophoresis. Genome Res. 7(11), 1094–1103.
Fennell J.P., Baker A.H., Dong Y., Zhu H. (2005) Single-strand conformational polymorphism analysis: basic principles and routine practice. Hypertension. 108, 149‒158.
Zheng X., Yang S., Zhang D., Zhong Z., Tang X., Deng K., Zhou J., Qi Y., Zhang Y. (2016) Effective screen of CRISPR/Cas9-induced mutants in rice by single-strand conformation polymorphism. Plant Cell Rep. 35(7), 1545–1554.
Zhu X., Xu Y., Yu S., Lu L., Ding M., Cheng J., Song G., Gao X., Yao L., Fan D., Meng S., Zhang X., Hu S., Tian Y. (2014) An efficient genotyping method for genome-modified animals and human cells generated with CRISPR/Cas9 system. Sci. Rep. 4, 6420.
Yu B., Sawyer N.A., Chiu C., Oefner P.J., Underhill P.A. (2006) DNA mutation detection using denaturing high-performance liquid chromatography (DHPLC). Curr. Protoc. Hum. Genet. 7(10), 1‒14.
Kc R., Srivastava A., Wilkowski J.M., Richter C.E., Shavit J.A., Burke D.T., Bielas S.L. (2016) Detection of nucleotide-specific CRISPR/Cas9 modified alleles using multiplex ligation detection. Sci. Rep. 6, 1–7.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология