

Молекулярная биология, 2020, T. 54, № 2, стр. 233-243
Мелатонин индуцирует апоптоз в преадипоцитах 3T3-L1
J. Lee a, Y.-M. Yoo b, Y. H. Lee a, C. H. Kim a, *
a Department of Biomedical Engineering, College of Health Sciences, Yonsei University
26493 Wonju, Gangwon-do, Republic of Korea
b Laboratory of Veterinary Biochemistry and Molecular Biology, College of Veterinary Medicine,
Chungbuk National University
28644 Cheongju, Chungbuk, Republic of Korea
* E-mail: chihyun@yonsei.ac.kr
Поступила в редакцию 07.12.2018
После доработки 28.03.2019
Принята к публикации 13.05.2019
Аннотация
Ожирение ‒ одно из распространенных заболеваний, вызывающее серьезные осложнения. Ингибирование пролиферации преадипоцитов рассматривается как перспективная стратегия для предупреждения ожирения и метаболических заболеваний. Мелатонин – это гормон шишковидной железы, который оказывает различное воздействие на клетки и ткани. В представленной работе изучена способность мелатонина индуцировать апоптоз в преадипоцитах 3T3-L1. Преадипоциты 3T3-L1 культивировали до слияния, а затем обрабатывали мелатонином в концентрациях 0, 10, 100 и 1000 мкМ в течение 1, 3 и 5 суток. Для определения жизнеспособности клеток использовали Cell viability kit. Белки маркеров клеточной смерти оценивали методом иммуноблотинга с использованием GAPDH в качестве контроля. Характерные для апоптотоза морфологические изменения с фрагментацией ядер наблюдали с помощью окрашивания DAPI. Обработка мелатонином снижала активацию фосфорилированных внеклеточных сигналрегулируемых киназ (р-ERK) при одновременном увеличении активации каспаз-3, -8 и -9. Кроме того, мелатонин не только увеличивал экспрессию Bcl-2-ассоциированного белка Х (Bax), но и снижал экспрессию белка Bcl-2 при увеличении дозы от 0 до 1000 мкМ. Воздействие повышенными концентрациями мелатонина также подавляло рост преадипоцитов. Эти эффекты ослаблялись лузиндолом, антагонистом рецептора мелатонина, и U0126, ингибитором активации p-ERK. Таким образом, мелатонин способен вызывать апоптоз преадипоцитов 3T3-L1 через подавление p-ERK.
Ожирение относится к одной из важнейших проблем здравоохранения во всем мире, так как приводит к различным серьезным осложнениям, таким как гипертония и сахарный диабет [1‒3]. Ожирение вызвано увеличением массы жировой ткани с повышением как объема, так и количества адипоцитов, на что влияют такие гормоны, как мелатонин [4, 5].
Преадипоциты ‒ это предшественники зрелых клеток белой жировой ткани [6, 7]. Преадипоциты 3T3-L1 считаются образцовой экспериментальной моделью, используемой в большинстве исследований на адипоцитах. Процесс дифференцировки преадипоцитов 3T3-L1 аналогичен процессу дифференцировки преадипоцитов человека [8, 9]. Используя именно эти преадипоциты, многие исследователи изучали различные химические вещества и гормоны, которые подавляют адипогенез и влияют на дифференцировку клеток [10‒12]. Один из факторов, вызывающих ожирение, ‒ пролиферация и дифференцировка преадипоцитов, поэтому апоптотическая гибель клеток в преадипоцитах может быть эффективным способом подавления или снижения адипогенеза, что, как оказалось, приводит к потере веса у мышей [13]. Апоптоз вызывает изменения в Bcl-2-ассоциированном белке Х (Bax) и белке 2 В-клеточной лимфомы (Bcl-2), а также в белках семейства каспаз, называемых апоптотическими белками. В исследованиях по апоптозу выявлено действие мелатонина на эти белки [14].
Мелатонин – это гормон, вырабатываемый шишковидной железой. Секреция мелатонина контролируется циркадным ритмом человека и наличием света или темноты [15]. Таким образом, концентрация мелатонина изменяется в зависимости от времени суток. Мелатонин имеет много физиологических функций, которые влияют на здоровье и старение путем контроля жировой массы и веса тела [16‒18]. Например, аномальные концентрации мелатонина могут вызвать аномальный циркадный ритм, в результате чего нарушается сон [19‒21]. У людей, которые регулярно работают по ночам, снижена секреция мелатонина и, как следствие, появляются проблемы со здоровьем, включая ожирение [22, 23]. Влияние мелатонина на клетки зависит от изменений его концентрации. При низких концентрациях мелатонин обладает антиапоптотическим эффектом, связанным с его способностью поглощать свободные радикалы [24, 25]. Напротив, в высоких концентрациях мелатонин подавляет дифференцировку клеток и индуцирует апоптоз в нескольких клеточных линиях. Например, известно, что мелатонин вызывает апоптоз в некоторых раковых клетках [26] и ингибирует дифференцировку и рост клеток глиомы [27].
Довольно хорошо исследовано влияние мелатонина на многие типы клеток и определены его различные терапевтические эффекты, однако влияние мелатонина на преадипоциты и ожирение пока не совсем понято [28‒30]. Для поиска оптимальных условий воздействия мелатонина необходимо проведение исследований апоптоза, индуцируемого этим гормоном в различных концентрациях и временных режимах, с использованием преадипоцитов. Таким способом можно исследовать физиологическую функцию мелатонина и в дальнейшем применить к зрелым адипоцитам. В проведенном исследовании изменяли дозу и продолжительность воздействия мелатонина, чтобы определить его влияние на апоптоз в преадипоцитах 3T3-L1.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Клеточная культура и обработка мелатонином. Преадипоциты 3T3-L1 (Корейский банк клеточных линий – Korean Cell Line Bank, Корея) культивировали в среде Игла в модификации Дульбекко (DMEM; “Lonza”, США), содержащей 5% телячьей сыворотки (“Gibco”, США), 1% пенициллина/стрептомицина (“Lonza”) при 37°С с 5% CO2 в инкубаторе с влажностью 100%. Для обработки мелатонином клетки высевали в 100-мм чашки в количестве 1 × 106 клеток на чашку и инкубировали в течение ночи. Клетки инкубировали при низкой концентрации телячьей сыворотки (1%) для минимизации антиапоптотического эффекта сыворотки [31] и обрабатывали мелатонином в концентрации 0, 10, 100 и 1000 мкМ (“Sigma”, США) в течение 1, 3 или 5 суток для индукции апоптоза. Для предотвращения апоптотического эффекта и его мелатонинового пути клетки предварительно обрабатывали соответственно лузиндолом (20 мкМ) в течение 5 ч и U0126 (10 мкМ) в течение 2 ч. Затем клетки обрабатывали мелатонином в концентрации 1000 или 0 мкМ. Все используемые агенты растворяли в диметилсульфоксиде (DMSO; “Sigma”), при этом все контрольные группы обрабатывали тем же объемом DMSO.
Анализ жизнеспособности клеток. Для оценки жизнеспособности клеток использовали набор EZ-Cytox (“DAEIL Lab”, Корея). Клетки высевали в 96-луночные микропланшеты в количестве 5 × 103 клеток на лунку, инкубировали в течение 24 ч, после чего обрабатывали мелатонином в концентрации 0, 10, 100 или 1000 мкМ в течение 1, 3 и 5 суток. По истечении этого времени в каждую лунку добавляли реагент WST и инкубировали микропланшет в течение 1 ч при 37°C. Поглощение измеряли на микропланшетном ридере при 450 нм. Для определения значения фонового сигнала в течение тех же временных интервалов инкубировали DMEM в отсутствие клеток. Процентное содержание жизнеспособных клеток (n, %) в каждом образце рассчитывали по формуле:
где A ‒ общий сигнал, Ab ‒ фоновый сигнал, Ac ‒ сигнал контроля.
Окрашивание DAPI. Характерные для апоптоза морфологические изменения: формирование апоптотических телец и аномальная морфология ядер ‒ наблюдали с помощью окрашивания DAPI (“Merck Millipore”, Германия). Клетки высевали в количестве 1 × 104 клетки/лунку в 4-луночные планшеты и инкубировали 24 ч, затем обрабатывали 0, 10, 100 и 1000 мкМ мелатонина в течение 1, 3 и 5 суток. Супернатант удаляли аспирацией, клетки фиксировали в 4%-ном растворе параформальдегида, после чего инкубировали с раствором DAPI (1 мкг/мл) в течение 5 мин в темноте. Для визуализации DAPI-окрашенных клеток использовали флуоресцентный микроскоп.
Иммуноблотинг. Клетки высевали по 1 × 106 клеток в чашки диаметром 100 мм и инкубировали в течение ночи, затем обрабатывали раствором 0, 10, 100 и 1000 мкМ мелатонина в течение 1, 3 и 5 суток. Клетки собирали, промывали холодным фосфатно-солевым буфером (PBS), а затем лизировали в буфере Трис-HCl (рН 7.4), содержащем ингибиторы протеазы и фосфатазы. Для определения концентрации белка в образцах использовали BCA assay (“Thermo Scientific”, США). Разделения клеточных белков (35 мкг) проводили электрофорезом в 9%-ном ПААГ в денатурирующих условиях (SDS-PAGE), белки переносили на мембрану из поливинилидендифторида (PVDF) (“Merck Millipore”). Мембраны инкубировали с антителами против следующих белков: внеклеточные сигналрегулируемые киназы (ERK) (1 : : 1000) (sc-514302; “Santa Cruz Biotechnology”, США), p-ERK (1 : 1000) (sc-81492; “Santa Cruz Biotechnology”), каспаза-9 (1 : 1000) (9504; “Cell Signaling Technology”, США), каспаза-8 (1 : 1000) (4790; “Cell Signaling Technology”), каспаза-3 (1 : 1000) (9662; “Cell Signaling Technology”), Bcl-2 (1 : 1000) (sc-23960; “Santa Cruz Biotechnology”), Bax (1 : 1000) (sc-20067; “Santa Cruz Biotechnology”) и GAPDH (1 : 1000) (sc-137179; “Santa Cruz Biotechnology”). Иммунореактивные белки визуализировали с помощью рентгеновской пленки, которую анализировали методом сканирования изображений.
Статистический анализ. Значимость различий определяли методом ANOVA с последующим применения критерия Тьюки. Анализ проводили с помощью программы Prism GraphPad v5.0 (“GraphPad Software Inc.”, США). Данные, полученные по результатам, по крайней мере, трех различных экспериментов, выражали в виде: среднее ± SD. Статистически значимыми считали значения р < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Влияние мелатонина на жизнеспособность клеток
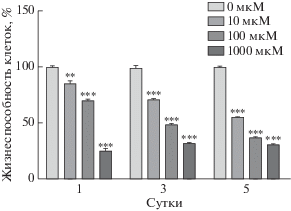
Влияние мелатонина на преадипоциты 3T3-L1 определяли с помощью анализа жизнеспособности клеток. Исследована зависимость жизнеспособности клеток от дозы мелатонина (от 0 до 1000 мкМ) и времени воздействия (1, 3 и 5 суток). Выявлено, что мелатонин снижает жизнеспособность преадипоцитов 3T3-L1 по мере увеличения его концентрации (рис. 1). Гибель клеток была значимо выше на 5 сутки воздействия мелатонина по сравнению с первыми сутками.
Рис. 1.
Жизнеспособность преадипоцитов 3T3-L1 после обработки мелатонином. Жизнеспособность клеток определяли с помощью набора Cell viability kit. Клетки обрабатывали различными дозами мелатонина в течение 1, 3 и 5 суток. Для сравнения приведены данные для необработанных клеток. Результаты представлены как процентное содержание жизнеспособных клеток; высота столбцов соответствует среднему значению результатов трех независимых экспериментов (отрезки с насечками представляют SD). **р < 0.01, ***р < 0.001 по сравнению с контролем.
Влияние мелатонина на гибель клеток
Морфологические изменения ядер преадипоцитов 3T3-L1 анализировали с помощью окрашивания DAPI. Окрашивание DAPI появилось после 5 суток обработки мелатонином (0 и 1000 мкМ). Выявлена фрагментация ядер при апоптозе, индуцированном в преадипоцитах 3T3-L1 высокими концентрациями мелатонина (рис. 2). Апоптотические изменения ядер при 5 сутках воздействия 1000-мкМ мелатонином были более значительными, чем в контрольных образцах (5 суток инкубации без мелатонина, т.е. 0 мкМ).
Рис. 2.
Исследование морфологии ядер с помощью окрашивания DAPI. Аномальные изменения ядер в преадипоцитах 3T3-L1 оценивали окрашиванием DAPI. Клетки с окрашенными ядрами анализировали с помощью флуоресцентной микроскопии (200×). На 5 сутки в преадипоцитах, обработанных 1000 мкМ мелатонина (б), было гораздо больше клеток с фрагментированными ядрами, чем в контроле (а). Масштабная линейка: 50 мкм.

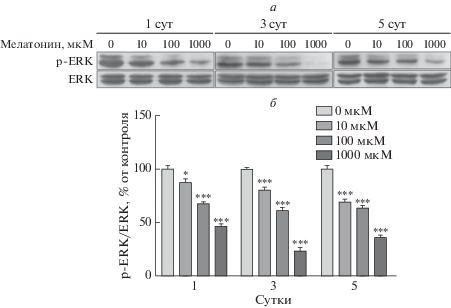
Для изучения влияния мелатонина на выживаемость и пролиферацию клеток определяли фосфорилирование ERK методом иммуноблотинга (рис. 3). Содержание p-ERK уменьшалось с каждым днем воздействия повышенных доз мелатонина. Изменение экспрессии p-ERK было сходно с состоянием жизнеспособности клеток.
Рис. 3.
Активация фосфорилированных киназ, регулируемых внеклеточными сигналами (p-ERK и ERK), мелатонином в преадипоцитах 3T3-L1. Клетки выращивали в среде DMEM, содержащей 1% телячьей сыворотки и 1% пенициллина/стрептомицина, а затем обрабатывали различными дозами мелатонина в течение 1, 3 и 5 суток Клеточные лизаты анализировали с помощью иммуноблотинга (а). Относительную экспрессию p-ERK рассчитывали, как описано в разделе “Экспериментальная часть”. Содержание p-ERK показано в процентах от аналогичной величины в контрольных образцах (б). Высота столбцов соответствует средним значениям результатов трех независимых экспериментов (отрезками показаны величины SD). *р < 0.05, ***р < 0.001 по сравнению с контролем.
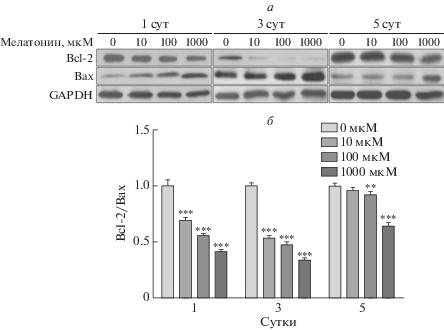
Также изучены изменения некоторых апоптотических белков, таких как каспазы, Bcl-2 и Bax. Содержание каспаз-9, -8 и -3 увеличивалось с ростом концентрации мелатонина и временем воздействия мелатонина (рис. 4). Кроме того, уровень Bcl-2 снижался с увеличением дозы мелатонина, в то время как Bax увеличивался (рис. 5). По сравнению с индивидуальными уровнями экспрессии Bcl-2 и Bax, соотношение Bcl-2/Bax четко указало на стимуляцию апоптоза в преадипоцитах 3T3-L1. На 1 и 3 сутки воздействия мелатонина выявлены наиболее выраженные апоптотические изменения, тогда как на 5 сутки существенных изменений не наблюдали. На основании этих результатов можно сделать вывод, что мелатонин способствует апоптозу в преадипоцитах 3T3-L1.
Рис. 4.
Активация каспаз под действием мелатонина в преадипоцитах 3Т3-L1. Клетки выращивали в среде DMEM, содержащей 1% телячьей сыворотки и 1% пенициллина/стрептомицина, а затем обрабатывали различными дозами мелатонина в течение 1, 3 и 5 суток. Клеточные лизаты анализировали с помощью иммуноблотинга (а). Относительное содержание каспазы-9 (б), -8 (в) и -3 (г) для каждой группы рассчитывали, как описано в разделе “Экспериментальная часть”. В качестве контроля нанесения использовали GAPDH. Высота столбцов соответствует средним значениям результатов трех независимых экспериментов (отрезками показаны величины SD). *р < 0.05, **р < 0.01, ***р < 0.001 по сравнению с контролем.
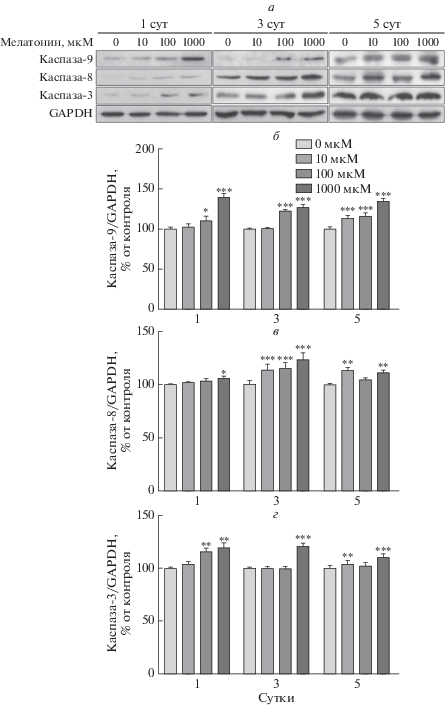
Рис. 5.
Активация белка Bcl-2 и Bcl-2-ассоциированного белка X (Bax) под действием мелатонина в преадипоцитах 3T3-L1. Клетки выращивали в DMEM, содержащей 1% телячьей сыворотки и 1% пенициллина/стрептомицина, а затем обрабатывали различными дозами мелатонина в течение 1 дня, 3 дней и 5 дней. Клеточные лизаты анализировали с помощью иммуноблотинга (а). Соотношение экспрессии Bcl-2/Bax на 1, 3 и 5 сутки рассчитывали, как описано в разделе “Экспериментальная часть” (б). В качестве контроля нанесения использовали GAPDH. Высота столбцов соответствует средним значениям результатов трех независимых экспериментов (отрезками показаны величины SD). **р < 0.01, ***р < 0.001 по сравнению с контролем.
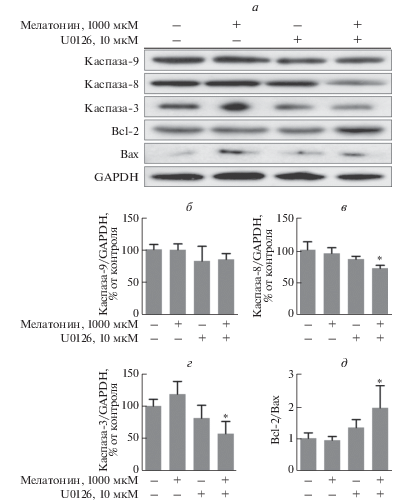
Антагонист рецептора мелатонина лузиндол подавлял индуцированную мелатонином активацию апоптотических белков (рис. 6). Активация каспаз-9, -8 и -3 мелатонином снижалась после предварительной обработки преадипоцитов лузиндолом. В то же время соотношение Bcl-2/Bax менялось в противопололожную сторону по сравнению с клетками, обработанными только мелатонином, и это еще раз подтверждает, что мелатонин оказывает апоптотическое действие на преадипоциты 3T3-L1. Аналогичным образом ингибитор р-ERK, U0126, снижал апоптотический эффект мелатонина, за исключением каспазы-9 (рис. 7). Следовательно, путь ERK оказывает обратное действие на апоптоз, индуцированный мелатонином.
Рис. 6.
Активация каспаз, Bcl-2 и Bax мелатонином в преадипоцитах 3T3-L1 и влияние предварительной обработки лузиндолом. Клетки выращивали в DMEM, содержащей 1% телячьей сыворотки и 1% пенициллина/стрептомицина. Группы лузиндола предварительно обрабатывали 20 мкМ лузиндола в течение 5 ч, а затем обрабатывали 1 мМ мелатонина в течение 1 суток. Клеточные лизаты анализировали иммуноблотингом (а). Содержание каспазы-9 (б), -8 (в), -3 (г) и соотношение Bcl-2/Bax (д) рассчитывали, как описано в разделе “Экспериментальная часть”. В качестве контроля нанесения использовали GAPDH. Высота столбцов соответствует средним значениям результатов трех независимых экспериментов (отрезками показаны величины SD). *р < 0.05, **р < 0.01, ***р < 0.001 по сравнению с мелатонином.
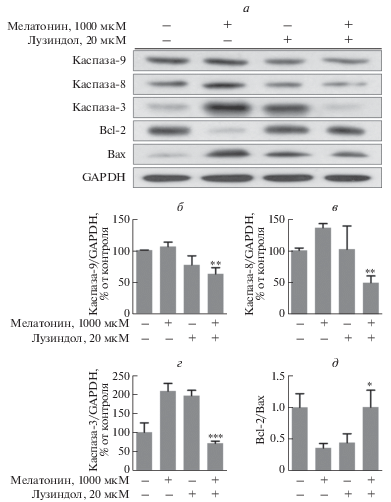
Рис. 7.
Активация каспаз, Bcl-2 и Bax мелатонином в преадипоцитах 3T3-L1 и влияние предварительной обработки U0126. Клетки выращивали в DMEM, содержащей 1% телячьей сыворотки и 1% пенициллина/стрептомицина. Группы U0126 предварительно обрабатывали 10 мкМ U0126 в течение 2 ч, а затем 1 мкМ мелатонина в течение 1 суток. Клеточные лизаты анализировали иммуноблотингом (а). Содержание каспазы-9 (б), -8 (в), -3 (г) и соотношение экспрессии Bcl-2/Bax (д) рассчитывали, как описано в разделе “Экспериментальная часть”. В качестве контроля нанесения использовали GAPDH. Высота столбцов соответствует средним значениям результатов трех независимых экспериментов (отрезками показаны величины SD). *p < 0.05 по сравнению с мелатонином.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Мелатонин – это гормон шишковидной железы, рецепторы которого находятся в различных тканях, включая жировую, и который регулирует жировой обмен [32, 33]. Мелатонин, выделяемый в ночное время и в условиях темноты, играет роль важного регулятора супрахиазматического ядра (SCN) ‒ одного из основных регуляторов циркадных ритмов. Мелатонин аффинно связывается со своими мембранными рецепторами 1A (MT1) и 1B (MT2) [34]. В ряде исследований показано, что мелатонин регулирует апоптоз в жировых клетках, а также может ингибировать адипогенез [35‒37].
Анализ жизнеспособности клеток показал, что мелатонин снижает выживаемость клеток в преадипоцитах 3T3-L1. Значительная гибель клеток происходила на пятые сутки обработки мелатонином, особенно при его высоких концентрациях. При окрашивании DAPI показано, что мелатонин индуцирует изменения в ядрах по типу апоптоза через 5 суток воздействия. Через 1 и 3 суток воздействия мелатонина фрагментации ядер не обнаружено. Апоптоз можно разделить на раннюю стадию, при которой происходит передача сигнала с активацией внутриклеточного ответа, и позднюю стадию, на которой происходит деградация ДНК [38, 39]. Нами показано, что 5-суточная обработка мелатонином индуцирует позднюю стадию апоптоза с фрагментацией ядер, а первый и третий день воздействия мелатонина соответствуют ранней стадии апоптоза.
Протеазы из семейства каспаз, такие как прокаспаза-3, -8 и -9, регулируют в клетках передачу сигнала с участием каспазного каскада. Эти ферменты действуют как инициаторы гибели клеток в большинстве путей, включая апоптоз [40]. Каспаза, активная форма прокаспаз, активируется рецепторами смерти, и сигналы стресса могут регулировать активацию следующей прокаспазы. Каспаза-3, активная форма прокаспазы-3, ‒ основной белок, ответственный за апоптоз, активируемый каспазой-8; этот процесс называется внешним путем [41]. Прокаспазы-8 и -9 также важные белки семейства каспаз и играют решающую роль в инициации апоптоза. Увеличение активности каспазы-9 может инициировать каспазу-3, которая, в свою очередь, активирует прокаспазу-9 по принципу обратной связи при передаче сигнала по внутреннему пути [42]. Мы убедились, что мелатонин вызывает апоптоз через активацию каспаз, причем дозозависимым образом. Показано, что мелатонин может индуцировать каспаза-3-зависимый апоптоз как внешнего, так и внутреннего пути.
Кроме того, нами выявлено, что мелатонин стимулирует митохондриальный путь апоптоза через участие членов семейства Bcl-2 ‒ важных белков митохондриальнозависимого апоптоза. В их число входят проапоптотический белок Bax и антиапоптотический белок Bcl-2 [43]. Показано, что соотношение Bcl-2 и Bax можно считать индикатором апоптоза, а сверхэкспрессию Bax использовать для регуляции апоптоза [44, 45]. В результате проведенного исследования выявлено значительное усиление экспрессии Bax в клетках, обработанных мелатонином. Так, показано, что в преадипоцитах 3T3-L1 мелатонин индуцирует апоптотическую гибель клеток через регуляцию митохондриального пути апоптоза.
Лузиндол, антагонист рецепторов мелатонина 1A и 1B, обычно используется в исследованиях in vitro для ингибирования ожидаемых эффектов, индуцированных мелатонином [46, 47]. С целью убедиться, что эти эффекты обусловлены мелатонином, а не другими факторами, клетки сначала обрабатывали лузиндолом в течение 5 ч, а затем добавляли мелатонин. Обнаружено, что активация апоптотических факторов: каспаз-9, -8, -3, – а также соотношение Bcl-2/Bax оказались разнонаправленными по сравнению с группой, получавшей только мелатонин. На основании этих данных по ингибирующему действию лузиндола можно считать, что мелатонин отвечает за апоптоз в преадипоцитах 3T3-L1. Протеинкиназы и внутриклеточные сигнальные молекулы, известные под названием ERK (киназы, регулируемые внеклеточными сигналами), выполняют различные регуляторные функции, в том числе в отношении таких клеточных процессов, как мейоз, митоз, клеточная пролиферация и апоптоз [48, 49]. Благодаря этим функциям ERK участвуют и в гибели клеток, а в случае подавления активации ERK может происходить индукция апоптоза [50]. ERK-опосредованный путь передачи сигналов влияет на адипогенез в преадипоцитах 3T3-L1 [51]. Активная форма ERK регулирует функции сигнального пути, что в конечном счете способствует выживанию клеток [52]. В этом исследовании нами показано, что обработка мелатонином приводит к ежедневному снижению экспрессии активной формы ERK на протяжении 5 суток воздействия. На этом основании можно говорить о том, что мелатонин препятствует выживанию преадипоцитов 3T3-L1.
Для более точного понимания роли ингибирования ERK в апоптозе, индуцированном мелатонином, клетки предварительно обрабатывали U0126, ингибитором p-ERK, в течение 2 ч [53]. Хотя и мелатонин, и U0126 обладают ингибирующим действием в отношении p-ERK, апоптотический эффект был ослаблен только в тех случаях, когда U0126 добавляли до обработки мелатонином, а не тогда, когда добавляли только U0126. В то же время для каспазы-9, которая активируется через внутренний путь апоптоза [42], не выявлено значимого снижения экспрессии под действием U0126. Следовательно, ингибирование ERK-пути как способа передачи апоптотических сигналов, индуцируемого мелатонином, связано скорее с внешними сигналами, чем с внутренними. Результаты, полученные нами на преадипоцитах, в основном совпадают с результатами исследований на других типах клеток. В клетках гепатокарциномы по мере увеличения продолжительности воздействия мелатонином выявили увеличение частоты остановки клеточного цикла и индукции апоптоза с повышением содержания Bax и активности каспаз [29]. Кроме того, в миелоидных клетках человека HL-60 мелатонин блокировал переход от фазы G1 к фазе S и индуцировал апоптоз с одновременным усилением активации каспаз [54]. Апоптотический сигналинг начинался с активации белков семейства каспаз и Bax.
В целом на основании полученных нами результатов можно говорить о том, что мелатонин ингибирует рост клеток и играет важную роль в апоптозе преадипоцитов 3T3-L1. Ожирение связано с гиперплазией жировой ткани, что обусловлено адипогенезом и пролиферацией преадипоцитов. Следовательно, снижение роста адипоцитов и индуцирование гибели клеток с помощью мелатонина может стать важной стратегией в лечении ожирения и сопутствующих заболеваний. Таким образом, мелатонин может предотвращать ожирение как фактор, способструющий апоптотической гибели преадипоцитов.
Исследование поддержано программой фундаментальных научных исследований (Basic Science Research Program) через Национальный исследовательский фонд Кореи (the National Research Foundation of Korea, NRF) и профинансировано Министерством образования (the Ministry of Education, NRF-2015R1D1A1A01060699).
Эта статья не содержит никаких исследований с участием человека или животных, выполненных кем-либо из авторов.
От всех участников исследования получено информированное согласие.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Devlin M.J., Yanovski S.Z., Wilson G.T. (2000) Obesity: what mental health professionals need to know. Am. J. Psychiatry. 157, 854‒866.
Janssen I., Katzmarzyk P.T., Ross R. (2002) Body mass index, waist circumference, and health risk: evidence in support of current National Institutes of Health guidelines. Arch. Intern. Med. 162, 2074‒2079.
Mokdad A.H., Ford E.S., Bowman B.A., Dietz W.H., Vinicor F., Bales V.S., Marks J.S. (2003) Prevalence of obesity, diabetes, and obesity-related health risk factors, 2001. JAMA. 289, 76‒79.
Barrett P., Bolborea M. (2012) Molecular pathways involved in seasonal body weight and reproductive responses governed by melatonin. J. Pineal Res. 52, 376‒388.
Stolk R.P., Meijer R., Mali W.P., Grobbee D.E., Graaf Y. (2003) Ultrasound measurements of intraabdominal fat estimate the metabolic syndrome better than do measurements of waist circumference. Am. J. Clin. Nutr. 77, 857‒860.
Lefterova M.I., Lazar M.A. (2009) New developments in adipogenesis. Trends Endocrinol. Metab. 20, 107‒114.
Seale P., Kajimura S., Spiegelman B.M. (2009) Transcriptional control of brown adipocyte development and physiological function ‒ of mice and men. Genes Dev. 23, 788‒797.
Green H., Meuth M. (1974) An established pre-adipose cell line and its differentiation in culture. Cell. 3, 127‒133.
Ailhaud G., Grimaldi P., Negrel R. (1994) Hormonal regulation of adipose differentiation. Trends Endocrinol. Metab. 5, 132‒136.
Li H., Lee J.H., Kim S.Y., Yun H.Y., Baek K.J., Kwon N.S., Yoon Y., Jeong J.H., Kim D.S. (2011) Phosphatidylcholine induces apoptosis of 3T3-L1 adipocytes. J. Biomed. Sci. 18, 91.
Xiao Y., Yuan T., Yao W., Liao K. (2010) 3T3-L1 adipocyte apoptosis induced by thiazolidinediones is peroxisome proliferator-activated receptor-gamma-dependent and mediated by the caspase-3-dependent apoptotic pathway. FEBS J. 277, 687‒696.
Wang N., Wang X., Shi M., Shi H., Yan X., Li H., Wang S., Wang Y. (2013) LMO4 modulates proliferation and differentiation of 3T3-L1 preadipocytes. FEBS Lett. 587, 3032‒3037.
Birk R.Z., Rubinstein M. (2006) IFN-alpha induces apoptosis of adipose tissue cells. Biochem. Biophys. Res. Commun. 345, 669‒674.
Perdomo J., Cabrera J., Estevez F., Loro J., Reiter R.J., Quintana J. (2013) Melatonin induces apoptosis through a caspase-dependent but reactive oxygen species-independent mechanism in human leukemia Molt-3 cells. J. Pineal Res. 55, 195‒206.
Pacchierotti C., Iapichino S., Bossini L., Pieraccini F., Castrogiovanni P. (2001) Melatonin in psychiatric disorders: a review on the melatonin involvement in psychiatry. Front. Neuroendocrinol. 22, 18‒32.
Prunet-Marcassus B., Desbazeille M., Bros A., Louche K., Delagrange P., Renard P., Casteilla L., Penicaud L. (2003) Melatonin reduces body weight gain in Sprague Dawley rats with diet-induced obesity. Endocrinology. 144, 5347‒5352.
Wolden-Hanson T., Mitton D.R., McCants R.L., Yellon S.M., Wilkinson C.W., Matsumoto A.M., Rasmussen D.D. (2000) Daily melatonin administration to middle-aged male rats suppresses body weight, intraabdominal adiposity, and plasma leptin and insulin independent of food intake and total body fat. Endocrinology. 141, 487‒497.
Barrenetxe J., Delagrange P., Martinez J.A. (2004) Physiological and metabolic functions of melatonin. J. Physiol. Biochem. 60, 61‒72.
Brzezinski A. (1997) Melatonin in humans. N. Engl. J. Med. 336, 186‒195.
Reiter R.J., Tan D.X., Korkmaz A., Ma S. (2012) Obesity and metabolic syndrome: association with chronodisruption, sleep deprivation, and melatonin suppression. Ann. Med. 44, 564‒577.
Spiegel K., Leproult R., Van Cauter E. (1999) Impact of sleep debt on metabolic and endocrine function. Lancet. 354, 1435‒1439.
Cipolla-Neto J., Amaral F.G., Afeche S.C., Tan D.X., Reiter R.J. (2014) Melatonin, energy metabolism, and obesity: a review. J. Pineal Res. 56, 371‒381.
Heslop P., Smith G.D., Metcalfe C., Macleod J., Hart C. (2002) Sleep duration and mortality: the effect of short or long sleep duration on cardiovascular and all-cause mortality in working men and women. Sleep Med. 3, 305‒314.
Sun F.Y., Lin X., Mao L.Z., Ge W.H., Zhang L.M., Huang Y.L., Gu J. (2002) Neuroprotection by melatonin against ischemic neuronal injury associated with modulation of DNA damage and repair in the rat following a transient cerebral ischemia. J. Pineal Res. 33, 48‒56.
Bułdak R.J., Pilc‑Gumuła K., Bułdak Ł., Witkowska D., Kukla M., Polaniak R., Zwirska‑Korczala K. (2015) Effects of ghrelin, leptin and melatonin on the levels of reactive oxygen species, antioxidant enzyme activity and viability of the HCT 116 human colorectal carcinoma cell line. Mol. Med. Rep. 12, 2275‒2282.
Joo S.S., Yoo Y.M. (2009) Melatonin induces apoptotic death in LNCaP cells via p38 and JNK pathways: therapeutic implications for prostate cancer. J. Pineal Res. 47, 8‒14.
Martin V., Herrera F., Carrera-Gonzalez P., Garcia-Santos G., Antolin I., Rodriguez-Blanco J., Rodriguez C. (2006) Intracellular signaling pathways involved in the cell growth inhibition of glioma cells by melatonin. Cancer Res. 66, 1081‒1088.
Jung-Hynes B., Reiter R.J., Ahmad N. (2010) Sirtuins, melatonin and circadian rhythms: building a bridge between aging and cancer. J. Pineal Res. 48, 9‒19.
Martin-Renedo J., Mauriz J.L., Jorquera F., Ruiz-Andres O., Gonzalez P., Gonzalez-Gallego J. (2008) Melatonin induces cell cycle arrest and apoptosis in hepatocarcinoma HepG2 cell line. J. Pineal Res. 45, 532‒540.
Haslam D.W., James W.P. (2005) Obesity. Lancet. 366, 1197‒1209.
Kntayya S., Ibrahim M., Mohd Ain N., Iori R., Ioannides C., Abdull Razis A. (2018) Induction of apoptosis and cytotoxicity by isothiocyanate sulforaphene in human hepatocarcinoma HepG2 cells. Nutrients. 10, 718.
Brydon L., Petit L., Delagrange P., Strosberg A.D., Jockers R. (2001) Functional expression of MT2 (Mel1b) melatonin receptors in human PAZ6 adipocytes. Endocrinology. 142, 4264‒4271.
Claustrat B., Brun J., Chazot G. (2005) The basic physiology and pathophysiology of melatonin. Sleep Med. Rev. 9, 11‒24.
Dubocovich M.L. (2007) Melatonin receptors: role on sleep and circadian rhythm regulation. Sleep Med. 3, 34‒42.
Alonso-Vale M.I.C., Peres S.B., Vernochet C., Far-mer S.R., Lima F.M. (2009) Adipocyte differentiation is inhibited by melatonin through the regulation of C/EBPb transcriptional activity. J. Pineal Res. 47, 221‒227.
Zhang L., Su P., Xu C., Chen C., Liang A., Du K., Peng Y., Huang D. (2010) Melatonin inhibits adipogenesis and enhances osteogenesis of human mesenchymal stem cells by suppressing PPARgamma expression and enhancing Runx2 expression. J. Pineal Res. 49, 364‒372.
Hong Y., Won J., Lee Y., Lee S., Park K., Chang K.T., Hong Y. (2014) Melatonin treatment induces interplay of apoptosis, autophagy, and senescence in human colorectal cancer cells. J. Pineal Res. 56, 264‒274.
Brauchle E., Thude S., Brucker S.Y., Schenke-Layland K. (2014) Cell death stages in single apoptotic and necrotic cells monitored by Raman microspectroscopy. Sci. Rep. 4, 4698.
Elmore S. (2007) Apoptosis: a review of programmed cell death. Toxicol. Pathol. 35, 495‒516.
Arya R., White K. (2015) Cell death in development: signaling pathways and core mechanisms. Semin. Cell Dev. Biol. 39, 12‒19.
Cohen G.M. (1997) Caspases: the executioners of apoptosis. Biochem. J. 326, 1‒16.
Fan T.J., Han L.H., Cong R.S., Liang J. (2005) Caspase family proteases and apoptosis. Acta Biochim. Biophys. Sin. 37, 719‒727.
Kroemer G., Reed J.C. (2000) Mitochondrial control of cell death. Nat. Med. 6, 513‒519.
Sun X., Zemel M.B. (2004) Role of uncoupling protein 2 (UCP2) expression and 1alpha, 25-dihydroxyvitamin D3 in modulating adipocyte apoptosis. FASEB J. 18, 1430‒1432.
Antonsson B., Martinou J.C. (2000) The Bcl-2 protein family. Exp. Cell Res. 256, 50‒57.
Adamczyk-Sowa M., Sowa P., Zwirska-Korczala K., Pierzchala K., Bartosz G., Sadowska-Bartosz I. (2013) Role of melatonin receptor MT 2 and quinone reductase II in the regulation of the redox status of 3T3-L1 preadipocytes in vitro. Cell Biol. Int. 37, 835‒842.
Kokkola T., Vaittinen M., Laitinen J.T. (2007) Inverse agonist exposure enhances ligand binding and G protein activation of the human MT1 melatonin receptor, but leads to receptor down-regulation. J. Pineal Res. 43, 255‒262.
Johnson G.L., Lapadat R. (2002) Mitogen-activated protein kinase pathways mediated by ERK, JNK, and p38 protein kinases. Science. 298, 1911‒1912.
Robinson M.J., Cobb M.H. (1997) Mitogen-activated protein kinase pathways. Curr. Opin. Cell Biol. 9, 180‒186.
Xia Z., Dickens M., Raingeaud J., Davis R.J., Greenberg M.E. (1995) Opposing effects of ERK and JNK-p38 MAP kinases on apoptosis. Science. 270, 1326‒1331.
Prusty D., Park B.H., Davis K.E., Farmer S.R. (2002) Activation of MEK/ERK signaling promotes adipogenesis by enhancing peroxisome proliferator-activated receptor(PPAR) and C/EBP gene expression during the differentiation of 3T3-L1 preadipocytes. J. Biol. Chem. 277, 46226‒46232.
Almeida R.D., Manadas B.J., Melo C.V., Gomes J.R., Mendes C.S., Graos M.M., Carvalho R.F., Carvalho A.P., Duarte C.B. 2005. Neuroprotection by BDNF against glutamate-induced apoptotic cell death is mediated by ERK and PI3-kinase pathways. Cell Death Differ. 12, 1329‒1343.
Cheung A., Newland P.L., Zaben M., Attard G.S., Gray W.P. (2012) Intracellular nitric oxide mediates neuroproliferative effect of neuropeptide Y on postnatal hippocampal precursor cells. J. Biol. Chem. 287, 20187‒20196.
Rubio S., Estevez F., Cabrera J., Reiter R.J., Loro J., Quintana J. (2007) Inhibition of proliferation and induction of apoptosis by melatonin in human myeloid HL-60 cells. J. Pineal Res. 42, 131‒138.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология