

Молекулярная биология, 2020, T. 54, № 2, стр. 321-332
Конструирование эффективного ингибитора ионного канала белка М2 вируса гриппа A
a Институт химической биологии и фундаментальной медицины Сибирского отделения Российской академии наук
630090 Новосибирск, Россия
b Новосибирский государственный университет
630090 Новосибирск, Россия
* E-mail: ynvorob@niboch.nsc.ru
Поступила в редакцию 02.04.2019
После доработки 19.09.2019
Принята к публикации 19.09.2019
Аннотация
Вирус гриппа А способен быстро инфицировать большую популяцию людей, поэтому разработка препаратов, способных ингибировать его репликацию, имеет исключительное значение. Важную роль в репликации вируса гриппа играет трансмембранный протонный канал, формируемый белком М2. Поэтому разумным подходом к созданию эффективного противовирусного препарата является конструирование молекулы, которая связывается в трансмембранном протонном канале М2, предотвращает диффузию протонов (H+) через канал и ингибирует цикл вируса гриппа А. Известные противовирусные препараты амантадин и римантадин связываются в ионном канале М2, однако они слабо влияют на репликацию вируса гриппа А. Для блокирования диффузии протонов через ионный канал М2 предложен новый класс положительно заряженных молекул, производных диазабициклооктана, имеющих постоянный заряд +2. Выполнено моделирование молекулярной динамики тепловых флуктуаций структуры белка М2 и ионизационных состояний остатков гистидина при физиологических значениях рН. Изучено связывание двух классов производных диазабициклооктана с ионным каналом белка M2. Определена оптимальная структура молекулы блокатора, которая наиболее эффективно связывается с протонным каналом белка М2 и блокирует диффузионный перенос протона через канал. Важное преимущество нового блокатора над амантадином и римантадином ‒ заряд +2, который создает положительный электростатический потенциальный барьер (в дополнение к стерическому) для блокирования транспорта протона H+ через ионный канал белка М2.
ВВЕДЕНИЕ
Вирус гриппа А ‒ это РНК-содержащий вирус, вызывающий пандемии (испанский грипп 1918 г., азиатский грипп 1957 г., гонконгский грипп 1968 г. и свиной грипп 2009 г.) [1]. Вирус гриппа быстро мутирует, поэтому существует острая необходимость в новых противовирусных препаратах. Разумным подходом к подавлению (нарушению) репродукции вируса гриппа А представляется воздействие на протонный канал, образованный трансмембранным доменом белка М2 (M2-TMD), например, блокирование ионного канала М2 и ингибирование протонного транспорта через него. Канал М2 образован аминокислотными остатками 24‒46 каждой из четырех α-спиралей и представляет собой тетрамер с центральным продольным каналом [2, 3]. Транспорт протонов через канал M2 незначительно меняется в области рН 5.4–7.0 [4]. Наиболее известными блокаторами ионного канала М2 являются амантадин и римантадин [5, 6]. Существуют блокаторы с более сложными структурами, например производные пикротоксина [7], производные адамантана и спироадамантиламин [8]. Моделирование методом молекулярной динамики (МД) показало, что профиль свободной энергии иона Н+ при движении вдоль ионного канала М2 имеет энергетический барьер ~5 ккал/моль [9]. Моделирование потенциальной энергии связывания амантадина методом МД с неявной моделью растворителя и окружающей среды, т.е. объема клеточной мембраны, предсказывает величину энергии связывания ~4 ккал/моль, что удовлетворительно соответствует экспериментальным данным [10]. Выход из протонного канала М2 контролируется остатками His37A (B, C, D) и Trp41A (B, C, D) четырех α-спиралей, формирующих этот канал. Закрытое состояние канала имеет достаточно узкий выход, формируемый Trp41A (B, C, D) с диаметром выхода несколько меньшим, чем у молекулы воды, 2.8 Å [11]. Методом ЯМР показано, что связывание амантадина с каналом М2 изменяет спектр конформационных флуктуаций и уменьшает их амплитуду [12]. Согласно данным мутагенеза и электрофизиологических экспериментов аминокислотные остатки, важные для транспорта протона и взаимодействия с амантадином, блокирующим канал М2, а именно, Val27, Аla30, Ser31 и Gly34, лежат на одной стороне α-спирали [13‒15]. Мутации этих остатков с большой вероятностью будут влиять на транспорт протона через канал М2. Таким образом, препараты на основе аминоадамантанов, включая амантадин и римантадин, в настоящее время не имеют широкого применения из-за устойчивости вируса к их связыванию и блокированию протонного канала в трансмембранном домене М2 вируса гриппа А [16‒18]. В настоящей работе предложен новый класс антивирусных препаратов, эффективно взаимодействующих с ионным каналом белка М2 вируса гриппа А, ‒ производные 1,4-диазабицикло-[2.2.2]октана (DABCO). Приведены результаты моделирования связывания и блокирования ионного канала М2, определена структура молекулы оптимального блокатора.
НОВЫЙ КЛАСС МОЛЕКУЛЯРНЫХ ИНГИБИТОРОВ ПРОТОННОГО КАНАЛА М2
Белок M2 вируса гриппа A представляет собой тетрамер, состоящий из четырех α-спиралей, которые образуют молекулярный канал для транспорта ионов водорода H+ через клеточную мембрану [17, 19‒27].
Чтобы затруднить и ослабить переход ионов H+ через ионный канал M2, этот канал должен быть блокирован. Эффективно блокировать (закрыть) канал М2 можно с помощью препарата, обладающего высоким сродством к внутренней поверхности канала М2. Один из классов молекул для адресного блокирования ионного канала М2 включает амантадин и римантадин (рис. 1а, б). При нейтральных значениях рН аминогруппа этих молекул находится в ионизированной форме ${\text{NH}}_{3}^{ + }$, так как рКа ионизации NH2-группы в ${\text{NH}}_{3}^{ + }$, составляет ~10.4 [28]. Свойства канала М2 изменяются в зависимости от величины pH. При pH > 7 канал M2 может находиться в двух состояниях ‒ закрытом и открытом. В закрытом состоянии канала М2 остатки Нis37A, His37B, His37C и His37D находятся в нейтральной форме (4H°). При pH < 7 открытое состояние ионного канала M2 представляет собой кислотно-активированную форму остатков гистидина с тетрапротонированным (4H+) гистидиновым тетрамером [18, 29, 30]. Отметим, что внутри ионного канала M2 pKa аминогруппы (${\text{NH}}_{3}^{ + }$) амантадина и римантадина может смещаться относительно pK0 вследствие локального электростатического потенциала.
Рис. 1.
Структуры: молекул амантадина (а), римантадина (б), 1,4-диазобицикло[2.2.2]октана (DABCO) (в).
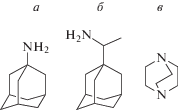
Ингибиторы ионного канала M2 нового класса основаны на молекуле DABCO (рис. 1в) [31‒33]. DABCO и его производные могут быть эффективными ингибиторами переноса протона через ионный канал М2 по двум причинам: (1) вследствие стерического блокирования канала в результате связывания молекулы препарата, как в случае амантадина и римантадина; (2) за счет электростатического блокирования канала путем создания электростатического потенциального барьера для диффузии протона H+ через канал М2, поскольку блокирующая молекула имеет положительный заряд (+2), распределенный по фрагменту DABCO.
МЕТОД МОДЕЛИРОВАНИЯ
Расчеты, описанные в работе, выполнены с помощью трех методов: (1) метода оптимизации потенциальной энергии и метода bioPASED при заданной температуре [34]; (2) метода глобального оптимального исчерпывающего молекулярного докинга молекулы лиганда на белок, hbDOCK [35]; (3) методов FAMBE и FAMBE-рН для расчета электростатической энергии, констант ионизации рКа и степени ионизации аминокислотных остатков молекулы белка как функции рН среды [36‒42]. Моделирование МД выполнено программным комплексом bioPASED [34] c использованием силового поля AMBER94 и AMBER-GAFF [43‒45] и модели неявной среды – модели гауссовских оболочек [46].
Докинг низкомолекулярных ингибиторов на белок М2 (PDB код 2KAD) выполнен с помощью метода иерархического слепого докинга hbDOCK [35]. Метод hbDOCK выполняет: (1) поиск всех возможных сайтов связывания на поверхности молекулы белка, доступных молекуле лиганда; (2) отбор потенциальных сайтов связывания по рангу их качества; и (3) глобальную оптимизацию для определения позиции/ориентации лиганда, конформации и энергии связывания методом МД симуляции отжига; (4) формирование списка позиций/ориентаций и значений энергии связывания в порядке убывания.
При встраивании в липидную мембрану белок М2 создает ионный канал, он окружен средой с диэлектрической проницаемостью, низкой по сравнению с диэлектрической проницаемостью воды ~80. Экспериментальные измерения показали, что диэлектрическая проницаемость липидной мембраны, т.е. среды вне молекулы белка M2, составляет ~30 [47]. Объемы молекулы белка М2 и ионных каналов рассматривают как объемы с диэлектрической проницаемостью, характерной для белков, DIN = 12‒16 [35, 39, 48‒50].
СТРУКТУРА ПРОТОННОГО КАНАЛА БЕЛКА М2
Атомная структура ионного канала M2 в комплексе с амантадином, содержащая все атомы водорода, PDB код 2KAD [16‒18], определенная методом ЯМР при температуре 243 К и рН 7.5, показана на рис. 2. Отметим, что ионный канал M2 образован четырьмя эквивалентными α-спиралями белка M2. Известны четыре структуры пустого канала М2 (PDB коды 2H95, 2L0J, 4QKL, 4QKM), полученные методами ЯМР и рентгеноструктурного анализа, табл. 1. Для наших исследований была выбрана структура PDB код 2KAD, полученная методом ЯМР [16‒18], поскольку она включает все атомы водорода и, следовательно, полную информацию об ионизационном состоянии остатков гистидина и типе таутомера нейтрального состояния. Структуры белка М2 в кристаллическом состоянии, полученные с помощью рентгеноструктурного анализа, как правило, не определяют атомы водорода и ионизационное состояние остатков гистидина.
Рис. 2.
Структура комплекса белка М2 с молекулой DABCO, связанной в ионном канале, метод ЯМР [16, 17] при T = 243 K, pH 7.5. Проекция вдоль оси канала, молекула DABCO в канале – CPK стиль; остатки Ser31A, Ser31B, Ser31C, Ser31D – CPK стиль, большие шары и толстые связи; остатки His37A, His37B, His37C, His37D – bonds стиль; остатки Trp 41A, Trp 41B, Trp 41C, Trp 41D ‒ bonds стиль, белый цвет, задний план.
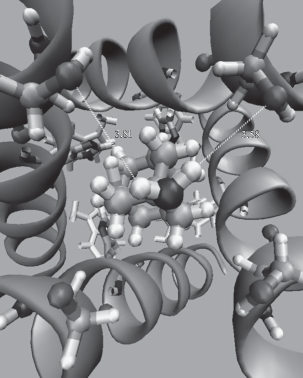
Таблица 1.
Экспериментальные структуры белка М2, формирующего ионный канала
| Код PDB | Год | Метод | Мутант | Т, К | рН | и.с.о.а | а.к.о.б | м.к.в | мод.г |
|---|---|---|---|---|---|---|---|---|---|
| 2H95 | 2006 | NMR | wild | 308 | 8.8 | HE2 | 26-43 | M2 | 1 |
| 2KAD | 2008 | NMR | wild | 243 | 7.5 | HE2 | 26-43 | M2 | 1 |
| 2L0J | 2010 | NMR | wild | 303 | 7.5 | HIS+/HE2 | 22-63 | M2 | 8 |
| 4QKL | 2014 | X-ray | wild | 273 | 8.0 | HE2 | 26-43 | M2 | 1 |
| 4QKM | 2014 | X-ray | wild | 273 | 5.5 | HIS+ | 26-43 | M2 | 1 |
Рассмотрим структуру белка М2 в деталях. Вход во внутренний объем ионного канала М2 формируется четырьмя остатками серина ‒ Ser31A, Ser31B, Ser31C и Ser31D, которые контролируют диффузию ионов Н+ через канал. Диагональ квадрата, образованного атомом Oγ Ser31A, Ser31B, Ser31C и Ser31D, т.е. пары расстояний Ser31A–Ser31B и Ser31B–Ser31C и т.д., равны 6.2 Å. Аминогруппа (NH2) амантадина в кристаллической структуре PDB 2KAD является нейтральной (рН 7.5) и образует слабые H-связи с ОγН- группой Ser31A и Ser31B, поскольку дистанция между атомами Oγ и NН2 амантадина ~4 Å (рис. 2). Отметим несколько особенностей структуры амантадина. (1) Связь C–NH2 в молекуле амантадина ориентирована вдоль оси ионного канала M2; (2) четыре остатка гистидина (His37A, His37B, His37C и His37D) лежат в одной плоскости, перпендикулярной к оси ионных каналов М2, как показано для экспериментальной ЯМР-структуры ионного канала М2 на рис. 2. Видно, что все четыре остатка ‒ His37A, His37B, His37C и His37D ‒ существуют в форме нейтральных таутомеров HE2 (HIE, как принято в структурном моделировании [36, 37]) в ЯМР-структуре 2KAD. Выход из канала М2 контролируется также и остатками триптофана (Trp41A, Trp41B, Trp41C и Trp41D), которые блокируют проникновение молекулы воды в канал М2 в закрытом состоянии [18].
Структура и энергия оптимизированной структуры изолированного белка M2 получены путем удаления молекулы амантадина из ионного канала M2 (PDB 2KAD) с последующей оптимизацией трехмерной структуры методом имитации отжига (рис. 2). Температура оптимизируемой структуры медленно повышалась от 0 до 300 К в течение 1 нс, а затем релаксировалась в течение 1 нс при 300 К. Далее были получены равновесные продуктивные МД траектории длительностью 25 нс при постоянных значениях рН 7.5 с периодическим уточнением ионизационных состояний всех аминокислотных остатков молекулы белка [45]. Аналогичным образом выполнены расчеты ионизации остатков гистидина в интервале рН 5.75‒8.25, в котором остатки гистидина переходят из ионизированной формы НIS+ в нейтральные таутомеры HIE и HID (табл. 2 и рис. 3).
Таблица 2.
Средняя заселенность ионизационных состояний остатков гистидина (His34A, His34B, His34C, His34D) белка M2
| pH | HIS+ a | HIEa | HIDa |
|---|---|---|---|
| 5.75 | 0.89б (0.02)в | 0.10б (0.02в) | 0.01б (0.01)в |
| 6.00 | 0.83 (0.03) | 0.16 (0.02) | 0.01 (0.01) |
| 6.25 | 0.76 (0.03) | 0.22 ( 0.02) | 0.02 (0.01) |
| 6.50 | 0.65 (0.04) | 0.33 (0.03) | 0.02 (0.01 ) |
| 6.75 | 0.57 (0.04) | 0.40 (0.03) | 0.03 (0.01) |
| 7.0 | 0.46 (0.04) | 0.50 (0.030) | 0.04 (0.01) |
| 7.25 | 0.35 (0.03) | 0.60 (0.02) | 0.05 (0.02) |
| 7.5 | 0.26 (0.03) | 0.67 (0.02) | 0.07 (0.02) |
| 7.75 | 0.17 (0.02) | 0.78 (0.02) | 0.05 (0.02) |
| 8.0 | 0.12 (0.01) | 0.83 (0.02) | 0.05 (0.02) |
| 8.25 | 0.06 (0.01) | 0.88 (0.02) | 0.06 (0.02) |
Рис. 3.
Средняя занятость (оккупация) ионизированного и нейтральных таутомерных состояний остатков His37A, His37B, His37C, His37C белка M2 при Т = 300 К вдоль равновесной МД траектории 25 нс; черный ‒ HIS+; серый штрих – таутомер HIE; штрих точка – таутомер HID.
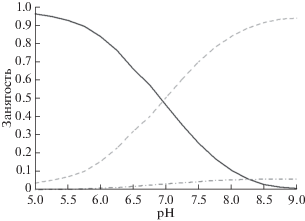
Средняя потенциальная энергия белка M2 вдоль равновесной МД траектории в ионизированном и нейтральном состоянии четырех остатков гистидина приведена в табл. 3. Видно, что различие средних потенциальных энергий 〈Epot〉MD белка М2 для ионизированного состояния HIS+ и нейтрального состояния HIE составляет ~6 ккал/моль. Средняя величина тепловых флуктуаций энергии таутомера HIE, оцененная по равновесной МД траектории длительностью 25 нс, равна ~12 ккал/моль (табл. 3). Заселенности заряженных и нейтральных состояний четырех остатков гистидина заметно флуктуируют вдоль МД траектории (рис. 4а, б). Однако средние значения долей заселенностей ионизированных и нейтральных форм His37A, His37B, His37C и His37D составляют 0.04‒0.06 (табл. 4). Состояния ионизации остатков гистидина, т.е. ионизированный HIS+ и нейтральный таутомер HIE при рН 6.5, реализуются с сопоставимыми вероятностями 0.64 и 0.32 соответственно (табл. 4). Средняя потенциальная энергия 〈Epot〉MD белка М2 с нейтральным таутомером HID значительно больше (на ~50 ккал/моль) по сравнению с энергиями состояний HIS+ и HIE остатков гистидина (табл. 3). Поэтому вероятность состояний HID мала ~0.02‒0.04. Средние ионизационные состояния остатков гистидина, рассчитанные по равновесной МД траектории длительностью 25 нс при рН 6.5, представлены в табл. 4. Согласно табл. 4, остатки гистидина белка M2 находятся в ионизированном состоянии HIS+ со средней вероятностью ~0.64, тогда как нейтральный таутомер HIE встречается с вероятностью ~0.32, а нейтральный таутомер HID ‒ с вероятностью ~0.04. Средняя структура нейтрального состояния ионного канала M2 незначительно отличается, в пределах амплитуды тепловых флуктуаций ~0.9 Å, от экспериментальной структуры канала М2, 2KAD (рис. 2). Можно отметить, что среднеквадратичное отклонение (RMSD) положений атомов Oγ остатков Ser31A, Ser31В, Ser31С, Ser31D структур канала M2 вдоль траектории MД не превышало 1.1 Å. Например, расстояния между атомами Oγ остатков Ser31A/Ser31C и Ser31B/Ser31D колеблются в диапазоне 8.4–9.5 Å. Проход через ионные каналы М2 контролируется остатками His37A, His37B, His37C, His37D и характеризуется флуктуациями попарных расстояний между ближайшими атомами Cε1 и Nε2 в парах His37A–His37B, His37B–His37C, His37C–His37D, His37D–His37A в диапазоне 4.5‒5.4 Å. Флуктуации значений рКа и вероятности встречаемости состояний HIS+, HIE всех остатков гистидина вдоль равновесной МД траектории длительностью 25 нс представлены в табл. 4. Величины долей ионизированных и нейтральных таутомеров гистидина имеют заметные флуктуации (рис. 4а, б), тогда как их средние значения практически равны (табл. 4). Зависимость средних долей фракций ионизационного и нейтральных состояний остатков гистидина от величины рН показана на рис. 3. Значения средней константы pKa остатков гистидина смещены на 0.3 единицы в сторону высоких значений рН, 〈pKa〉 ~ 6.8, (pK0 6.5) (табл. 4). Следовательно, остатки гистидина находятся в отрицательном электростатическом потенциале, который стабилизирует ионизированное состояние H+. Небольшая разность между энергиями заряженного HIS+ и нейтрального HIE состояний остатков гистидина (~8 ккал/моль), и значительные тепловые флуктуации энергий, ~15 и 12 ккал/моль, для состояний НIS+ и HIE (табл. 3) способствуют процессу переноса протона через “гистидиновый затвор”, т.е. через квартет His37A, His37B, His37C, His37D ионного канала М2.
Таблица 3.
Энергии молекулы белка M2 для ионизированного и нейтральных состояний остатков гистидина при pH 6.5 и Т = 300 K
| Состояние ионизации |
〈Epot〉MDa | 〈EVDW〉MDб | 〈ECOL〉MDв | 〈EHbond〉MDг | 〈ESolv〉MDд |
|---|---|---|---|---|---|
| HIS+ | –1344.5 (15.2) | –423.7 (13.6) | –495.1 (6.6) | –331.2 (7.0) | –98.3 (7.1) |
| HIE | –1336.5 (12.1) | –416.8 (10.9) | –489.5 (4.2) | –323.2 ( 5.6) | –106.1 (8.3) |
| HID | –1289.7 (10.9) | –413.7 (9.3) | –477.2 (4.4) | –294.7 (5.5) | –133.2 (7.2) |
Рис. 4.
Фракции (доли) остатков гистидина белка М2 вдоль равновесной МД траектории в 25 нс при рН 6.5, Т = 300 К в различных ионизационных состояниях: ионизированная форма HIS+ (а); нейтральный таутомер HIE (б). Черный цвет ‒ остатки His37A, темно-серый ‒ His37B, серый ‒ His37C, светло-серый ‒ HIS37D.
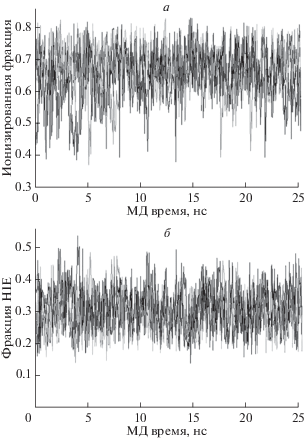
Таблица 4.
Средние величины pKa и фракций ионизированных и нейтральных таутомеров остатков гистидина белка М2 вдоль равновесной траектории в 25 нс при pH 6.5, T = 300 K
| Остаток | HIS+ а | HIDа | HIEа | pKaб |
|---|---|---|---|---|
| His37A His37B His37C His37D |
0.65в (0.04)г 0.63 (0.06) 0.66 (0.03) 0.62 (0.05) |
0.03в (0.01)г 0.04 (0.02) 0.05 (0.01) 0.05 (0.03) |
0.32в (0.03)г 0.33 (0.03) 0.29 (0.02) 0.32 (0.04) |
6.80в (0.14)г 6.80 (0.09) 6.82 (0.16) 6.78 (0.18) |
| Среднее | 0.64 | 0.04 | 0.32 | 6.80 |
ОСНОВНОЕ СОЕДИНЕНИЕ НОВОГО СЕМЕЙСТВА ИНГИБИТОРОВ ИОННОГО КАНАЛА M2
Молекула DABCО (рис. 1в) практически равновелика в трех направлениях и имеет размер, близкий поперечному размеру молекул амантадина и римантадина (рис. 1а, б), известных как ингибиторы ионного канала белка M2 [16, 17]. Исчерпывающий докинг выполнен как для внешней молекулярной поверхности белка М2, так и для внутренней молекулярной поверхности ионного канала М2. На рис. 5а, б приведен результат докинга, т.е. оптимальная позиция и ориентация молекулы DABCО на молекуле белка М2. Эта структура показывает, что (1) ось между атомами N1-N2 молекулы DABCO перпендикулярна продольной оси канала М2; (2) образуются две практически идеальные водородные Н-связи между атомами N1, N2 молекулы DABCO и атомом Оγ остатков Ser31A и Ser31B (или Ser31C и Ser31D) с геометрией триплетов атомов Оγ(Ser31A)···Н–N1 и Оγ-Ser31B···Н–N2, вовлеченных в образование Н-связей, близких к оптимальной. Суммарная энергия Н-связей двух экваториальных атомов азота N1, N2 молекулы DABCO с OH-группами двух остатков Ser31A, Ser31B составляет ~8 ккал/моль. В альтернативной эквивалентной ориентации молекула DABCO повернута на 90° вокруг центральной оси ионного канала М2, создавая две Н-связи с Ser31С и Ser31D (рис. 5б). Две эквивалентные ориентации молекулы DABCO увеличивают свободную энергию связывания на величину kT × ln2 ~ 0.4 ккал/моль. Полная энергия связи и удельный вклад различных типов атом‒атомных взаимодействий приведены в табл. 5. Видно, что главные вклады в энергию связывания вносят ван-дер-ваальсовы взаимодействия, H-связи и дегидратация молекулы лиганда. Молекула DABCO может связываться с внутренней поверхностью ионного канала M2, т.е. в канале (IN) и с молекулярной внешней поверхностью вне ионного канала (OUT) белка M2. Разность между оптимальными энергиями связывания в канале и вне канала равна энергии зазора, EGAP= EIN‒EOUT, определяет долю молекул блокатора, связанную в канале, пропорциональную величине ехр(–ЕGAP/kT), определяющей эффективность блокирования канала М2. Т.е. энергия зазора EGAP является важным параметром эффективности блокирования ионного канала M2 и переноса протона по каналу. Из представленных в табл. 5 данных видно, что энергетическая щель EGAP для DABCО равна 2.4 ккал/моль, а соответствующая свободная энергия связывания на 0.4 ккал/моль выше с учетом двух эквивалентных ориентаций молекулы DABCО в канале М2.
Рис. 5.
Молекула DABCО (а) и молекула DABCО, связанная в канале М2 (б). Показаны водородные связи между атомами Н–N1(N2) и Oγ остатков Ser31A, Ser31B с длиной связи, 2.13 и 2.28 Å соответственно.
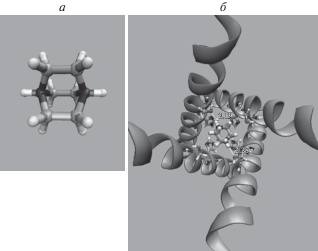
Таблица 5.
Энергии связывания производных диазабициклооктана в ионном канале и вне канала белка М2
| Молекула | Тип связывания | ePLa | eVDWб | eColб | eHbб | eSolvб | eGeoб |
|---|---|---|---|---|---|---|---|
| DABCО | В каналев | –9.0 | –8.4 | 4.9 | –9.2 | –8.1 | 11.8 |
| Вне каналаг | –6.6 | –4.2 | 3.1 | –4.8 | –11.4 | 10.7 | |
| Щель {Ein‒Eout}д | –2.4 | ||||||
| Номер в ряду эффективностие | 3/4 | ||||||
| DABCOF4 | В каналев | –24.3 | –8.0 | –6.1 | –9.3 | –13.0 | 12.1 |
| Вне каналаг | –24.2 | –2.1 | –11.0 | –4.9 | –18.4 | 12.2 | |
| Щель {Ein‒Eout}д | –0.1 | ||||||
| Номер в ряду эффективностие | 6 | ||||||
| DABCOF8 | В каналев | –41.9 | –6.0 | –21.5 | –9.6 | –18.4 | 13.6 |
| Вне каналаг | –47.1 | 1.6 | –30.4 | –5.0 | –28.0 | 14.6 | |
| Щель {Ein‒Eout}д | 5.2 | ||||||
| Номер в ряду эффективностие | 7 | ||||||
| DABCOF12 | В каналев | –59.3 | –3.5 | –42.4 | –9.4 | –20.7 | 16.8 |
| Вне каналаг | –65.7 | 3.7 | –49.6 | –4.9 | –32.5 | 17.7 | |
| Щель {Ein‒Eout}д | 6.4 | ||||||
| Номер в ряду эффективностие | 8 | ||||||
| DABCOB | В каналев | –16.9 | –15.3 | 1.6 | –8.8 | –7.5 | 13.1 |
| Вне каналаг | –13.8 | –11.5 | –0.6 | –5.0 | –9.2 | 12.4 | |
| Щель {Ein‒Eout}д | –3.1 | ||||||
| Номер в ряду эффективностие | 1 | ||||||
| DABCOT | В каналев | –16.6 | –16.6 | 1.6 | –8.3 | –6.3 | 13.0 |
| Вне каналаг | –13.9 | –16.7 | 0.8 | –5.0 | –7.0 | 14.0 | |
| Щель {Ein‒Eout}д | –2.8 | ||||||
| Номер в ряду эффективностие | 2 | ||||||
| DABCON | В каналев | –20.5 | –19.6 | 2.1 | –7.8 | –7.8 | 12.6 |
| Вне каналаг | –18.1 | –13.3 | –2.9 | –4.9 | –9.2 | 12.1 | |
| Щель {Ein‒Eout}д | –2.4 | ||||||
| Номер в ряду эффективностие | 3/4 | ||||||
| DABCONM | В каналев | –17.1 | –20.6 | 2.4 | –7.2 | –6.7 | 14.4 |
| Вне каналаг | –17.5 | –19.3 | 2.3 | –4.5 | –10.1 | ||
| Щель {Ein‒Eout}д | 0.4 | 14.1 | |||||
| Номер в ряду эффективностие | 5 |
a ePL – полная энергия связывания лиганда в ионном канале белка M2. б Энергии различных типов взаимодействий: eVDW – ван-дер-ваальсовы, eCoul ‒ электростатические, eHb ‒ водородные связи, eSolv ‒ взаимодействие с окружающей средой, eGeo ‒ энергия деформации оптимальной структуры лиганда, ккал/моль. в Оптимальная энергия связывания в канале М2. г Максимальная энергия связывания вне канала M2. д Величина щели ΔGAP = Ein–Eout между энергиями связи in и out протонного канала M2. е Место в ряду эффективностей.
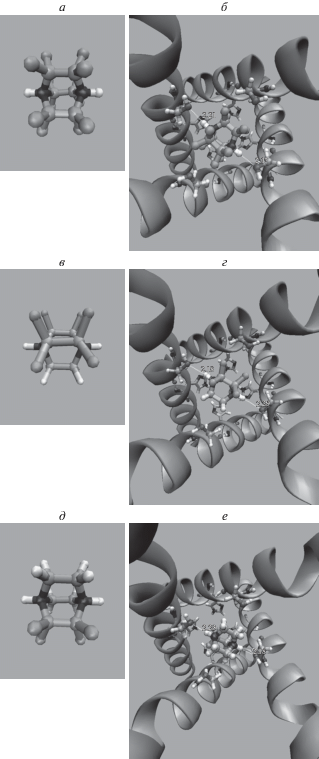
Фторпроизводные диазабициклооктана
Оптимизировать энергию связывания молекулы ингибитора ионного канала М2 можно путем модификаций, влияющих на ван-дер-ваальсовы взаимодействия препарата с внутренней поверхностью ионного канала М2. Это может быть достигнуто путем замены атомов водорода молекулы DABCО на атомы фтора, который имеет значительно большую энергию ван-дер-ваальсовых взаимодействий с ионным каналом M2. Нами рассмотрены три фторпроизводных исходной молекулы DABCO, а именно, тетрафтордиазабициклооктан (DABCOF4), октафтордиазабициклооктан (DABCOF8) и додекафтордиазабициклооктан (DABCOF12), которые содержат четыре, восемь и 12 атомов фтора (рис. 6а, в, д) соответственно. Оптимизированные комплексы F-производных, DABCOF4, DABCOF8 и DABCOF12, в ионном канале М2 показаны на рис. 6б, г, е. Фторпроизводные создают две сильные Н‑связи между атомами N1/N2 молекулы препарата с атомом Oγ остатков Ser31A и Ser31B или Ser31C, Ser31D. Энергии связывания фторпроизводных DABCOF4, DABCOF8 и DABCOF12 с ионным каналом M2 приведены в табл. 5. Показано, что энергия связи в канале M2 значительно больше, чем у основного соединения DABCО. Однако энергия зазора между энергиями связывания в канале и вне канала М2, т.е. параметр EGAP ~ 0.1 ккал/моль (табл. 5). Это свидетельствует о том, что у производных DABCOF4 доли связывания в канале и вне канала практически равны. Энергии зазора EGAP являются положительными, ~5–6 ккал/моль у производных DABCOF8 и DABCOF12 (табл. 5), т.е. связывание вне канала это основной тип, а внутри канала М2 оно достаточно мало. Таким образом, несмотря на большую энергию связывания в канале М2 энергии связывания F-производных вне канала сопоставимы или заметно больше. Преимущественное связывание вне канала обусловлено большой энергией гидратации F-производных ‒ DABCOF4, DABCOF8 и DABCOF12, следовательно, дегидратация F-производных при внутриканальном связывании крайне невыгодна.
Полициклические производные диазабициклооктана
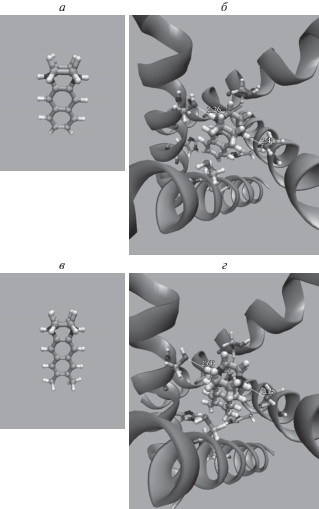
Полициклические производные DABCО ‒ диазабициклооктанбензол (DABCOB) (рис. 7а) и диазабициклооктантолуол (DABCOТ) (рис. 7в) соответственно. Молекула DABCOТ получена заменой экваториальных атомов водорода во фрагменте бензола в молекуле DABCOB на метильные группы (СН3). Более высокомолекулярные производные ‒ это диазабициклооктаннафталин (DABCON) (рис. 8а), и молекула диазабициклооктанметилнафталина (DABCONM) (рис. 8в), полученная заменой экваториальных атомов водорода на метильные группы (СН3) во фрагменте нафталина.
Рис. 7.
Свободные формы DABCOВ и DABCOТ (а, в) и оптимальная позиция этих производных, связанных в канале М2 (б, г).
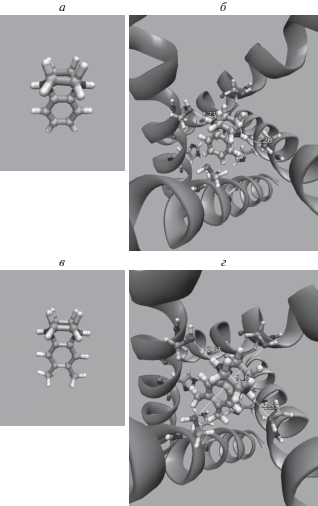
Рис. 8.
Свободная форма DABCON и DABCONМ (а, в) и оптимальная позиция этих молекул, связанных в канале М2 (б и г).
Конструирование молекул ингибиторов с использованием жесткого полициклического фрагмента более предпочтительно, чем с использованием гибкого фрагмента за счет меньшей потери конформационной энтропии при ограничении конформационной подвижности молекулы препарата при связывании внутри ионного канала М2. Результаты докинга бициклических и трициклических производных DABCO на белок М2 показаны на рис. 7б, г и рис. 8б, г. Молекулы ингибиторов DABCOB, DABCOT, DABCON и DABCON, связанные в канале М2, стабилизированы водородными связями между атомами Н–N1(N2) и Oγ остатков Ser31A, Ser31B с длиной связи в пределах 2.1–2.6 Å. Энергии связывания бициклических и трициклических производных DABCO с ионным каналом M2 представлены в табл. 5. Видно, что бициклические производные DABCOB и DABCOТ имеют энергию связи на ~7 ккал/моль больше энергии связывания ведущего соединения DABCO.
Трициклические производные DABCON и DABCONM демонстрируют дальнейшее увеличение энергии связывания с ионным каналом M2 на ~3.5 ккал/моль по сравнению с бициклическими производными DABCOB и DABCOBM (табл. 5). Тем не менее, замещение двух периферических атомов водорода в трициклическом производном DABCON на метильные группы CH3 с образованием молекулы DABCONM не приводит к дальнейшему увеличению энергии связывания. Самая большая молекула DABCONM характеризуется ослаблением ван-дер-ваальсовых взаимодействий с ионным каналом М2 по сравнению с молекулой DABCON (на ~3 ккал/моль, табл. 5).
Можно сделать вывод, что наиболее эффективным блокатором ионного канала M2 является молекула DABCOB (табл. 5), энергия связывания которой с ионным каналом M2 больше ~16.9 ккал/моль, тогда как энергия связывания ведущего соединения DABCO составляет ~9.0 ккал/моль. Разность энергий связывания в каналe и вне канала у DABCOB равна ~3.1 ккал/моль, т.е. она на 0.7 ккал/моль больше энергии связывания ведущего соединения DABCO (табл. 5). Отношение фракций молекул DABCOB, связанных вне (Cout) и внутри канала (Cin), достаточно мало, а именно, Сout/Сin, составляет ~0.01, т.е. эта молекула является эффективным блокатором ионного канала М2.
ЗАКЛЮЧЕНИЕ
Блокирование ионного канала M2 вируса гриппа А путем связывания внутри канала молекулы производного DABCO представляется разумной стратегией ингибирования переноса протона H+ через ионный канал М2 и деактивации вируса гриппа А. Рассмотрены семь структур молекул блокаторов, производных DABCO, и детально исследовано их связывание с белком М2. Показано, что наилучшее блокирование ионного канала обеспечивает молекула DABCOВ. Энергия связывания молекулы DABCOВ в канале М2 достаточно высока ~16.9 ккал/моль. Разность энергий связывания вне и внутри канала М2 более 3 sккал/моль. Заметная разность энергий связывания вне и внутри ионного канала М2 обеспечивает преимущественное связывание внутри канала, так что отношение фракций молекул, связанных вне и внутри канала, составляет ~ 0.01, т.е. молекулы препарата будут связаны внутри канала М2 и будут блокировать транспорт протона Н+.
Вторая важная особенность DABCO и его производных ‒ положительный заряд +2, локализованный на атомах азота. Этот заряд создает положительный электростатический потенциальный барьер, оцениваемый величиной ~3‒4 ккал/моль, для диффузии протонов H+ через канал М2. Такой положительный электростатический потенциальный барьер отсутствует или незначителен у амантадина и римантадина как в нейтральном состоянии, при высоких значениях рН > 7, так и в заряженном состоянии при низких значениях рН. Нейтральность амантадина и римантадина доказана методом ЯМР при температуре 243 К и pН 7.5 [16, 17]. Сочетание стерических и электростатических потенциальных барьеров представляет собой дополнительный фактор повышения эффективности блокирования транспорта ионов Н+ через ионный канал M2 при внутриканальном связывании молекулы DABCOВ.
Автор выражает глубокую признательность Федоровой О.С. за интерес к работе.
Исследования выполнены при поддержке Российского фонда фундаментальных исследований (№ 18-04-00005-a) и Минобрнауки РФ (№ 0309-2019-0001).
Настоящая статья не содержит каких-либо исследований, выполненных с использованием биологических материалов.
Автор заявляет об отсутствии конфликта интересов.
Список литературы
World Health Organization. Influenza. http:// www.who.int/en/news-room/fact-sheets/detail/influenza-(seasonal)
Stouffer A.L., Acharya R., Salom D., Levine A.S., Costanzo L.D., Soto1 C.S., Tereshko V., Nanda V., Stayrook S., DeGrado W.F. (2008) Structural basis for the function and inhibition of an influenza virus proton channel. Nature. 451, 596‒599. https://doi.org/10.1038/nature06528
Hu F., Luo W., Hong M. (2010) Mechanisms of proton conduction and gating in influenzaM2 proton channels from solid-state NMR. Science. 330(6003), 505‒508. https://doi.org/10.1126/science.1191714
Wanka L., Iqbal K., Peter R., Schreiner P.R. (2013) The lipophilic bullet hits the targets: medicinal chemistry of adamantane derivatives. Chem. Rev. 113, 3516–3604.https://doi.org/10.1021/cr100264t
Moffat J.S., Vijayvergiya V., Gao P.F., Cross T.A., Woodbury D.J., Busath D.D. (2008) Proton transport through influenza A virus M2 protein reconstituted in vesicles. Biophys. J. 94, 434–445. https://doi.org/10.1529/biophysj.107.109082
Hari Z.S., Moorthy N., Poongavanam V., Pratheepa V. (2014) Viral M2 ion channel protein: a promising target for anti-influenza drug discovery. Mini-Rev. Med. Chem. 14(10), 819–830. https://doi.org/10.2174/138955751410141020150822
Olsen R.W. (2006) Picrotoxin-like channel blockers of GABAA receptors. Proc. Natl. Acad. Sci. USA. 103, 6081–6082. www.pnas.org/cgi/doi/10.1073/pnas.0601121103
Thomaston J.L., Polizzi N.F., Konstantinidi A., Wang J., Kolocouris A., DeGrado W.F. (2018) Inhibitors of the M2 proton channel engage and disrupt transmembrane networks of hydrogen-bonded waters. J. Am. Chem. Soc. 140, 15219−15226. https://doi.org/10.1021/jacs.8b06741
Mustafa M., Henderson D.J., Busath D.D. (2009) Free-energy profiles for ions in the influenza M2-TMD channel. Proteins. 76, 794–807. https://doi.org/10.1002/prot.22376
Homeyer N., Ioannidis H., Kolarov F., Gauglitz G., Zikos C., Kolocouris A., Gohlke H. (2016) Interpreting thermodynamic profiles of aminoadamantane compounds inhibiting the M2 proton channel of influenza A by free energy calculations. J. Chem. Inf. Model. 56, 110‒126. https://doi.org/10.1021/acs.jcim.5b00467
Nishimura K., Kim S., Zhang L., Cross T.A. (2002) The closed state of a H+ channel helical bundle combining precise orientational and distance restraints from solid state NMR. Biochemistry. 41, 13170‒13177. https://doi.org/10.1021/bi0262799
Cady S.D., Hong M. (2008) Amantadine-induced conformational and dynamical changes of the influenza M2 transmembrane proton channel. Proc. Natl. Acad. Sci. USA. 105, 1483–1488.https://doi.org/10.1073pnas.0711500105
Holsinger L.J., Nichani D., Pinto L.H., Lamb R.A. (1994) Influenza A virus M2 ion channel protein: a structure-function analysis. J. Virol. 68, 1551–1563. https://www.ncbi.nlm.nih.gov/pubmed/7508997
Wang J., Kim S., Kovacs F., Cross T.A. (2001) Structure of the trans membrane region of the M2 protein H+ channel. Protein Sci. 10, 2241–2250. http://www.proteinscience.org/cgi/doi/10.1101/ps.17901
Sakai Y., Kawaguchi A., Nagata K., Hirokawa H. (2018) Analysis by metadynamics simulation of binding pathway of influenza virus M2 chan. Microbiol. Immunol. 62, 34–43.https://doi.org/10.1111/1348-0421.12561nelblockers
Cady S.D., Mishanina T.V., Hong M. (2009) Structure of amantadine-bound M2 transmembrane peptide of influenza A in lipid bilayers from magic-angle-spinning solid-state NMR: the role of Ser31 in amantadine binding. J. Mol. Biol. 385, 1127‒1141. https://doi.org/10.1016/j.jmb.2008.11.022
Cady S.D., Schmidt-Rohr K., Wang J., Soto C.S., Degrado W.F., Hong M. (2010) Structure of the amantadine binding site of influenza M2 proton channels in lipid bilayers. Nature. 463, 689–692. https://doi.org/10.1038/nature08722
Intharathep P., Laohpongspaisan C., Rungrotmongkol T., Loisruangsin A., Malaisree M., Decha P., Aruksakunwong O., Chuenpennit K., Kaiyawet N., Sompornpisut P., Pianwanit S., Hannongbu S. (2008) How amantadine and rimantadine inhibit proton transport in the M2 protein channel. J. Mol. Graphics Modelling. 27, 342–348. https://doi.org/10.1016/j.jmgm.2008.06.002
Mould J.A., Li H.C., Dudlak C.S., Lear J.D., Pekosz A., Lamb R.A., Pinto L.H. (2000) Mechanism for proton conduction of the M2 ion channel of influenza A virus. J. Biol. Chem. 275, 8592–8599. https://doi.org/10.1074/jbc.275.12.8592
Schnell J.R., Chou J.J. (2008) Structure and mechanism of the M2 proton channel of influenza A virus. Nature. 451, 591‒595. https://doi.org/10.1038/nature06531
Acharya R., Carnevale V., Fiorin G., Levine B.G., Polishchuk A.L., Balannik V., Samish I., Lamb R.A., Pinto L.H., DeGrado W.F., Klein M.L. (2010) Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc. Natl. Acad. Sci. USA. 107, 15075–15080. https://doi.org/10.1073/pnas.1007071107
Roux B., Schulten K. (2004) Computational studies of membrane channels. Structure. 12, 1343–1351. https://doi.org/10.1016/j.str.2004.06.013
Thomaston J.L., Alfonso-Prieto M., Woldeyes R.A., Fraser J.S., Klein M.L., Fiorin G., DeGrado W.F. (2015) High-resolution structures of the M2 channel from influenza A virus reveal dynamic pathways for proton stabilization and transduction. Proc. Natl. Acad. Sci. USA. 112, 14260–14265. https://doi.org/10.1073/pnas.1518493112
Hong M., DeGrado W.F. (2012) Structural basis for proton conduction and inhibition by the influenza M2 protein. Protein Sci. 21, 1620–1633. https://doi.org/10.1002/pro.2158
Hu F., Schmidt-Rohr K., Hong M. (2012) NMR detection of pH-dependent histidine water proton exchange reveals the conduction mechanism of a transmembrane proton channel. J. Am. Chem. Soc. 134, 3703–3713. https://doi.org/10.1021/ja2081185
Chen H., Wu Y., Voth G.A. (2007) Proton transport behavior through the influenza A M2 channel: Insights from molecular simulation. Biophys. J. 93, 3470–3479. https://doi.org/10.1529/biophysj.107.105742
Acharya R., Carnevale V., Fiorin G., Levine B. G., Polishchuk A.L., Balannik V., Samish I., Lamb R. A., Pin-to L.H., DeGrado W. F., Klein M.L. (2010) Structure and mechanism of proton transport through the transmembrane tetrameric M2 protein bundle of the influenza A virus. Proc. Natl. Acad. Sci. USA. 107, 15075–15080. https://doi.org/10.1073/pnas.1007071107
Du Q.-S., Wang S.-Q., Chen D., Meng J.-Z., Huang R.-B. (2014) In depth analysis on the binding sites of adamantane derivatives in HCV (Hepatitis C Virus) p7 channel based on the NMR structure. PLoS One. 9(4), e93613. https://doi.org/10.1371/journal.pone.0093613
Smondyrev A.M., Voth G.A. (2002) Molecular dynamics simulation of proton transport through the influenza A virus M2 channel. Biophys. J. 83, 1987‒1996. https://doi.org/10.1016/S0006-3495(02)73960-X
Chen H., Wu Y., Voth G.A. (2007) Proton transport behavior through the influenza A M2 channel: insights from molecular simulation. Biophys. J. 93, 3470–3479.https://doi.org/10.1529/biophysj.107.105742
Baghernejad B. (2010) 1,4-Diazabicyclo[2.2.2]octane (DABCO) as a useful catalyst in organic synthesis. Eur. J. Chem. 1, 54‒60. https://doi.org/10.5155/eurjchem.1.1.54-60.2
Cecchi L., DeSarlo F., Machetti F. (2006) 1,4-Diazabicyclo[2.2.2]octane (DABCO) as an efficient reagent for the synthesis of isoxazole derivatives from primary nitro compounds and dipolarophiles: the role of the base. Eur. J. Organic Chem. 21, 4852–4860. https://doi.org/10.1002/ejoc.200600475
Zhang K., Drummey K.J., Moon N.G., Chiang W D., Long T.E. (2016) Styrenic DABCO salt-containing monomers for the synthesis of novel charged polymers. Polymer Chem. 7, 3370-3374. https://doi.org/10.1039/C6PY00426A
Попов А.В., Воробьев Ю.Н. (2010) Программа GUI-BioPASED для моделирования молекулярной динамики биополимеров с графическим пользовательским интерфейсом. Молекуляр. биология. 44(4), 735–742.
Vorobjev Y.N. (2010) Blind docking method combining search of low-resolution binding sites with ligand pose refinement by molecular dynamics-based global optimization. J. Comput. Chem. 31, 1080–1092. http://bison.niboch.nsc.ru/index.html
Vorobjev Y.N., Hermans J. (1997) SIMS, computation of a smooth i.nvariant molecular surface. Biophys. J. 73, 722–732. https://doi.org/10.1016/S0006-3495(97)78105-0
Vorobjev Y.N., Scheraga H.A. (1997) A fast adaptive multigrid boundary element method for macromolecular electrostatic computations in a solvent. J. Comput. Chem. 18, 569–583. https://doi.org/10.1002/(SICI)1096- 987X(199703)18:4<569::AID-JCC10>3.0.CO;2-B
Vorobjev Y.N., Almagro, J.C., Hermans J. (1998) Discrimination between native and intentionally misfolded conformations of proteins: ES/IS, a new method for calculating conformational free energy that uses both dynamics simulations with an explicit solvent and an implicit solvent continuum model. Proteins. 32, 399–413. https://doi.org/10.1002/(SICI)1097-0134(19980901)32:4<399::AID-PROT1>3.0.CO;2-C
Vorobjev Y.N., Vila J.A., Scheraga H.A. (2008) FAMBEpH: A fast and accurate method to compute the total solvation free energies of proteins. J. Phys. Chem. B, 112, 11122–11136. https://doi.org/10.1021/jp709969n
Vorobjev Y.N. (2011) Advances in implicit models of water solvent to compute conformational free energy and molecular dynamics of proteins a constant pH. Adv. Protein Chem. Structural Biol. 85, 281–322. https://doi.org/10.1002/jcc.22909
Vorobjev Y.N. (2012) Potential of mean force of water-proton bath and molecular dynamic simulation of proteins at constant pH. J. Comput. Chem. 33, 832–842. https://doi.org/10.1002/jcc.22909
Vorobjev Y.N., Scheraga H.A., Vila J.A. (2018) Coupled molecular dynamics and continuum electrostatic method to compute the ionization pKa’s of proteins as a function of pH. Test on a large set of proteins. J. Biomol. Struct. Dynamics 36, 563‒574. https://doi.org/10.1080/07391102.2017.1288169
Cornell W.D., Cieplak P., Bayly C.I., Gould I.R., Merz K.M., Ferguson D.M., Spellmeyer D.C., Fox T., Caldwell J.W., Kollman P.A. (1995) A second generation for the simulation of proteins, nucleic acids and organic molecules. J. Am. Chem. Soc. 117, 5179–5197. https://doi.org/10.1021/ja00124a002
Wang J., Cieplak P., Kollman P.A. (2000) How well does a restrained electrostatic potential (RESP) model perform in calculating conformational energies of organic and biological molecules? J. Comput. Chem. 21, 1049–1074.https://doi.org/10.1002/1096-987X(200009)21:12%3C1049::AID-JCC3%3E3.0.CO;2-F
Wang J., Wolf R.M., Caldwell J.W., Kollman P.A., Case D.A. (2004) Development and testing of a general amber force fields. J. Comput. Chem. 25, 1157–1174. https://doi.org/10.1002/jcc.20035
Lazaridis T., Karplus M. (1999) Effective energy function for proteins in solution. PROTEINS: Structure, Function, Genetics. 35, 133–152.
Mostafa M.F., Youssef A.A.A. (2001) Dielectric permittivity and ac conductivity investigation for the new model lipid bilayer material: (CH2)10 (NH3)2CdCl4 Z. Naturforsch. 56a, 568‒578.https://doi.org/10.1515/zna-2001-0806
Isom D.G., Castaneda C.A., Cannon B.R. (2011) Large shifts in pKa values of lysine residues buried inside a protein. Proc. Natl. Acad. Sci. USA. 108, 5260–5265. https://doi.org/10.1073/pnas.1010750108
Li C., Li L., Zhang J., Alexov E. (2012) Highly efficient and exact method for parallelization of grid-based algorithms and its implementation in DelPhi. J. Comput. Chem. 33, 1960−1966. https://doi.org/10.1002/jcc.23033
Li L., Li C., Zhang Z., Alexov E. (2013) On the dielectric “Constant” of proteins: smooth dielectric function for macromolecular modeling and its iplementation in DelPhi. J. Chem. Theory Comput. 9, 2126−2136.https://doi.org/10.1021/ct400065j
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология