

Молекулярная биология, 2020, T. 54, № 3, стр. 487-496
Поддержание плазмидной экспрессии in vivo зависит главным образом от содержания CpG-мотивов в векторе и трансгене
А. В. Брутер a, b, М. В. Калашникова a, b, А. П. Притыко a, А. В. Белявский a, *
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
b Национальный медицинский исследовательский центр онкологии им. Н.Н. Блохина,
Министерства здравоохранения Российской Федерации
115478 Москва, Россия
* E-mail: abelyavs@yahoo.com
Поступила в редакцию 29.10.2019
После доработки 12.12.2019
Принята к публикации 12.12.2019
Аннотация
Генная терапия с использованием плазмидных векторов ‒ безопасная и относительно недорогая терапевтическая стратегия, однако быстрое подавление экспрессии трансгена в тканях значительно снижает ее эффективность. Ранее мы создали плазмидный вектор pMBR2 с низким содержанием CpG-мотивов, поддерживающий длительную экспрессию трансгенов в мезенхимальных стволовых клетках in vitro. В данной работе проанализирована долговременная экспрессия секретируемой щелочной фосфатазы мыши (mSEAPwt) и ее варианта, лишенного CpG-динуклеотидов (mSEAP0), в мышцах задних конечностей и печени мышей. Анализ экспрессии проводили путем измерения уровня mSEAP в крови в течение 1 года. Введение конструкции pMBR2-mSEAP0 привело к повышению уровня mSEAP в мышцах конечностей более чем в 2.5 раза в течение первых 2 мес., причем превышение исходного уровня сохранялось до конца эксперимента. Контрольная конструкция pCDNA3.1-mSEAP0 обеспечивала существенно более низкий уровень экспрессии. Уровень mSEAP, экспрессируемой конструкцией pMBR2-mSEAPwt, снизился приблизительно до 40% через 6 мес. и после этого оставался примерно постоянным. В печени мышей уровень экспрессии трансгена конструкцией pMBR2-mSEAP0 уменьшился примерно в 2 раза за первые 18 недель, после чего продолжал медленно снижаться ‒ до 17% к концу эксперимента. В случае pMBR2-mSEAPwt уровень экспрессии трансгена в печени снизился до 18% и далее оставался на уровне примерно 10%. В конструкции pCDNA3.1-mSEAP0 экспрессия трансгена через 2 недели резко упала до 5% и оставалась близкой к нулю в течение эксперимента. Таким образом, для длительной экспрессии трансгенов плазмидными векторами в печени содержание CpG как в векторе, так и в трансгене должно быть существенно снижено, тогда как для продолжительной экспрессии в скелетных мышцах достаточно минимизации содержания CpG-мотивов в векторе. Наши данные также свидетельствуют о том, что локализация S/MAR-элементов внутри транскрипционной единицы, в отличие от локализации вне ее, приводит к значительному снижению уровня секретируемых, но не цитоплазматических белков.
ВВЕДЕНИЕ
Считается, что вирусные векторы пригодны для экспрессии терапевтических генов в клетках и органах, однако за последнее время обнаруживается все больше фактов, свидетельствующих о проблемах их использования в генной терапии. Так, на этапе трансдукции вирусные частицы нередко способны вызывать сильный иммунный ответ [1], особенно при повторном применении. Иммунный ответ может угрожать жизни пациента, а позднее с высокой вероятностью приводить к элиминации клеток, несущих трансген [2]. При интеграции вектора в геном хозяина гены, обладающие критически важными функциями, например онкосупрессоры, могут быть повреждены или деактивированы [3, 4]. Недавно обнаружили нейротоксичность высоких доз аденоассоциированных вирусов [5]. Очевидно, что требования к безопасности выше при терапии врожденных генетических дефектов у новорожденных и детей. Кроме того, использование вирусных векторов накладывает ограничения на размер трансгена. И, наконец, производство таких векторов технологически сложно и дорого.
Плазмидные векторы лишены большинства из этих недостатков. Они нетоксичны, не встраиваются в геном хозяина, не вызывают развития опасного для жизни иммунного ответа, могут содержать вставку значительного размера, а их производство относительно дешево. Основными недостатками этих векторов являются низкая эффективность доставки и ограниченная длительность экспрессии. Низкую эффективность доставки сейчас пытаются преодолевать с помощью электропорации или гидродинамических методов. Однако плазмидные векторы еще не оптимизированы в достаточной степени, чтобы достичь значительной длительности экспрессии трансгена во многих органах-мишенях.
Среди потенциальных органов-мишеней для генной терапии важное место занимают печень и скелетные мышцы. В печени синтезируются многие белки (например, фенилаланингидроксилаза, альфа-1-антитрипсин, факторы свертывания крови), мутации в которых вызывают серьезные наследственные болезни. Кроме того, клетки поджелудочной железы плохо поддаются генетической модификации, поэтому печень рассматривают как перспективную цель для генной терапии сахарного диабета [6]. Генетическая модификация клеток печени сыграла важную роль в определенном прогрессе в генной терапии фенилкетонурии [7], гемофилии [8] и альфа-1-антитрипсиновой недостаточности [9].
Генетическая модификация миоцитов может использоваться для лечения миопатий у детей и новорожденных [10], восстановления кровоснабжения нижних конечностей у взрослых пациентов при диабете [11]. Мышцы доступны для инъекции, электропорации [12] или гидродинамической доставки плазмид [13, 14]. Последние два метода существенно повышают эффективность доставки плазмид, что делает мышцы привлекательной мишенью для экспрессии белков с системным терапевтическим эффектом, например, эритропоэтина при анемии, вызванной почечной недостаточностью [15]. Кроме того, гепатоциты и миоциты ‒ это долгоживущие, медленно делящиеся клетки, что способствует сохранению числа молекул плазмидных векторов, количество которых в интенсивно размножающихся клетках быстро снижается в результате разведения.
Задача достижения длительной экспрессии трансгена в мышцах животных уже в существенной мере выполнена. Опубликованы результаты исследований, согласно которым использование стандартных плазмид позволяет обеспечить долговременную экспрессию трансгена и коррекцию фенотипа в экспериментах продолжительностью от 4 до 12 мес. [15‒17]. Однако добиться долговременной экспрессии трансгена в печени оказалось гораздо труднее, решить эту задачу с помощью стандартных плазмид не удалось [16, 18‒20]. Даже при использовании специально сконструированных векторов (линейных кассет, миниколец, плазмид со сниженным содержанием CpG и плазмид с инсуляторами) уровень экспрессии трансгена падал на порядок или более в течение первых недель после доставки [7, 21‒27]. Однако в некоторых работах установлено, что удаление CpG-мотивов, добавление инсуляторов или S/MAR-элементов может уменьшить сайленсинг трансгена in vivo [23, 24, 27]. Ранее мы сконструировали плазмидный вектор со S/MAR-элементами и низким содержанием CpG, который позволяет значительно повысить длительность экспрессии трансгена в мезенхимальных стволовых клетках (МСК) in vitro [28]. В данной работе мы использовали этот вектор для экспрессии трансгена in vivo в скелетных мышцах задних конечностей и в печени мышей. Исследовано также влияние содержания CpG-мотивов в векторе и трансгене на кинетику экспрессии трансгена.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Конструирование репортерных плазмид. Конструирование векторов pMBR1 и pMBR2, и их вариантов, экспрессирующих люциферазу светлячка, описано нами ранее [28]. Для изучения влияния конфигурации вектора на экспрессию люциферазы мы вырезали открытую рамку считывания люциферазы Lucia (“Invivogen”, США) из плазмиды pSELECT-Lucia (“Invivogen”) и встроили ее в векторы pMBR1 и pMBR2 по сайтам NcoI и NheI.
В качестве репортерного трансгена в экспериментах in vivo мы выбрали mSEAP, кодирующий секретируемую щелочную фосфатазу мыши [29], которая секретируется в кровоток, не вызывает иммунного ответа и имеет низкий фон (т.е., низкий эндогенный уровень экспрессии). Векторы конструировали с использованием лишенной CpG версии гена mSEAP (mSEAP0), а также гена mSEAP дикого типа (mSEAPwt), содержащего 69 CpG-мотивов.
Фрагмент, соответствующий открытой рамке считывания (ОРС) mSEAP0, был вырезан из плазмиды pCpG-mSEAP (“Invivogen”) по сайтам BsrGI и NheI и вставлен в вектор pMBR2 по тем же сайтам. mSEAP0 для встраивания в вектор pCDNA3.1 разрезали с помощью рестриктаз Acc65I и XbaI.
Фрагмент, соответствующий ОРС mSEAPwt, амплифицировали с помощью высокоточной ДНК-полимеразы KAPA HiFi и праймеров mSEAPwt-S1 (TCTACTTACATGTGGGGAGCCTGCTTGC) и mSEAPwt-A1 (ATATTTGCTAGCT-CTAGCCCGGGCTCACTGCAC), содержащих сайты PscI и NheI соответственно. В качестве матрицы использовали клон MGC (MGC 60698), содержащий кДНК mSEAP. Амплифицированный фрагмент разрезали полностью по сайту PscI и частично по NheI, очищали в геле и вставляли в вектор pMBR2 по сайтам NcoI и NheI. Фрагмент, соответствующий ОРС mSEAP, вырезали из полученной конструкции pMBR2-mSEAPwt по сайтам BsrGI и NheI и встраивали в вектор pCDNA3.1 по совместимым сайтам Acc65I и XbaI.
Всего для исследований in vivo были подготовлены четыре конструкции: pMBR2-mSEAP0 (13 CpG), pcDNA3.1-mSEAP0 (329 CpG), pMBR2-mSEAPwt (82 CpG), pcDNA3.1-mSEAPwt (398 CpG).
Доставка плазмид in vivo. Протокол исследования утвержден этическим комитетом в области исследований на животных НМИЦ онкологии им. Н.Н. Блохина. Мышей содержали в условиях 12 ч/12 ч циклов темнота/свет и постоянного доступа к воде и пище. Использовали самок мышей C57BL/6 в возрасте 8‒10 недель на момент начала эксперимента.
Плазмидные конструкции для доставки in vivo выделяли с помощью Plasmid Mega Kit (“Qiagen”, Германия). Плазмиды вводили посредством множественных инъекций в мышцы задних конечностей с последующей электропорацией при напряженности поля 600 В/см и длительности импульса 50 мс. Чтобы исключить из рассмотрения разницу в иммунном ответе на репортерный белок или плазмидную ДНК как возможную причину различий в кинетике экспрессии, выбирали количества ДНК, которые давали схожие изначальные уровни экспрессии mSEAP, а для компенсации различий в количестве ДНК добавляли пустой вектор. Таким образом, для инъекций в заднюю конечность одной мыши с последующей электропорацией использовали 30 мкг pMBR2-mSEAP0, 100 мкг pMBR2-mSEAPwt и 200 мкг pCDNA3.1-mSEAP0. Общее количество конструкций pMBR2-mSEAP0 и pMBR2-mSEAPwt доводили до 200 мкг с помощью пустого вектора pMBR2. В каждую конечность вводили равные объемы плазмид. Уровень экспрессии плазмиды pCDNA3.1-mSEAPwt не превышал заметно фоновые значения, поэтому в долговременных экспериментах in vivo эту конструкцию не использовали.
Для доставки в печень одной мыши использовали 15 мкг pMBR2-mSEAP0, 150 мкг pMBR2-mSEAPwt и 300 мкг pCDNA3.1-mSEAP0. В случае конструкций pMBR2-mSEAP0 и pMBR2-mSEAPwt общую массу плазмид доводили до 300 мкг с помощью пустого вектора pMBR2. Плазмидную ДНК для ее гидродинамической доставки в печень растворяли в 2 мл физиологического раствора, после чего 2 мл полученного раствора вводили в хвостовую вену мыши в течение 15 с.
Измерение mSEAP. Кровь (20 мкл) забирали из глазного синуса мыши, далее кровь инкубировали на льду в течение 15 мин и центрифугировали в течение 5 мин при 1000 g. Полученную сыворотку (10 мкл) использовали для измерения активности mSEAP с помощью системы Phospha-Light™ SEAP Reporter Gene Assay (“ThermoFisher Scientific”, США) согласно инструкции производителя. Сигнал измеряли с помощью люминометра 20/20n (“Turner BioSystems”, США).
Измерение активности люциферазы. Электропорацию МСК человека и измерение активности несекретируемой люциферазы светлячка проводили, как описано нами ранее [28]. Для измерения активности секретируемой люциферазы Lucia среду, используемую для культивирования клеток, заменяли на аналогичную среду без сыворотки. Через 4 ч среду собирали и измеряли активность люциферазы с помощью набора QUANTI-LucTM (“Invivogen”).
Статистический анализ. Статистический анализ проводили с использованием смешанной линейной модели (процедура MIXED в SAS), значимым считали p ≤ 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Выбор вектора для исследований in vivo
Для мониторинга экспрессии гена плазмидными конструкциями in vivo в качестве репортерного белка мы выбрали секретируемую щелочную фосфатазу мыши (mSEAP). Этот белок имеет два важных преимущества перед прочими репортерными белками, обычно используемыми в исследованиях экспрессии гена in vivo. Во-первых, он не вызывает развития иммунного ответа у мышей, при котором возможно уничтожение клеток, экспрессирующих трансген; во-вторых, его активность можно обнаружить в периферической крови, что позволяет анализировать уровень экспрессии трансгена у одних и тех же мышей на протяжении длительного периода времени.
Согласно [30, 31], размещение S/MAR-элементов внутри транскрипционной кассеты повышает стабильность экспрессии трансгена и ее поддержания в трансфицированных клетках в виде эписомы, поэтому изначально мы создали два типа векторов, а именно pMBR1 и pMBR2. В векторе pMBR1 S/MAR находится внутри транскрипционной кассеты между стоп-кодоном и сайтом полиаденилирования SV40 (SV40 polyA), а в векторе pMBR2 ‒ снаружи, сразу после SV40 polyA. На рис. 1 показаны карты плазмидных конструкций на основе векторов pMBR1, pMBR2 и pCDNA3.1, использованных в нашей работе.
Рис. 1.
Карты плазмидных конструкций на основе векторов pMBR1, pMBR2 и pCDNA3.1, экспрессирующих цитоплазматический белок – люциферазу светлячка (Luc0), и секретируемые белки – люциферазу Lucia и щелочную фосфатазу mSEAP.
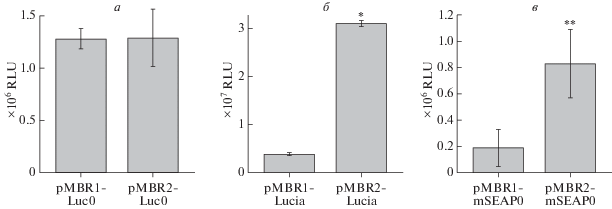
В первоначальных экспериментах анализ экспрессии несекретируемой люциферазы светлячка in vitro в МСК не выявил различий между векторами (рис. 2а). Однако в дополнительных экспериментах in vitro было установлено, что уровень экспрессии секретируемой люциферазы Lucia конструкцией pMBR2-Lucia значительно выше по сравнению с конструкцией pMBR1-Lucia (рис. 2б). Сходные результаты получены и в пилотных экспериментах in vivo в мышцах с использованием mSEAP в качестве репортерного белка. Вектор pMBR2 обеспечивал значительно более высокий уровень экспрессии mSEAP, чем pMBR1 (рис. 2в). Таким образом, размещение S/MAR-элемента внутри транскрипционной кассеты в векторе pMBR1 привело к значительному сокращению продукции двух секретируемых репортерных белков по сравнению с вектором pMBR2, но не повлияло на продукцию цитоплазматической люциферазы светлячка.
Рис. 2.
Сравнение экспрессии трансгенов векторами pMBR1 и pMBR2. а ‒ Оба вектора с одинаковой эффективностью экспрессируют люциферазу светлячка. б и в ‒ Вектор pMBR2 обеспечивает более эффективную экспрессию секретируемых белков ‒ люциферазы Lucia в МСК человека (б) и mSEAP в мышцах мышей (в), чем pMBR1. RLU – относительные единицы люминесценции. На каждой диаграмме приведены данные, усредненные по трем повторам и представленные в виде среднее ± стандартное отклонение. * p < 0.0001, ** p < 0.05.
Исходя из упомянутых первичных результатов, получены конструкции на основе pMBR2 и контрольного вектора pcDNA3.1, экспрессирующие mSEAP. Мы планировали протестировать in vivo четыре варианта плазмид ‒ pMBR2-mSEAP0, pcDNA3.1-mSEAP0, pMBR2-mSEAPwt и pcDNA3.1-mSEAPwt. Однако в случае конструкции pcDNA3.1-mSEAPwt даже начальные люминесцентные сигналы были очень слабыми и не превышали фонового уровня у контрольных мышей, которым плазмиды не вводили. Таким образом, эта конструкция не подходит для долговременного исследования экспрессии. После доставки плазмид в мышцы и печень у мышей отбирали образцы венозной крови через 3, 7 и 14 дней, и каждые 2 недели в дальнейшем. По прошествии 2 мес. кровь отбирали 1 раз в 4 недели, а после 6 мес. – 1 раз в 6 недель.
Экспрессия mSEAP в скелетных мышцах
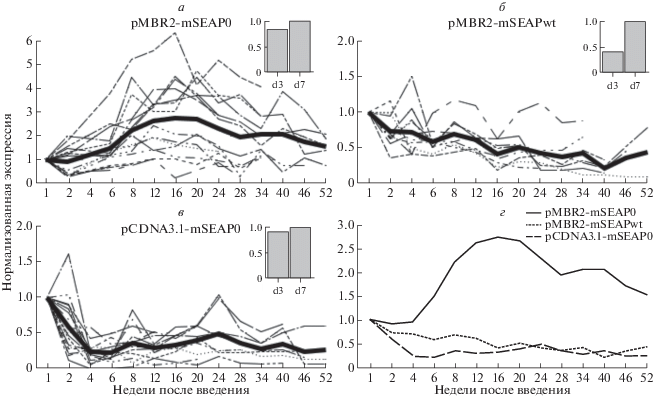
Плазмиду pMBR2-mSEAP0 вводили путем электропорации 18 мышам, pCDNA3.1-mSEAP0 ‒ 17 и pMBR2-mSEAPwt ‒ 11 мышам. Уровень экспрессии трансгена после доставки плазмиды значительно варьировал у разных животных. По всей вероятности, это обусловлено ограниченной воспроизводимостью результатов инъекций плазмид в мышцы мышей, имеющих малые размеры. Для адекватного сравнения результатов в дальнейшем мы использовали данные, нормированные на результат 7-го дня (в этот день уровень экспрессии большинства плазмид был максимальным).
В течение первого месяца после электропорации картины изменения экспрессии плазмид pMBR2-mSEAP0 и pMBR2-mSEAPwt были довольно сходными (рис. 3). Затем уровень экспрессии pMBR2-mSEAP0 начал расти, в то время как экспрессия pMBR2-mSEAPwt оставалась примерно на том же уровне с тенденцией к медленному убыванию. Уровень экспрессии pMBR2-mSEAP0 поднимался до 16-й недели и достигал примерно 275% от уровня на 7-й день. После этого экспрессия стала медленно снижаться, но на 52-й неделе все еще составляла около 150% от уровня 7-го дня. Экспрессия pcDNA3.1-mSEAP0 начала быстро падать после первой недели, а после первого месяца стала медленно расти до 24-й недели. Следует отметить, что экспрессия pMBR2-mSEAPwt значительно превосходила экспрессию pcDNA3.1-mSEAP0 в течение 12 недель. После 16-й недели уровень экспрессии обеих плазмид был довольно сходным.
Рис. 3.
Изменение экспрессии mSEAP векторами pMBR2 и pCDNA3.1 в мышцах задних конечностей мышей со временем. На оси ординат приведен относительный уровень экспрессии трансгена, нормированный на уровень, измеренный на 7-й день после введения. На врезках изображены изменения относительного уровня экспрессии mSEAP в течение первой недели. а ‒ pMBR2-mSEAP0; б ‒ pMBR2-mSEAPwt; в ‒ pcDNA3.1-mSEAP0. Пунктиром показаны значения для отдельных мышей, сплошной линией – среднее значение в группе. г – Сравнение графиков средних значений для трех плазмид.
Экспрессия трансгена в печени
Плазмида pMBR2-mSEAP0 была введена гидродинамическим способом 11 мышам, pMBR2-mSEAPwt ‒ 10 и pCDNA3.1-mSEAP0 ‒ 9 мышам. Результаты длительного мониторинга экспрессии изображены на рис. 4, где уровень экспрессии нормирован на результат 7-го дня. Уровень экспрессии трансгена со всех трех плазмид начал падать после введения, что согласуется с опубликованными данными. В случае pMBR2-mSEAP0 падение остановилось после 2 недель и оставалось на уровне 50‒60% от результата 7-го дня (30‒40% от уровня к 3-му дню) вплоть до 14-й недели. После этого экспрессия равномерно снижалась до 17% от результата 7-го дня (11% от уровня 3-го дня) к 52-й недели.
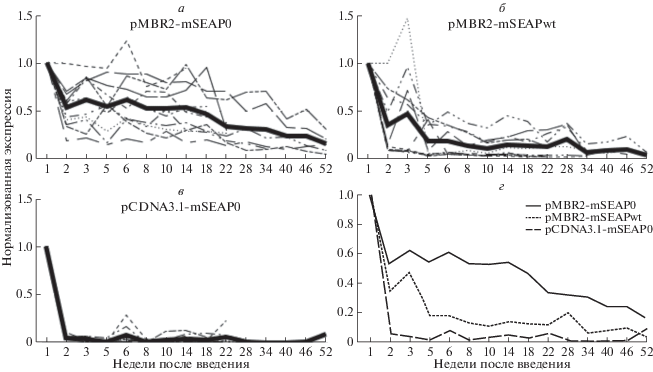
В то же время, падение уровня экспрессии было гораздо более заметным в случае двух других плазмид. Экспрессия с плазмиды pMBR2-mSEAPwt за 5 недель снизилась до 18% от уровня к 7-му дню (9% от 3-го дня) и стабильно составляла 10‒15% (5‒10% от уровня 3-го дня), и к концу периода наблюдения медленно упала до 5% (2% от результата 3-го дня). Экспрессия трансгена с плазмиды pCDNA3.1-mSEAP0 резко снизилась примерно до 5% (1% от результата 3-го дня) после 2 недель эксперимента и оставалась большую часть времени на уровне, близком к нулю.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Полученные нами с помощью вектора pMBR2 in vivo данные хорошо согласуются с более ранними результатами, согласно которым длительная экспрессия трансгенов в МСК связана с уменьшением содержания CpG-мотивов как в векторе, так и в трансгене (люцифераза светлячка) [28]. Наши данные также соответствуют результатам других исследований, показывающих, что уменьшение количества CpG-мотивов в плазмидной конструкции замедляет падение уровня экспрессии трансгена со временем. Так, Yew N.S. и соавт. показали, что плазмидная конструкция, лишенная CpG-мотивов, может длительно поддерживать стабильную экспрессию трансгена в легком. Однако в печени результаты, полученные с использованием той же плазмиды, были довольно скромными: через 2 недели уровень экспрессии снизился более чем на порядок, хотя и оставался после этого на том же уровне в течение 1.5 мес. [26, 32]. В наших экспериментах плазмидная конструкция pMBR2-mSEAP0 поддерживала заметно более высокий уровень экспрессии трансгена в печени мышей по сравнению с ранее опубликованными работами [33‒35]. Можно предположить, что это связано прежде всего с более низким содержанием CpG в нашей конструкции. Так, в работе [33] вектор был лишен CpG-мотивов, тогда как трансген содержал значительное их число, и, наоборот, в исследовании [34] трансген не содержал CpG-мотивов, однако бактериальный участок начала репликации включал в себя большое их количество.
Механизм снижения экспрессии, связанный с присутствием CpG-мотивов, остается неясным. У млекопитающих ДНК, содержащая неметилированные CpG, вызывает иммунный ответ, опосредованный рецепторами TLR9, представленными на поверхности дендритных клеток, макрофагов, естественных киллерных клеток и других клеток иммунной системы [36, 37]. Тем не менее, рецепторы TLR9 вряд ли могут быть основной причиной сайленсинга плазмиды. Как и ожидалось, у мышей без этих рецепторов не развивался усиленный иммунный ответ на ДНК, содержащую неметилированные CpG [38]. При этом отсутствие TLR9 не влияло на кинетику экспрессии трансгена [32]. Кроме того, не наблюдалась элиминация плазмид или клеток, содержащих плазмиды [24, 39]. Следовательно, должны существовать иные механизмы, отвечающие за подавление экспрессии плазмидных конструкций, содержащих CpG.
Другой вероятный механизм падения экспрессии трансгена ‒ транскрипционный сайленсинг посредством de novo метилирования CpG-мотивов, ведущего к образованию неактивного хроматина. Однако в одних работах выявлена корреляция между de novo метилированием и уровнем экспрессии трансгена [25, 26], но не обнаружена в других [27].
Согласно исследованиям группы Kay М.А., на сайленсинг трансгена влияют не CpG-мотивы в векторе или вставке, а бактериальные последовательности, ковалентно связанные с транскрипционной кассетой. В своих первых работах они использовали лишенные бактериальных последовательностей линейные экспрессионные кассеты, вырезанные из плазмид, или миникольца, полученные из плазмид с помощью рекомбиназ [25, 40, 41]. Развивая свою гипотезу, они пришли к заключению, что даже стандартные плазмиды способны поддерживать долговременную экспрессию трансгена, если экспрессионная кассета ограничена инсуляторами, отделяющими ее от бактериальных последовательностей. Предположили, что в эукариотических клетках супрессированный гетерохроматин формируется вокруг бактериальных последовательностей, после чего распространяется по ДНК. Инсуляторы сдерживают распространение супрессированного хроматина, и не происходит сайленсинг трансгена. Однако экспрессия трансгена в печени даже лучшими векторами в этих исследованиях снижалась в 5‒10 раз в течение первых дней после введения [35].
Стоит заметить, что конструкция, использованная в данном исследовании, содержит S/MAR-элемент. Показано, что S/MAR-элементы обладают некоторыми свойствами инсуляторов, в частности, они способны оградить транскрипционную кассету от супрессированного гетерохроматина. Они также могут содействовать ассоциации плазмиды с хромосомами и способствовать длительному поддержанию плазмиды в виде эписомы [30]. Однако S/MAR выполняют эту функцию, если находятся внутри транскрипционной кассеты, в то время как в векторе pMBR2 этот элемент локализован вне кассеты.
В наших исходных экспериментах S/MAR-элемент, локализованный внутри транскрипционной кассеты вектора pMBR1, негативно воздействовал на экспрессию секретируемых репортерных белков Lucia и mSEAP, но не на несекретируемую люциферазу светлячка. Этот результат можно связывать с различиями в путях ядерного экспорта мРНК, кодируюших цитоплазматические и секретируемые белки. РНК секретируемых белков экспортируются из ядра посредством альтернативного пути ALREX, зависящего от свойств нетранслируемой области [42]. Можно предположить, что S/MAR-элемент, расположенный внутри 3'-нетранслируемой области мРНК, кодирующей секретируемые белки, влияет на ядерный экспорт, тем самым снижая общую эффективность экспрессии.
Подводя итог, можно заключить, что комбинация двух подходов ‒ исключения большинства CpG-мотивов из вектора и трансгена, и введения S/MAR-элемента ‒ позволила создать конструкцию, поддерживающую долговременную экспрессию трансгена в скелетных мышцах и, что наиболее важно, в печени. При этом на стабильность плазмидной экспрессии значительное негативное влияние оказывает присутствие CpG-мотивов как в векторе, так и в трансгене. Таким образом, метилирование остатков цитозина в плазмидах, по-видимому, играет решающую роль в постепенном подавлении экспрессии трансгена. Следует отметить, что не-CpG-метилирование цитозинов тоже приводит к подавлению экспрессии гена [43]. Можно предположить, что наблюдаемое нами снижение уровня экспрессии конструкции pMBR2-mSEAP0 в мышцах и печени мышей вызвано в значительной степени не-CpG-метилированием.
Эксперименты, представленные в данной работе, выполнены при поддержке Российского научного фонда (проект № 18-14-00300).
Эксперименты на животных проведены согласно рекомендациям ARRIVE и полностью удовлетворяли директиве Европейского союза n2010/63/UE об исследованиях на животных.
Авторы сообщают об отсутствии конфликта интересов.
Список литературы
Raper S.E., Chirmule N., Lee F.S., Wivel N.A., Bagg A., Gao G.P., Wilson J.M., Batshaw M.L. (2003) Fatal systemic inflammatory response syndrome in a ornithine transcarbamylase deficient patient following adenoviral gene transfer. Mol. Genet. Metab. 80(1‒2), 148–158.
Manno C.S., Pierce G.F., Arruda V.R., Glader B., Ragni M., Rasko J.J., Ozelo M.C., Hoots K., Blatt P., Konkle B., Dake M., Kaye R., Razavi M., Zajko A., Zehnder J., Rustagi P.K., Nakai H., Chew A., Leonard D., Wright J.F., Lessard R.R., Sommer J.M., Tigges M., Sabatino D., Luk A., Jiang H., Mingozzi F., Couto L., Ertl H.C., High K.A., Kay M.A. (2006) Successful transduction of liver in hemophilia by AAV-Factor IX and limitations imposed by the host immune response. Nat. Med. 12(3), 342–347.
Howe S.J., Mansour M.R., Schwarzwaelder K., Bartholomae C., Hubank M., Kempski H., Brugman M.H., Pike-Overzet K., Chatters S.J., de Ridder D., Gilmour K.C., Adams S., Thornhill S.I., Parsley K.L., Staal F.J., Gale R.E., Linch D.C., Bayford J., Brown L., Quaye M., Kinnon C., Ancliff P., Webb D.K., Schmidt M., von Kalle C., Gaspar H.B., Thrasher A.J. (2008) Insertional mutagenesis combined with acquired somatic mutations causes leukemogenesis following gene therapy of SCID-X1 patients. J. Clin. Invest. 118(9), 3143–3150.
Davé U.P., Akagi K., Tripathi R., Cleveland S.M., Thompson M.A., Yi M., Stephens R., Downing J.R., Jenkins N.A., Copeland N.G. (2009) Murine leukemias with retroviral insertions at Lmo2 are predictive of the leukemias induced in SCID-X1 patients following retroviral gene therapy. PLoS Genet. 5(5), e1000491.
Hinderer C., Katz N., Buza E.L., Dyer C., Goode T., Bell P., Richman L.K., Wilson J.M. (2018) Severe toxicity in nonhuman primates and piglets following high-dose intravenous administration of an adeno-associated virus vector expressing human SMN. Hum. Gene Ther. 29(3), 285‒298.
Handorf A.M., Sollinger H.W., Alam T. (2015) Insulin gene therapy for type 1 diabetes mellitus. Exp. Clin. Transplant. 13(Suppl. 1), 37‒45.
Viecelli H.M., Harbottle R.P., Wong S.P., Schlegel A., Chuah M.K., VandenDriessche T., Harding C.O., Thöny B. (2014) Treatment of phenylketonuria using minicircle-based naked-DNA gene transfer to murine liver. Hepatology. 60(3), 1035‒1043.
High K.A. (2011) Gene therapy for haemophilia: a long and winding road. J. Thromb. Haemost. 9(Suppl. 1), 2‒11.
Gruntman A.M., Flotte T.R. (2017) Therapeutics: gene therapy for alpha-1 antitrypsin deficiency. Methods Mol. Biol. 1639, 267‒275.
Robinson-Hamm J.N., Gersbach C.A. (2016) Gene therapies that restore dystrophin expression for the treatment of Duchenne muscular dystrophy. Hum. Genet. 135(9), 1029‒1040.
Rissanen T.T., Vajanto I., Ylä-Herttuala S. (2001) Gene therapy for therapeutic angiogenesis in critically ischaemic lower limb - on the way to the clinic. Eur. J. Clin. Invest. 31(8), 651‒666.
Murakami T., Sunada Y. (2011) Plasmid DNA gene therapy by electroporation: principles and recent advances. Curr. Gene Ther. 11(6), 447‒456.
Herweijer H., Wolff J.A. (2007) Gene therapy progress and prospects: hydrodynamic gene delivery. Gene Ther. 14(2), 99‒107.
Wells D.J. (2004) Opening the floodgates: clinically applicable hydrodynamic delivery of plasmid DNA to skeletal muscle. Mol. Ther. 10(2), 207‒208.
Sun J., Wang Y., Yang J., Du D., Li Z., Wei J., Yang A. (2012) Long-term and stable correction of uremic anemia by intramuscular injection of plasmids containing hypoxia-regulated system of erythropoietin expression. Exp. Mol. Med. 44(11), 674‒683.
Morrissey D., van Pijkeren J.P., Rajendran S., Collins S.A., Casey G., O’Sullivan G.C., Tangney M. (2012) Control and augmentation of long-term plasmid transgene expression in vivo in murine muscle tissue and ex vivo in patient mesenchymal tissue. J. Biomed. Biotechnol. 2012, 379845.
Yamano S., Dai J., Hanatani S., Haku K., Yamanaka T., Ishioka M., Takayama T., Yuvienco C., Khapli S., Moursi A.M., Montclare J.K. (2014) Long-term efficient gene delivery using polyethylenimine with modified Tat peptide. Biomaterials. 35(5), 1705‒1715.
Yokoo T., Kamimura K., Suda T., Kanefuji T., Oda M., Zhang G., Liu D., Aoyagi Y. (2013) Novel electric power-driven hydrodynamic injection system for gene delivery: safety and efficacy of human factor IX delivery in rats. Gene Ther. 20(8), 816‒823.
Pergolizzi R.G., Jin G., Chan D., Pierre L., Bussel J., Ferris B., Leopold P.L., Crystal R.G. (2006) Correction of a murine model of von Willebrand disease by gene transfer. Blood. 108(3), 862‒869.
De Meyer S.F., Vandeputte N., Pareyn I., Petrus I., Lenting P.J., Chuah M.K., VandenDriessche T., Deckmyn H., Vanhoorelbeke K. (2008) Restoration of plasma von Willebrand factor deficiency is sufficient to correct thrombus formation after gene therapy for severe von Willebrand disease. Arterioscler. Thromb. Vasc. Biol. 28(9), 1621‒1626.
Magnusson T., Haase R., Schleef M., Wagner E., Ogris M. (2011) Sustained, high transgene expression in liver with plasmid vectors using optimized promoter-enhancer combinations. J. Gene Med. 13(7‒8), 382‒391.
Cim A., Sawyer G.J., Zhang X., Su H., Collins L., Jones P., Antoniou M., Reynes J.P., Lipps H.J., Fabre J.W. (2012) In vivo studies on non-viral transdifferentiation of liver cells towards pancreatic β cells. J. Endocrinol. 214(3), 277‒288.
Wong P., Argyros O., Coutelle C., Harbottle R.P. (2011) Non-viral S/MAR vectors replicate episomally in vivo when provided with a selective advantage. Gene Ther. 18(1), 82‒87.
Chen Z.Y., Yant S.R., He C.Y., Meuse L., Shen S., Kay M.A. (2001) Linear DNAs concatemerize in vivo and result in sustained transgene expression in mouse liver. Mol. Ther. 3(3), 403‒410.
Gracey Maniar L.E., Maniar J.M., Chen Z.Y., Lu J., Fire A.Z., Kay M.A. (2013) Minicircle DNA vectors achieve sustained expression reflected by active chromatin and transcriptional level. Mol. Ther. 21(1), 131‒138.
Yew N.S., Zhao H., Przybylska M., Wu I.H., Tousignant J.D., Scheule R.K., Cheng S.H. (2002) CpG-depleted plasmid DNA vectors with enhanced safety and long-term gene expression in vivo. Mol. Ther. 5(6), 731‒738.
Argyros O., Wong S.P., Niceta M., Waddington S.N., Howe S.J., Coutelle C., Miller A.D., Harbottle R.P. (2008) Persistent episomal transgene expression in liver following delivery of a scaffold/matrix attachment region containing non-viral vector. Gene Ther. 15(24), 1593–1605.
Bruter A.V., Kandarakov O.F., Belyavsky A.V. (2018) Persistence of plasmid-mediated expression of transgenes in human mesenchymal stem cells depends primarily on CpG levels of both vector and transgene. J. Gene Med. 20(2‒3), e3009.
Maelandsmo G.M., Ross P.J., Pavliv M., Meulenbroek R.A., Evelegh C., Muruve D.A., Graham F.L., Parks R.J. (2005) Use of a murine secreted alkaline phosphatase as a non-immunogenic reporter gene in mice. J. Gene Med. 7(3), 307‒315.
Jenke A.C., Stehle I.M., Herrmann F., Eisenberger T., Baiker A., Bode J., Fackelmayer F.O., Lipps H.J. (2004) Nuclear scaffold/matrix attached region modules linked to a transcription unit are sufficient for replication and maintenance of a mammalian episome. Proc. Natl. Acad. Sci. USA. 101(31), 11322–11327.
Haase R., Argyros O., Wong S.P., Harbottle R.P., Lipps H.J., Ogris M., Magnusson T., Vizoso Pinto M.G., Haas J., Baiker A. (2010) pEPito: a significantly improved non-viral episomal expression vector for mammalian cells. BMC Biotechnol. 10, 20.
Bazzani R.P., Pringle I.A., Connolly M.M., Davies L.A., Sumner-Jones S.G., Schleef M., Hyde S.C., Gill D.R. (2016) Transgene sequences free of CG dinucleotides lead to high level, long-term expression in the lung independent of plasmid backbone design. Biomaterials. 93, 20‒26.
Kanda G.N., Miyamoto S., Kobayashi M., Matsuoka I., Harashima H., Kamiya H. (2014) Anatomy of plasmid DNAs with anti-silencing elements. Int. J. Pharm. 464(1‒2), 27‒33.
Hodges B.L., Taylor K.M., Joseph M.F., Bourgeois S.A., Scheule R.K. (2004) Long-term transgene expression from plasmid DNA gene therapy vectors is negatively affected by CpG dinucleotides. Mol. Ther. 10(2), 269‒278.
Chen Z.Y., Riu E., He C.Y., Xu H., Kay M.A. (2008) Silencing of episomal transgene expression in liver by plasmid bacterial backbone DNA is independent of CpG methylation. Mol. Ther. 16(3), 548‒556.
Hemmi H., Takeuchi O., Kawai T., Kaisho T., Sato S., Sanjo H., Matsumoto M., Hoshino K., Wagner H., Takeda K., Akira S. (2000) A Toll-like receptor recognizes bacterial DNA. Nature. 408(6813), 740‒745.
Hyde S.C., Pringle I.A., Abdullah S., Lawton A.E., Davies L.A., Varathalingam A., Nunez-Alonso G., Green A.M., Bazzani R.P., Sumner-Jones S.G., Chan M., Li H., Yew N.S., Cheng S.H., Boyd A.C., Davies J.C., Griesenbach U., Porteous D.J., Sheppard D.N., Munkonge F.M., Alton E.W., Gill D.R. (2008) CpG-free plasmids confer reduced inflammation and sustained pulmonary gene expression. Nat. Biotechnol. 26(5), 549–551.
Tan Y., Liu F., Li Z., Li S., Huang L. (2001) Sequential injection of cationic liposome and plasmid DNA effectively transfects the lung with minimal inflammatory toxicity. Mol. Ther. 3(5 Pt 1), 673‒682.
Pringle I.A., Raman S., Sharp W.W., Cheng S.H., Hyde S.C., Gill D.R. (2005) Detection of plasmid DNA vectors following gene transfer to the murine airways. Gene Ther. 12(15), 1206‒1214.
Chen Z.Y., He C.Y., Ehrhardt A., Kay M.A. (2003) Minicircle DNA vectors devoid of bacterial DNA result in persistent and high-level transgene expression in vivo. Mol. Ther. 8(3), 495‒500.
Chen Z.Y., He C.Y., Kay M.A. (2005) Improved production and purification of minicircle DNA vector free of plasmid bacterial sequences and capable of persistent transgene expression in vivo. Hum. Gene Ther. 16(1), 126‒131.
Akef A., Zhang H., Masuda S., Palazzo A.F. (2013) Trafficking of mRNAs containing ALREX-promoting elements through nuclear speckles. Nucleus. 4(4), 326‒340.
Patil V., Ward R.L., Hesson L.B. (2014) The evidence for functional non-CpG methylation in mammalian cells. Epigenetics. 9(6), 823–828.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология