

Молекулярная биология, 2020, T. 54, № 4, стр. 667-679
Роль остатков цистеина во взаимодействии никующей эндонуклеазы BspD6I с ДНК
Л. А. Абросимова a, *, А. Р. Самсонова b, Т. А. Перевязова c, А. К. Юнусова c, Р. И. Артюх c, Е. А. Романова d, Л. А. Железная c, Т. С. Орецкая d, Е. А. Кубарева d
a Химический факультет Московского государственного университета им. М.В. Ломоносова
119991 Москва, Россия
b SABNP, University of Evry, INSERM U1204, Université Paris-Saclay
91025 Evry, France
c Институт теоретической и экспериментальной биофизики Российской академии наук
142290 Московская обл., Пущино, Россия
d Научно-исследовательский институт физико-химической биологии им. А.Н. Белозерского
Московского государственного университета им. М.В. Ломоносова
119991 Москва, Россия
* E-mail: abrludmila@gmail.com
Поступила в редакцию 05.03.2020
После доработки 05.03.2020
Принята к публикации 16.03.2020
Аннотация
Никующие эндонуклеазы (НЭ) представляют собой немногочисленное малоизученное семейство эндонуклеаз рестрикции. Эти ферменты узнают в двухцепочечной ДНК определенную последовательность, но катализируют гидролиз только одной цепи. Необходимость конструирования НЭ с новой специфичностью для решения практических задач биотехнологии обусловливает актуальность изучения механизма их действия. Один из современных методов исследования белково-нуклеиновых взаимодействий ‒ флуоресцентная спектроскопия. Эта технология основана на введении флуорофоров в белки, главным образом, через остатки Cys, используя высокую реакционную способность их тиольной группы. Для реализации этого подхода необходимо четко понимать роль остатков Cys в функционировании нативного белка и возможных последствий их модификации. Нами проведен анализ сближенности с ДНК остатков Cys никующей эндонуклеазы BspD6I методом ковалентного связывания. В реакции использовали дикий тип фермента и его мутантную форму BspD6I(C11S/C160S), а также модифицированные ДНК-дуплексы, содержащие 2-пиридилдисульфидную группу при С2'-атоме углеводного фрагмента в различных положениях олигонуклеотидной цепи. Впервые показано, что остатки Cys НЭ находятся в непосредственной близости от ДНК в процессе связывания, в том числе на стадии формирования неспецифического комплекса. Замены Cys11Ser и Cys160Ser в N-концевом домене белка приводили к незначительному снижению гидролитической активности фермента. Создание бесцистеинового варианта НЭ BspD6I и изучение его свойств позволит получить дополнительную информацию о функциональной значимости остатков Cys этого уникального фермента.
ВВЕДЕНИЕ
В результате интенсивного развития молекулярно-биологических и биотехнологических методов все чаще возникает потребность в сайтспецифических эндонуклеазах, вносящих разрыв в определенное положение только одной из цепей двухцепочечной (дцДНК) [1]. Эти ферменты получили название никующих эндонуклеаз (НЭ) (от англ. nicking endonucleases, nick – разрыв, или “ник”). НЭ рассматривают как ферменты с высоким потенциалом использования в генной терапии. Они используются при конструировании химерных белков с заданной специфичностью [2]. Такие химерные белки могут стимулировать гомологичную рекомбинацию для устранения повреждений в ДНК [3]. Однако механизм действия НЭ практически не изучен, что ограничивает возможность их практического применения.
Объектом представленного исследования была НЭ BspD6I из термофильного организма Bacillus species [4]. Она представляет собой большую субъединицу гетеродимерной эндонуклеазы рестрикции (ЭР) BspD6I [5, 6]. НЭ BspD6I узнает в дцДНК последовательность 5'‑GAGTC‑3'/ 5'‑GACTC‑3', которая часто встречается в промоторах фаговых генов, например, бактериофага T7. НЭ BspD6I катализирует гидролиз “верхней” цепи ДНК после четвертого нуклеотида от участка узнавания в сторону 3'-конца: 5'‑GAGTCNNNN↓‑3'/ 5'‑NNNNGACTC‑3'. Согласно базе данных REBASE (http://rebase.neb.com/cgi-bin/iso-get?Nt. BspD6I), к настоящему моменту известно пять изошизомеров НЭ BspD6I, гидролизующих последовательность 5'‑GAGTC‑3'/5'‑GACTC‑3', и предсказано существование еще двух изошизомеров этого фермента.
Качалова (Kachalova) с соавт. [7] разрешили кристаллическую структуру НЭ BspD6I (PDB-код 2EWF) в отсутствие ДНК-субстрата. НЭ BspD6I имеет три домена: i) предположительно ДНК-связывающий, состоящий из двух структурных субдоменов, D1 (1–116, 280–300 а.о.) и D2 (117–279 а.о.), ii) линкерный (301–381 а.о.) и iii) каталитический (382–604 а.о.). Разделенные в пространстве домены, предположительно отвечающие за узнавание и катализ, позволяют рассматривать НЭ BspD6I как удобный объект для внесения замен в ДНК-связывающий (узнающий) домен с целью получения ферментов с новой специфичностью.
В наших предшествующих работах исследовано явление олигомеризации НЭ BspD6I при повышенных концентрациях белка в растворе [8], разработаны подходы, позволяющие регулировать активность этого фермента [9, 10], определены термодинамические и кинетические параметры его взаимодействия с субстратом и установлены потенциально формирующие контакты с белком группы атомов в узнаваемой нуклеотидной последовательности и в последовательности ДНК, примыкающей к расщепляемому межнуклеотидному узлу [6, 10]. Однако на сегодняшний день недостаточно данных для понимания роли отдельных аминокислотных остатков в функционировании фермента, так как пока не разрешена структура его комплекса с ДНК.
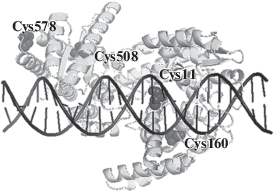
В C-концевом домене НЭ локализован каталитический мотив. Это аминокислотные остатки Pro455, Asp456, Glu469, Val470 и Glu482. В результате анализа свойств мутантных форм НЭ BspD6I с единичными заменами Asp456Ala и Glu418Ala подтверждена существенная роль этих аминокислотных остатков в катализе [11]. На основании кристаллической структуры BspD6I создана модель комплекса НЭ с ДНК [12]. В состав BspD6I входит 4 остатка Cys: в N-концевом домене в позициях 11 и 160, в С-домене в позициях 508 и 578. Согласно модели белково-нуклеинового комплекса [12], остатки Cys в N-концевом домене сближены с ДНК (рис. 1). Рядом с предсказанным каталитическим центром НЭ расположен остаток Cys508, в то время как Cys578 удален и, по-видимому, не влияет на взаимодействие с ДНК. Оценить сближенность того или иного остатка Cys c ДНК позволяет метод ковалентного связывания (“кросслинкинг”) белка с ДНК-дуплексом, содержащим пиридилдитиогруппу. В основе взаимодействия лежит реакция тиолдисульфидного обмена [13‒21]. Такой подход по-прежнему остается относительно недорогим, надежным и обратимым методом конъюгации ДНК, РНК и их аналогов между собой и с другими биомолекулами и удобным инструментом исследования их структуры и функции [22].
Рис. 1.
Расположение остатков Cys в комплексе никующей эндонуклеазы BspD6I с ДНК согласно модели, предложенной в работе [12].
Следует отметить, что остатки Cys, благодаря их способности образовывать дисульфидные связи и координировать ионы металлов, как правило, важны для поддержания структуры белков. С одной стороны, высокая реакционная способность остатков Cys определяет их ключевую роль в реализации окислительно-восстановительного потенциала и присутствие в составе активных центров белков [23, 24]. Так, наряду с остатками Arg и Lys, остатки Cys могут контактировать с углевод-фосфатным остовом ДНК в составе белково-нуклеиновых комплексов [25, 26]. Классический пример ‒ остаток Cys62 ДНК-связывающего центра субъединицы р50 фактора транскрипции NF-κВ человека. Согласно данным РСА и “кросс-линкинга”, Cys62 сближен с 3'-фосфатными группами 6 и 7 нуклеотидных остатков в κВ-участке из энхансера генов легких цепей иммуноглобулинов [27‒30]. С другой стороны, именно благодаря своей редкой встречаемости в последовательностях белков и способности тиольной группы специфически взаимодействовать с соединениями, содержащими галоацетильную, дисульфидную или малеимидную группу, остатки Сys чрезвычайно привлекательны как мишень для введения флуорофоров в молекулу белка [31]. Флуоресцентное мечение белков по остаткам Cys в последнее время получило широкое распространение в связи с развитием подходов к исследованию механизмов функционирования биомолекул и комплексов, основанных на методах флуоресцентного резонансного переноса энергии и флуоресцентной микроскопии одиночных молекул [20, 32‒34]. Использование такого подхода требует понимания изначальной роли остатков Cys в функционировании белка и того, как модификация повлияет на функциональную активность фермента.
В результате проведенного исследования впервые выяснена роль остатков цистеина НЭ BspD6I во взаимодействии с ДНК методом “кросслинкинга” с использованием дуплексов, содержащих 2-пиридилдисульфидную группу при С2'-атоме углеводного фрагмента в заданном положении олигонуклеотидной цепи. Показано, что остатки цистеина, как N-концевого ДНК-связывающего, так и С-концевого каталитического доменов НЭ BspD6I, сближены с ДНК в процессе образования фермент-субстратного комплекса. При этом замена остатков Сys N-концевого домена на Ser не приводила к существенному нарушению функций белка.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Олигодезоксирибонуклеотиды, реагенты и буферные растворы. Олигодезоксирибонуклеотиды с 2'-аминогруппой были синтезированы в группе доцента Т.С. Зацепина (Сколковский институт науки и технологий, Москва). Немодифицированные и флуоресцентно меченные олигодезоксирибонуклеотиды – коммерческие препараты производства ООО “Синтол” (Россия) и ЗАО “Евроген” (Россия).
Использовали N-сукцинимидил-3-(2-пиридилдитио)пропионат (SPDP); полиэтиленгликоль c молекулярной массой 5 кДа, содержащий остаток малеимида (PEG-Mal); канамицин (Km); хлорамфеникол (Cam); изопропил-β-D-1-тиогалактопиранозид (IPTG) фирмы “Sigma-Aldrich” (США); додецилсульфат натрия (SDS) – “Amresco” (США); трео-2,3-дигидрокси-1,4-димеркаптобутан (1,4-дитиотреит, DTT) – “AppliChem” (Германия); наборы белков-маркеров молекулярной массы PageRuler™ (10–200 кДа) – “Thermo Fisher Scientific” (США); бромфеноловый синий (BPB) – “Reanal” (Венгрия); Ni-NTA Agarose – “Novagen” (Германия); миниколонки для гель-фильтрации NAP-5 – “GE HealthCare” (США); триптон – “VWR Life Science” (США); дрожжевой экстракт – ООО “Хеликон” (Россия).
При проведении экспериментальных работ использовали следующие буферные растворы: А (10 мМ Трис-HCl, pH 7.8, 150 мМ KCl, 10 мМ CaCl2), Б (10 мМ Трис-HCl, pH 7.8, 150 мМ KCl, 10 мМ MgCl2), В (20 мМ HEPES, pH 7.5, 100 мM NaCl, 0.1 мM EDTA, 50% (m/V) глицерин), Г (50 мМ Трис-HCl, pH 8.3, 50 мМ борная кислота, 200 мМ EDTA, 50% (m/V) глицерин), Д (20 мМ HEPES, pH 7.5, 5 мМ MgCl2, 125 мМ KCl, 1 мМ АDP), ТАЕ (40 мМ Tris-CH3COOH, pH 7.5, 1 мМ EDTA), TBE (50 мМ Трис-HCl, pH 8.3, 50 мМ борная кислота, 1 мМ EDTA), TГ (25 мМ Трис-HCl, pH 8.3, 250 мМ глицин, 0.1% (m/V) SDS), LAP (50 мМ Трис-HCl, pH 6.8, 2.5% (m/V) SDS, 10% (m/V) глицерин, 0.01% (m/V) BPB), буфер CD (10 мМ K-фосфатный буфер, рН 7.6, 150 мM KCl, 0.1 мM EDTA), ‒ а также культуральную среду LB (10 г/л триптон, 10 г/л NaCl, 5 г/л дрожжевой экстракт).
Конструирование гена мутантной формы НЭ ‒ BspD6I(С11S/C160S). На основе плазмиды pET28b, кодирующей НЭ BspD6I дикого типа с дополнительной гексагистидиновой последовательностью (His6-tag) на С-конце [35], получали плазмиды с мутантным геном, кодирующим фермент, в N-концевом домене которого два остатка Cys заменены на Ser. На первом этапе получали ген белка с заменой Cys11Ser, затем проводили мутагенез этого гена, приводящий к замене Cys160Ser в белке.
Сайт-направленный мутагенез гена НЭ BspD6I дикого типа для замены Cys11Ser проводили с использованием праймеров 11F (прямого) ‒ 5'‑GGTATGTTTCTTCTTCA-CCTAGAAGTCCAG‑3') и 11R (обратного) ‒ 5'‑CTGGACTTCTAGGTGAA-GAAGAAACATACC‑3') ‒и набора QuikChange (“Agilent Technologies”, США) по протоколу фирмы-производителя. ДНК амплифицировали с помощью ДНК-полимеразы Pfu Turbo (“Agilent Technologies”). Условия ПЦР: 30 с при 95°С (1 цикл); 30 с при 95°С, 1 мин при 55°С, 7 мин при 68°С (16 циклов). Для получения гена НЭ BspD6I(С11S/C160S) плазмиду с геном, кодирующим мутантную форму НЭ BspD6I(С11S), подвергали мутагенезу с использованием праймеров 160F (5'‑CTTTAACAGCAACAAATAATAATCAGGTGGA‑3') и 160R (5'‑AGAACATAGCAATATCTAATTTCTGAGGCCGCCT‑3') и ДНК-полимеразы VELOCITY (“Bioline”, Великобритания) по протоколу, предложенному для этого фермента фирмой-производителем. Условия ПЦР: 2 мин при 95°С (1 цикл); 30 с при 95°С, 30 с при 60°С, 4 мин при 72°С (30 циклов); 10 мин при 72°С (1 цикл). В обоих случаях после проведения ПЦР в реакционную смесь добавляли ЭР DpnI (10 ед. акт.) для гидролиза матричной (метилированной) плазмиды и инкубировали 1 ч при 37°С. Полученной плазмидой трансформировали штамм NovaBlue(DE3) клеток Escherichia coli, содержащий также плазмиду pRARE с геном метилтрансферазы SscL1I, которая “защищает” ДНК от гидролиза НЭ BspD6I [35]. Клетки выращивали на агаризованной среде LB с антибиотиками Km (40 мкг/мл) и Cam (10 мкг/мл). Наличие в колониях плазмиды рЕТ28b, кодирующей мутантные формы НЭ BspD6I, проверяли методом ПЦР с праймерами к гену НЭ BspD6I: Nick1 (5'‑GCGCCATGGCTAAAAAAGTTAATTG‑3') и Nick2 (5'‑GCGGTCGACAAACCTTACCTCCTTC‑3'). Целевые колонии, несущие плазмиду с геном НЭ BspD6I(С11S/C160S), выращивали в 5 мл среды LB в присутствии антибиотиков Km (40 мкг/мл) и Cam (10 мкг/мл). Плазмиду выделяли с помощью набора GeneJET Plasmid Miniprep фирмы “Thermo Fisher Scientific”. Фрагмент плазмиды, содержащий мутации, амплифицировали методом ПЦР, используя праймеры Nick3 (5'‑AAA-GTCGACAAATGAAA-ACTGTGGTAAAC‑3') и T7 (5'‑TAATACGACTCACTATAGGG‑3'), отделяли от побочных продуктов методом электрофореза в 0.8%-ном агарозном геле в буфере ТАЕ при напряженности поля 10 В/см и выделяли из геля с использованием набора реагентов Gel extraction (“Qiagen”, США). Наличие необходимых нуклеотидных замен подтверждали анализом первичной структуры продукта ПЦР на секвенаторе ABI Prism310 Genetic Analyzer фирмы “Applied Biosystems” в Институте белка РАН.
Выделение белков. Клетки E. coli NovaBlue(DE3), содержащие плазмиды pRARE с геном метилтрансферазы SscL1I и рЕТ28b с НЭ BspD6I или ее мутантной формой, выращивали при 37°С до оптической плотности А590 = 0.6 о.е. Среда LB содержала 40 мкг/мл Km и 10 мкг/мл Cam. Экспрессию НЭ в клеточной культуре индуцировали добавлением 0.7 М IPTG, затем клетки инкубировали в течение 12 ч при 20°С до поздней логарифмической фазы при интенсивной аэрации. Дальнейшую процедуру выделения и очистки НЭ BspD6I дикого типа и мутантной формы проводили по одинаковой схеме. Полученную из бактериальной суспензии биомассу разбавляли буфером для лизиса клеток (0.02 M K-фосфатный буфер, pH 7.5, 7 мM β-меркаптоэтанол, 1 мM EDTA, 10 мM КCl, 1 мM фенилметилсульфонилфторид). Раствор перемешивали до получения однородной суспензии, которую обрабатывали ультразвуком. Лизат отделяли от неразрушенных компонентов клеток центрифугированием (18 000 × g, 30 мин, 4°C). Далее НЭ BspD6I и НЭ BspD6I(С11S/C160S) выделяли методом колоночной хроматографии низкого давления на приборе BioLogic LP System фирмы “Bio-Rad” в две стадии. Сначала лизат клеток наносили на колонку с фосфоцеллюлозой Р11 Whatman фирмы “GE HealthCare” объемом 5 мл, уравновешенную буфером КХ (0.02 M K-фосфатный буфер, pH 7.5, 7 мM β-меркаптоэтанол), содержащим 10 мM КCl. Разделение проводили в 50 мл линейного градиента концентрации КCl (0.1–1.0 M в буфере KX) со скоростью 30 мл/ч; объем собираемых фракций – 0.5 мл.
Наличие целевого белка во фракциях определяли методом электрофореза по Лэммли [36]. Электрофорез проводили в электродном буфере TГ в пластинах размером 10 × 8 см при напряженности поля 18 В/см в камере Mini-PROTEAN TetraSystem фирмы “Bio Rad”. Разделяющий гель: 12% акриламида, 0.4‒0.5% N,N'-метиленбисакрил-амида, 0.1% SDS, 375 мМ Трис-НСl (рН 8.8). Концентрирующий гель: 4% акриламида, 0.1‒0.2% N,N'-метиленбисакриламида, 0.1% SDS, 130 мМ Трис-НСl (рН 6.5). Пробы наносили на гель в объеме 10 мкл в буфере LAP без DTT. Перед нанесением на гель пробы прогревали в течение 5 мин при 95°С и затем помещали в баню со льдом. Для визуализации зон белка гель окрашивали раствором PageBlue фирмы “Thermo Fisher Scientific” на основе кумасси бриллиантового синего G-250.
Фракции, содержащие целевой белок, диализовали против буфера КХ с 10 мМ КСl, а затем наносили на колонку с Ni-NTA-агарозой фирмы “Qiagen” объемом 1 мл. Элюцию проводили ступенчатым градиентом концентрации имидазола со скоростью 30 мл/ч в буфере КХ: при 30 мМ концентрации имидазола очищали препарат белка от примесей, затем при 150 мМ концентрации имидазола элюировали целевой белок. Препараты очищенных НЭ BspD6I и НЭ BspD6I(С11S/C160S) диализовали против буфера для хранения (10 мM Трис-HCl, pH 7.5, 150 мМ KCl, 0.1 мМ EDTA, 0.1 мM DTT, 50% глицерин (V/V)) и использовали в дальнейшей работе или хранили при ‒20°C.
Концентрацию препаратов НЭ BspD6I и НЭ BspD6I(С11S/C160S) определяли спектрофотометрически, используя коэффициент экстинкции ε280 = 77 240 М–1 см–1. Концентрация дикого типа НЭ BspD6I составляла 6.15 мг/мл, мутантной формы НЭ BspD6I(С11S/C160S) – 1.5 мг/мл.
Препарат MutS был любезно предоставлен инженером НИИ физико-химической биологии им. А.Н. Белозерского (МГУ им. М.В. Ломоносова) М.В. Монаховой.
Спектры кругового дихроизма НЭ BspD6I и НЭ BspD6I(C11S/C160S) регистрировали на спектрометре Chirascan (“Applied Photophysics Ltd.”, Великобритания) при 20°С в цилиндрической кювете с длиной оптического пути 0.05 см. Образцы НЭ находились в буфере CD.
Реакцию НЭ BspD6I и НЭ BspD6I(C11S/ C160S) с PEG-Mal проводили в буфере А без DTT при 37°С в течение 15 мин. Концентрация белков составляла 10 мкМ. Добавляли 200-кратный избыток PEG-Mal. За образованием конъюгатов следили методом гель-электрофореза по Лэммли [36] (см. пункт “Выделение белков”).
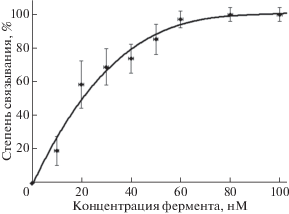
Комплексообразование НЭ BspD6I и НЭ BspD6I(C11S/C160S) с ДНК-дуплексом I (рис. 2) проводили в течение 30 мин при 37°С в 10 мкл буфера А, дополнительно содержащего 1 мМ DTT, 15% глицерина, 0.1 мг/мл BSA. При подборе условий реакции ковалентного связывания белков с ДНК-дуплексом I реакционные смеси не содержали DTT и BSA, концентрация ДНК-дуплекса составляла 0.5 мкМ, концентрацию НЭ BspD6I или НЭ BspD6I(C11S/C160S) варьировали от 0.1 до 5 мкМ. Для определения кажущейся константы диссоциации (Kd) комплекса НЭ BspD6I(C11S/ C160S) с ДНК-дуплексом I (10 нМ) использовали возрастающие концентрации фермента в диапазоне 10−100 нМ. За образованием ДНК-белковых комплексов следили методом электрофореза в плоском 200 × 200 × 1.5 мм ПААГ, содержащем 7% акриламида и N,N '-бисакриламида (акриламид : : N,N '-бисакриламид = 29 : 1). Электрофорез проводили в буфере ТВЕ при напряженности поля 15 В/см. Флуоресцентно меченные ДНК детектировали в геле с помощью прибора Typhoon FLA 9500 (“GE Healthcare”). Количественный анализ флуоресцентных зон проводили в программе ImageQuant (“GE Healthcare”). Степень связывания ДНК-субстрата ферментом рассчитывали как отношение интенсивности зон, соответствующих комплексу, к суммарной интенсивности всех зон в дорожке. Считали, что значению Kd комплекса НЭ BspD6I(C11S/C160S) с ДНК-дуплексом I соответствует концентрация белка, при которой 50% меченного карбоксифлуоресцеином (FAM) ДНК-субстрата находится в комплексе с ферментом. Стандартную ошибку определяли в программе Origin по формуле SE = s/n0.5, где SE – стандартная ошибка, s – стандартное отклонение, n – число независимых экспериментов по комплексообразованию (равнялось 3).
Рис. 2.
Используемые в работе ДНК-дуплексы, содержащие флуоресцентную метку FAM на 3'-конце “верхней” цепи. Участок узнавания НЭ BspD6I выделен жирным шрифтом. U ‒ остаток 2'-дезокси-2'-[3-(2-пиридилдитио)пропион-амидо]уридина.

“Кросслинкинг” белков с модифицированными ДНК-дуплексами. На первой стадии модифицировали олигонуклеотиды, содержащие аминогруппу в составе остатка 2'-амино-2'-дезоксиуридина, реагентом сукцинимидил-3-(2-пиридилдитио)пропионатом (SPDP). Реакционную смесь, содержащую 800 пкмоль олигонуклеотида и 2.4 ммоль SPDP (3000-кратный избыток) в 50 мкл Na-боратного буфера (pH 8.5), выдерживали при 37°С в течение 24 ч. Модифицированные олигонуклеотиды отделяли от избытка реагентов на колонках Illustra NAP-5 (“GE Healthcare”) согласно протоколу производителя и затем осаждали ацетоном в присутствии 0.4 М раствора LiClO4. На второй стадии формировали ДНК-дуплекс добавлением комплементарной цепи c последующим нагреванием при 95°С в течение 5 мин и медленным охлаждением до комнатной температуры. Далее проводили реакцию “кросслинкинга” модифицированных ДНК-дуплексов (0.5 мкМ) с НЭ BspD6I и НЭ BspD6I(C11S/C160S) (1 мкМ) при температуре 37°С в течение 30 мин в 20 мкл буфера А. “Кросслинкинг” модифицированного ДНК-дуплекса IV (0.5 мкМ) с белком MutS (2.1 мкМ), входящим в систему репарации неканонических пар нуклеотидов E. coli, проводили в 20 мкл буфера Д в тех же условиях инкубации. Продукты реакции “кросслинкинга” анализировали электрофорезом в ПААГ параллельно в неденатурирующих (7% ПААГ, см. пункт “Комплексообразование НЭ BspD6I и НЭ BspD6I(C11S/C160S) с ДНК-дуплексом I”) и денатурирующих (см. пункт “Выделение белков”) условиях. Однако для “удерживания” в геле олигонуклеотидов, имеющих значительно меньшую молекулярную массу по сравнению с белками, был использован модифицированный вариант SDS-ПААГ. В качестве дополнительного нижнего слоя разделяющего геля применяли “полосу-ловушку”, содержащую 0.375 М Трис-НСl (рН 8.8), 20% акриламида, 1% N,N '-метиленбисакриламида, 0.1% SDS [37].
Анализ эндонуклеазной активности НЭ BspD6I и НЭ BspD6I(C11S/C160S) проводили в течение 30 мин при 37°C в 10 мкл буфера Б, дополнительно содержащего 1 мМ DTT, 0.1% BSA и 15% глицерина. Использовали 1, 10, 20, 100 нМ фермент и 10 нМ ДНК-дуплекс. Перед нанесением в реакционные смеси добавляли 2 мкл буфера Г и выдерживали 3 мин при температуре 95°C. Продукты гидролиза разделяли в 20%-ном ПААГ размером 200 × 200 × 1 мм (акриламид : N,N '-бисакриламид = 19 : 1) с 7 М мочевиной при напряженности поля 30 В/см в буфере TBE. Флуоресценцию зон, содержащих ДНК, детектировали на приборе Typhoon FLA 9500 и проводили их анализ с помощью программы ImageQuant. Степень расщепления ДНК-субстрата ферментом рассчитывали как отношение интенсивности зон, соответствующих продуктам гидролиза, к суммарной интенсивности всех зон (исходная ДНК и продукты реакции).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Исследование ковалентного связывания остатков Cys НЭ BspD6I с пиридилдисульфидными производными ДНК
Для подтверждения сближенности остатков Сys в НЭ BspD6I с ДНК был использован метод ковалентного связывания НЭ и ее мутантной формы с модифицированными ДНК-дуплексами, содержащими 2-пиридилдитиогруппу в 2'-положении выбранного нуклеотида в цепи ДНК. Ранее такая модификация ДНК была использована нами для получения конъюгата с субъединицей p50 фактора транскрипции NF-κB человека [16]. Известно, что 2-пиридилдитиогруппа селективно взаимодействует с остатками Cys белка по реакции тиолдисульфидного обмена в водном буферном растворе при рН 7‒8 (рис. 3). Метелёв и соавт. [16] показали, что единственным направлением протекания этой реакции является атака тиольной группы остатка Cys белка по атому серы, сближенному с 2'-пропионамидной группировкой.
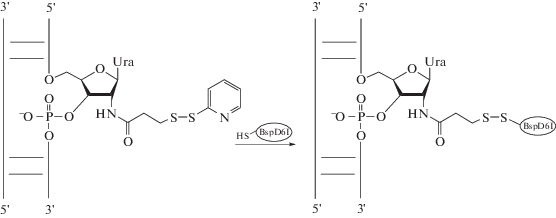
В качестве аналогов субстрата НЭ BspD6I мы выбрали 26-звенные дуплексы (рис. 2), так как ранее было показано, что для эффективного связывания и гидролиза ДНК этим ферментом необходимо, чтобы длина фланкирующих участков составляла не меньше 4 п.н. до участка узнавания (со стороны 5'-конца “верхней” цепи) и хотя бы 3 п.н. после места гидролиза [6]. Реакционноспособную группу вводили в “верхнюю” гидролизуемую цепь ДНК участка узнавания НЭ BspD6I вместо остатка Т (дуплекс II) или непосредственно с 5'-стороны от гидролизуемой ферментом межнуклеотидной связи (дуплекс III). В качестве контроля использовали ДНК-дуплекс IV с 2-пиридилдитиогруппой, не содержащий участок узнавания НЭ, а также ДНК-дуплекс I без реакционноспособной группировки.
Для получения 2-пиридилдитиосодержащих ДНК (дуплексы II–IV) использовали 26-звенные олигодезоксирибонуклеотиды-предшественники с включением в заданное положение олигомерной цепи единичного остатка 2'-амино-2'-дезоксиуридина [16], а на 3'-конец ‒ флуоресцентной метки FAM. Из 2'-амино-2'-дезоксиуридинсодержащих и комплементарных им олигонуклеотидов были сформированы дуплексы, содержащие участок узнавания НЭ BspD6I. Эти дуплексы гидролизуются ферментом с эффективностью 80−97% по сравнению с немодифицированным дуплексом I (данные не приведены). Таким образом, введение модификации по 2'-положению рибозы в участок узнавания НЭ BspD6I или рядом с расщепляемым межнуклеотидным узлом не препятствует функционированию фермента.
2'-Аминогруппу олигонуклеотидов модифицировали SPDP. На одном конце модифицирующий реагент содержал сукцинимидную группу, способную взаимодействовать с NH2-группами при рН 8.3, на другом – 2-пиридилдитиогруппу – потенциальный реагент, взаимодействующий при рН 7.5 с SH-группами остатков цистеина белка (рис. 3). SPDP − самый “короткий” коммерчески доступный “кросслинкер” c 2-пиридилдитиогруппой. Его длина составляет 6.8 Å. Небольшая длина делает реакционноспособную группу менее конформационно подвижной, что может приводить к снижению выхода конъюгата, однако позволяет определить сближенные аминокислоту в белке и нуклеотид в ДНК в процессе формирования ДНК-белкового комплекса. Реакционноспособные ДНК-дуплексы II‒IV формировали, гибридизуя модифицированный олигонуклеотид с комплементарной цепью.
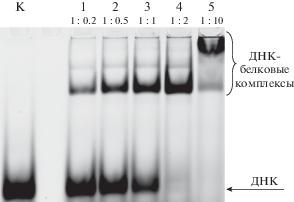
Для проведения реакции “кросслинкинга” подбирали условия, соответствующие эффективному комплексообразованию НЭ BspD6I с ДНК. Оптимальная температура функционирования НЭ BspD6I 55°C [4], однако мы изучали ее свойства при 37°С для возможности сравнения с большинством других эндонуклеаз рестрикции. Ионы Mg2+ служат естественным кофактором ЭР и необходимы для катализа гидролиза ДНК. Ионы Ca2+ способствуют образованию фермент-субстратного комплекса, но такой комплекс не продуктивен [38], поэтому традиционно эффективность комплексообразования ЭР с ДНК изучают в присутствии ионов Ca2+ для предотвращения гидролиза субстратов. НЭ BspD6I инкубировали с ДНК-субстратом I (0.5 мкМ) в буфере А в течение 30 мин. Концентрацию НЭ варьировали в диапазоне 0.5‒5.0 мкМ (рис. 4).
Рис. 4.
Анализ продуктов комплексообразования ДНК-дуплекса I (0.5 мкМ), содержащего меченную FAM “верхнюю” цепь, и НЭ BspD6I. Электрофорез проведен в 7%-ном ПААГ в неденатурирующих условиях. Дорожка К ‒ исходная ДНК; дорожки 1–5 – реакционные смеси содержат соответственно 0.1, 0.25, 0.5, 1.0 и 5.0 мкМ НЭ BspD6I. Над дорожками указано соотношение концентраций ДНК-дуплекса и НЭ BspD6I. Условия реакции: 30 мин, 37°С.
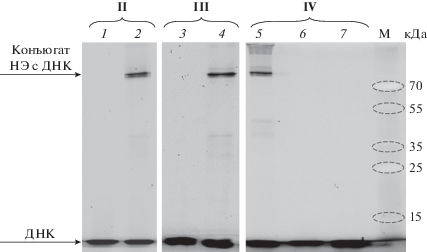
Эффективное связывание НЭ BspD6I с субстратом I регистрировали уже при двукратном избытке фермента. Реакцию тиолдисульфидного обмена фермента с ДНК-дуплексами II–IV проводили при температуре 37°C в течение 30 мин в буфере, не содержащем DTT (рис. 5). Концентрация ДНК составляла 0.5 мкМ, концентрация фермента – 1 мкМ. Продукты реакции анализировали методом гель-электрофореза в ПААГ, содержащем SDS (см. пункт “Кросслинкинг” белков с модифицированными ДНК-дуплексами” раздела “Экспериментальная часть”). Детекцию продуктов реакции проводили по флуоресценции метки FAM, находящейся в ДНК. Как видно из рис. 5, зона конъюгата выше зоны маркерного белка с массой 70 кДа, что согласуется с ожидаемым результатом. Mолекулярная масса НЭ BspD6I – 70.8 кДа, а “пришиваемой” цепи ДНК – 7.9 кДа.
Рис. 5.
Анализ продуктов ковалентного связывания НЭ BspD6I c ДНК-дуплексами II−IV (номера дуплексов указаны над дорожками), а также белка MutS с ДНК-дуплексом IV в 12%-ном SDS-ПААГ. Зоны, содержащие ДНК, детектировали по флуоресценции FAM. Дорожки 1, 3, 7 – реакционные смеси содержат только исходную ДНК; дорожки 2, 4, 5 – ДНК-дуплекс II, III или IV (0.5 мкМ) соответственно и НЭ BspD6I (1 мкМ); дорожка 6 – MutS (2.1 мкМ) и ДНК-дуплекс IV (0.5 мкМ). М – маркер молекулярной массы белков, кДа. Условия реакции: 30 мин, 37°С.
Продукт ковалентного связывания регистрировали при инкубации НЭ со всеми реакционноспособными дуплексами II–IV (рис. 5, табл. 1). Таким образом, можно говорить о сближенности c ДНК, по крайней мере, одного из четырех остатков Cys в процессе образования белково-нуклеинового комплекса. В случае дуплекса IV, не содержащего участок узнавания НЭ BspD6I, также присутствует зона конъюгата ДНК-белок. Ранее методом “торможения в геле” в условиях комплексообразования уже было показано высокое сродство НЭ BspD6I к неспецифическому ДНК-лиганду [8]. Известно, что ЭР после первичного контакта с ДНК “скользят” по ней в поисках участка узнавания [38]. Как следствие, всегда присутствует некоторый уровень фонового, неспецифического связывания, особенно если ДНК-субстрат достаточно длинный и в нем имеются участки, похожие по последовательности на участок узнавания ЭР. Отметим, что субъединица p50 NF-κВ, характеризующаяся высоким сродством к неспецифической ДНК, также взаимодействовала как с дуплексом, содержащим 2-пиридилдисульфидную группировку в κВ-участке, так и с реакционноспособной ДНК произвольной последовательности [16].
Таблица 1.
Выход конъюгатов, образующихся при взаимодействии НЭ BspD6I и НЭ BspD6I(C11S/C160S) с ДНК-дуплексами II–IV
| ДНК-дуплекс | НЭ BspD6I дикого типа, % | НЭ BspD6I(C11S/C160S), % |
|---|---|---|
| II | 16 | 6 |
| III | 18 | 5 |
| IV | 10 | 5 |
В качестве контроля был использован гомодимерный белок системы репарации MutS E. coli, способный формировать комплекс с ДНК любой последовательности и содержащий 7 остатков Cys на мономер. Однако при его инкубации с ДНК-дуплексом IV конъюгат не образовывался (рис. 5), что свидетельствует, во-первых, об отсутствии контактов остатков Cys белка MutS с ДНК, а, во-вторых, о возможности использовать эту реакцию для зондирования таких взаимодействий в процессе функционирования белка.
На основании полученных данных невозможно вычленить, какой из остатков Cys взаимодействует с реакционноспособной группировкой дуплексов II–IV. Можно лишь предположить, что в реакцию с модифицированными ДНК-фрагментами могут вступать остатки Cys N-концевого домена НЭ BspD6I (в позициях 11 и/или 160), отвечающего за связывание ДНК. Образование конъюгата в случае ДНК-дуплекса III, содержащего модификацию рядом с гидролизуемым НЭ межнуклеотидным узлом, позволяет говорить и о возможном участии в комплексообразовании с ДНК остатков Cys С-концевого домена, ответственного за катализ. Остаток Cys508 расположен рядом с каталитическим центром фермента и тоже может вносить свой вклад в “кросслинкинг”.
Для дальнейшего выяснения роли остатков Cys фермента BspD6I во взаимодействии с ДНК использовали мутантную форму НЭ BspD6I(C11S/ C160S), в которой оба остатка Cys в N-домене, предположительно сближенные с участком узнавания фермента в белково-нуклеиновом комплексе [12], заменены на Ser. Относительная оценка выходов ДНК-белковых конъюгатов с учетом возможности неспецифического связывания позволяет сделать следующие предположения. Если только остатки Cys N-концевого ДНК-связывающего домена вовлечены во взаимодействие с ДНК, то НЭ BspD6I(C11S/C160S) не будет образовывать конъюгаты с реакционноспособными дуплексами II‒IV. В том случае, если НЭ BspD6I(C11S/ C160S) будет ковалентно связываться с ДНК, можно говорить о сближенности Cys508 и/или Сys578 каталитического С-домена с дуплексом.
Структурно-функциональная характеристика мутантной формы НЭ BspD6I(C11S/C160S)
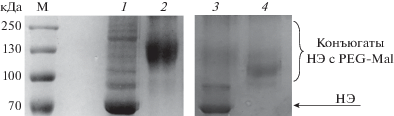
Наличие остатков Cys в белках и их доступность для химических реагентов можно подтвердить с помощью качественной реакции с полиэтиленгликолем, содержащим малеимидную группу (PEG-Mal, 5 кДа). Увеличение массы белка за счет образования конъюгата детектировали методом гель-электрофореза по Лэммли. Реакцию проводили с использованием 200-кратного избытка PEG-Mal по отношению к белку (15 мин, 37°С). Из рис. 6 видно образование конъюгатов разной молекулярной массы в случае НЭ BspD6I дикого типа и мутантной формы BspD6I(C11S/C160S), что свидетельствует о меньшем числе остатков Cys в указанной мутантной форме. При добавлении в реакционную смесь DTT (после 15 мин инкубации) тиоэфирная связь, образующаяся при взаимодействии PEG-Mal с тиогруппой Cys, разрушается с образованием исходного белка (данные не приведены).
Рис. 6.
Анализ в 12%-ном SDS-ПААГ продуктов взаимодействия PEG-Mal (5 кДа, 200-кратный избыток) c НЭ BspD6I дикого типа (10 мкМ) или мутантной формой ‒ НЭ BspD6I(C11S/C160S) (10 мкМ). Гель окрашен раствором Кумасси G-250. М – маркер молекулярной массы, кДа. НЭ BspD6I дикого типа и НЭ BspD6I(C11S/C160S) в отсутствие PEG-Mal (дорожки 1 и 3 соответственно) и в его присутствии (дорожки 2 и 4 соответственно).
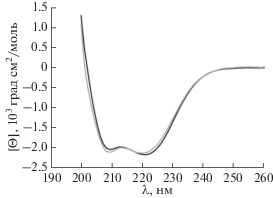
При создании мутантных форм белков остаток Cys часто заменяют на Ser, что в большинстве случаев позволяет сохранить вторичную структуру фермента ввиду подобия свойств этих аминокислотных остатков. Влияние аминокислотных замен на структуру НЭ BspD6I(C11S/C160S) оценивали методом кругового дихроизма (КД). Спектры КД записывали для мутантной формы и для белка дикого типа в диапазоне длин волн 200–260 нм при 20°С, буфер CD (рис. 7). Видно, что характер спектров КД существенно не меняется при замене Сys11 и Сys160 на Ser, что свидетельствует о сохранении в целом вторичной структуры НЭ BspD6I(C11S/C160S) по сравнению с исходным белком.
Рис. 7.
Спектры кругового дихроизма НЭ BspD6I дикого типа (кривая черного цвета) и мутантной формы BspD6I(C11S/C160S) (кривая серого цвета).
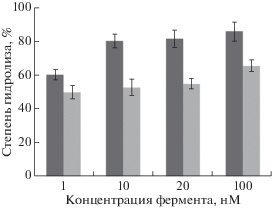
Функциональную активность НЭ BspD6I(C11S/ C160S) определяли по способности белка вносить одноцепочечный разрыв в 26-звенный субстрат I (рис. 8). Реакцию проводили при 37°С в течение 30 мин. Один из продуктов гидролиза представляет собой меченный FAM 8-звенный олигонуклеотид, детектируемый методом электрофореза в 20%-ном ПААГ, содержащем 7 М мочевину. В этих экспериментах использовали следующие концентрации фермента: 1, 10, 20, 100 нМ ‒ при постоянной концентрации ДНК-дуплекса 10 нМ. Значения степени гидролиза на гистограмме (рис. 9) соответствуют усредненным значениям трех независимых экспериментов. Из представленных данных можно заключить, что мутантная форма НЭ BspD6I(C11S/C160S) достаточно эффективно гидролизует субстрат, хотя по сравнению с белком дикого типа она снижена в 1.5 раза.
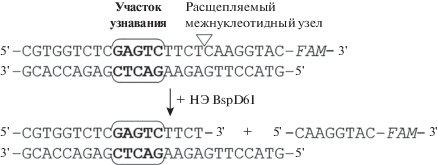
Рис. 9.
Результаты гидролиза ДНК-дуплекса I под действием НЭ BspD6I дикого типа (темно-серые столбцы) и мутантной формы НЭ BspD6I(C11S/C160S) (серые столбцы). Приведены средние значения степени гидролиза ДНК по результатам трех экспериментов. Условия реакции: 30 мин, 37°C.
Анализ модели комплекса НЭ BspD6I с ДНК [12] предсказывает взаимодействие Cys11 с ДНК в районе аденозина “нижней” цепи участка узнавания (5'‑GAGTC‑3'/3'‑CTCAG‑5'). Возможно, пониженная гидролитическая активность НЭ BspD6I(C11S/C160S) связана с участием этого аминокислотного остатка в образовании комплекса с ДНК-субстратом. Константу диссоциации комплекса НЭ BspD6I(C11S/C160S) с дуплексом I определяли методом “торможения” в геле (см. пункт “Комплексообразование НЭ BspD6I и НЭ BspD6I(C11S/C160S) с ДНК-дуплексом I” раздела “Экспериментальная часть”). Значение кажущейся Kd рассчитывали как концентрацию фермента, необходимую для связывания 50% субстрата. Для этого проводили реакцию комплексообразования в буфере А при 37°C, концентрация ДНК составляла 10 нМ, а концентрацию BspD6I(C11S/C160S) варьировали в пределах 10‒100 нМ (рис. 10). Значение Kd составило 35 ± 9 нМ. Ранее аналогичным способом было рассчитано значение Kd комплекса субстрата I с НЭ BspD6I дикого типа, составившее 8 ± 2 нМ [10]. Исходя из этих данных, можно заключить, что замена остатков Cys в положениях 11 и 160 снижает эффективность комплексообразования НЭ и ДНК примерно в 4 раза, что свидетельствует о возможном участии остатков Cys11 и Cys160 в связывании с ДНК.
Взаимодействие НЭ BspD6(C11S/C160S) с пиридилдисульфидными производными ДНК
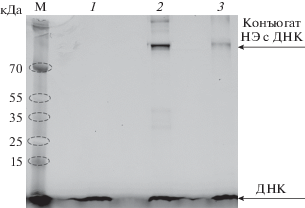
Реакцию “кросслинкинга” мутантной формы НЭ, BspD6I(C11S/C160S), с ДНК проводили в тех же условиях, что и в случае НЭ дикого типа. Пример геля с анализом продуктов реакции “кросслинкинга” обоих ферментов с ДНК-дуплексом II, содержащим участок узнавания НЭ BspD6I, приведен на рис. 11. Мутантная форма НЭ BspD6I(C11S/C160S), не содержащая остатков Cys в N-концевом домене, взаимодействовала с дуплексами II−IV с образованием конъюгатов (рис. 11, табл. 1). Следовательно, Cys508 и/или Cys578 С‑концевого каталитического домена сближен(ы) с ДНК на стадии комплексообразования. Отметим, что априори не ожидалось участие каталитического С-домена в связывании ДНК-субстрата, так как для ЭР FokI, наиболее близкой по структуре к НЭ BspD6I, выявлено, что каталитический домен контактирует с ДНК только на стадии гидролиза субстрата [39].
Рис. 11.
Анализ продуктов ковалентного связывания НЭ BspD6I дикого типа (дорожка 2) и мутантной формы НЭ BspD6I(C11S/C160S) (дорожка 3) c ДНК-дуплексом II в 12%-ном SDS-ПААГ. Зоны, содержащие ДНК, детектировали по флуоресценции FAM. М – маркер молекулярной массы белков, кДа. Дорожка 1 − исходная ДНК. Концентрация ДНК-дуплекса – 0.5 мкМ, концентрация белков – 1 мкМ. Условия реакции: 30 мин, 37°C.
Мутантная форма НЭ BspD6I(C11S/C160S) образовывала конъюгат с дуплексом IV, не содержащим узнаваемого ферментом участка. Следовательно, один или оба остатка Cys каталитического домена сближены с ДНК уже на стадии неспецифического связывания. Сходный выход получен для мутантной формы НЭ BspD6I(C11S/ C160S) и дуплексов II и III ‒ с различной локализацией реакционноспособной группировки, ‒ что также подтверждает гипотезу об участии остатков Cys С-концевого домена НЭ в первичном сканировании ДНК.
Отметим, что выход ковалентного комплекса воспроизводимо выше для дикого типа НЭ по сравнению с мутантной формой (табл. 1). Возможно, это связано с меньшим сродством НЭ BspD6I(C11S/C160S) к ДНК (см. выше). Однако нельзя исключить и непосредственное участие остатков Cys11 и/или Cys160 в катализируемом белком расщеплении ДНК, что неизбежно повышает выход продукта реакции НЭ дикого типа в сравнении с BspD6I(C11S/C160S). В целом полученные данные согласуются с существующей моделью взаимодействия НЭ BspD6I с ДНК [12], несмотря на то, что при ее построении не учтен обнаруженный нами позднее изгиб ДНК на 66 ± 4 градусов в комплексе с ферментом [6].
Таким образом, методом “кросслинкинга” впервые показано, что остатки цистеина НЭ BspD6I находятся в непосредственной близости от ДНК уже на стадии формирования неспецифического белково-нуклеинового комплекса. Продемонстрировано, что остатки цистеина С-концевого каталитического домена НЭ BspD6I также сближены с ДНК. Охарактеризованы функциональные особенности мутантной формы НЭ BspD6I, не содержащей остатки цистеина в N-концевом домене. Установлено, что замены Сys11Ser и Cys160Ser в 4 раза снижают эффективность связывания НЭ с ДНК, при этом гидролитическая активность фермента уменьшается лишь в 1.5 раза.
Анализ белок-белковых и нуклеиново-белковых взаимодействий в сложных комплексах биомолекул ‒ одна из основных задач функциональной протеомики. Хорошо зарекомендовал себя подход, заключающийся в создании мутантных форм белков, содержащих единственный остаток Cys в заданном положении полипептидной цепи. Последующая модификация таких одноцистеиновых вариантов коммерчески доступными молекулами флуорофора или “кросслинкера” c малеимидной группировкой позволяет зондировать их взаимодействие с окружающими биомолекулами-партнерами. Важное условие этого подхода заключается в сохранении функциональной активности белка, в котором заменены все входящие в него остатки Cys, как базового инструмента для создания мутантных форм с единственным остатком Cys [20, 40]. Создание мутантной формы НЭ BspD6I, не содержащей остатков Cys, и исследование свойств такого фермента позволит оценить применимость этого подхода для исследования белок-белковых взаимодействий в гетеродимерной эндонуклеазе рестрикции BspD6I.
Авторы благодарят доцента Т.С. Зацепина (Сколковский институт науки и технологий, Москва) за любезно предоставленные олигодезоксирибонуклеотиды с 2'-аминогруппой и инженера НИИ физико-химической биологии имени А.Н. Белозерского (МГУ им. М.В. Ломоносова) М.В. Монахову за препарат белка MutS.
Работа выполнена при финансовой поддержке гранта Российского Научного Фонда (№ 18-74-00049).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Абросимова Л.А., Кисиль О.В., Романова Е.А., Орецкая Т.С., Кубарева Е.А. (2019) Никующие эндонуклеазы как уникальные инструменты в биотехнологии и генетической инженерии. Биоорганическая химия. 45, 451–471.
Gabsalilow L., Schierling B., Friedhoff P., Pingoud A., Wende W. (2013) Site-and strand-specific nicking of DNA by fusion proteins derived from MutH and I-SceI or TALE repeats. Nucleic Acids Res. 41, e83.
Van Nierop G. P., de Vries A. A. F., Holkers M., Vrijsen K.R., Gonçalves M. A. (2009) Stimulation of homology-directed gene targeting at an endogenous human locus by a nicking endonuclease. Nucleic Acids Res. 37, 5725–5736.
Железная Л.А., Перевязова Т.А., Альджанова Д.В., Матвиенко Н.И. (2001) Сайт-специфическая никаза из штамма Bacillus species D6. Биохимия. 66, 595–600.
Юнусова А.К., Рогулин Е.А., Артюх Р.И., Железная Л.А., Матвиенко Н.И. (2006) Биохимия. 71, 815–820.
Abrosimova L.A., Kubareva E.A., Migur A.Y., Gavshina A.V., Ryazanova A.Y., Norkin M.V., Perevyazova T.A., Wende W., Hianik T., Zheleznaya L.A., Oretskaya T.S. (2016) Peculiarities of the interaction of the restriction endonuclease BspD6I with DNA containing its recognition site. Biochim. Biophys. Acta ‒ Proteins and Proteomics. 1864, 1072–1082.
Kachalova G.S., Rogulin E.A., Artyukh R.I., Pere-vyazova T.A., Zheleznaya L.A., Matvienko N.I., Bartunik H.D. (2005) Crystallization and preliminary crystallographic analysis of the site-specific DNA nickase Nb.BspD6I. Acta Crystallogr. Sect. F Struct. Biol. Cryst. Commun. 61, 332–334.
Секерина С.А., Гришин А.В., Рязанова А.Ю., Артюх Р.И., Рогулин Е.А., Юнусова А.К., Орецкая Т.С., Железная Л.А., Кубарева Е.А. (2012) Олигомеризация сайт-специфической никазы BspD6I при повышенных концентрациях белка. Биоорган. химия. 38, 431–438.
Абросимова Л.А., Мигур А.Ю., Кубарева Е.А., Венде В., Железная Л.А., Орецкая Т.С. (2015) Разработка ингибиторов никующей эндонуклеазы BspD6I на основе синтетических фрагментов ДНК. Известия ВУЗов. Прикладная химия и биотехнология. 2, 48–59.
Abrosimova L.A., Migur A.Y., Kubareva E.A., Zat-sepin T.S., Gavshina A.V., Yunusova A.K., Perevyazova T.A., Pingoud A., Oretskaya T.S. (2018) A study on endonuclease BspD6I and its stimulus-responsive switching by modified oligonucleotides. PLoS One. 13, e0207302.
Юнусова А.К., Артюх Р.И., Перевязова Т.А., Абросимова Л.А., Качалова Г.С., Железная Л.А. (2017) Малая субъединица эндонуклеазы рестрикции R.BspD6I активна только в присутствии каталитически активной большой субъединицы. MEDLINE.RU. 18, 200–208.
Kachalova G.S., Rogulin E.A., Yunusova A.K., Artyukh R.I., Perevyazova T.A., Matvienko N.I., Zhelez-naya L.A., Bartunik H.D. (2008) Structural analysis of the heterodimeric type IIS restriction endonuclease R.BspD6I acting as a complex between a monomeric site-specific nickase and a catalytic subunit. J. Mol. Biol. 384, 489–502.
Verdine G.L., Norman D.P.G. (2003) Covalent trapping of protein-DNA complexes. Annu. Rev. Biochem. 72, 337–366.
Metelev V.G., Kubareva E.A., Vorob’eva O.V., Romanenkov A.S., Oretskaya T.S. (2003) Specific conjugation of DNA binding proteins to DNA templates through thiol-disulfide exchange. FEBS Lett. 538, 48–52.
Воробьева О.В., Романенков А.С., Метелев В.Г., Карягина А.С., Лаврова Н.В., Орецкая Т.С., Кубарева Е.А. (2003) Ковалентное связывание Cys142 метилтрансферазы SsoII с ДНК-дуплексами, содержащими фосфорилдисульфидную межнуклеотидную группу. Молекуляр. биология. 37, 906–915.
Metelev V., Romanenkov A., Kubareva E., Zubin E., Polouchine N., Zatsepin T., Molochkov N., Oretskaya T. (2006) Structure-based cross-linking of NF-κB p50 homodimer and decoy bearing a novel 2′-disulfide trapping site. IUBMB Life. 58, 654–658.
Романенков А.С., Кисиль О.В., Зацепин Т.С., Ямскова О.В., Карягина А.С., Метелев В.Г., Орецкая Т.С., Кубарева Е.А. (2006) ДНК-метилтрансфераза SsoII как бифункциональный белок: особенности взаимодействия с промоторной областью генов системы рестрикции-модификации SsoII. Биохимия. 1, 1648–1658.
Heinze R.J., Sekerina S., Winkler I., Biertümpfel C., Oretskaya T.S., Kubareva E., Friedhoff P. (2012) Covalently trapping MutS on DNA to study DNA mismatch recognition and signaling. Mol. Biosyst. 8, 1861–1864.
Monakhova M., Ryazanova A., Hentschel A., Viryasov M., Oretskaya T., Friedhoff P., Kubareva E. (2015) Chromatographic isolation of the functionally active MutS protein covalently linked to deoxyribonucleic acid. J. Chromatogr. A. 1389, 19–27.
Monakhova M., Ryazanova A., Kunetsky V., Li P., Shilkin E., Kisil O., Rao D.N., Oretskaya T., Friedhoff P., Kubareva E. (2020) Probing the DNA-binding center of the MutL protein from the Escherichia coli mismatch repair system via crosslinking and Förster resonance energy transfer. Biochimie. 171, 43–54.
Stasińska A.R., Putaj P., Chmielewski M.K. (2020) Disulfide bridge as a linker in nucleic acids’ bioconjugation. Part II: a summary of practical applications. Bioorg. Chem. 95, 103518.
Stasińska A.R., Putaj P., Chmielewski M.K. (2019) Disulfide bridge as a linker in nucleic acids' bioconjugation. Part I: an overview of synthetic strategies. Bioorg. Chem. 92, 103223.
Leichert L.I., Jakob U. (2006) Global methods to monitor the thiol-disulfide state of proteins in vivo. Antioxid. Redox Signal. 8, 763–772.
Holliday G.L., Mitchell J.B., Thornton J.M. (2009) Understanding the functional roles of amino acid residues in enzyme catalysis. J. Mol. Biol. 390, 560–577.
Luscombe N.M., Laskowski R.A., Thornton J.M. (2001) Amino acid-base interactions: a three-dimensional analysis of protein-DNA interactions at atomic level. Nucleic Acids Res. 29, 2860–2874.
Luscombe N.M., Thornton J.M. (2002) Protein-DNA interactions: amino acid conservation and the effects of mutations on binding specificity. J. Mol. Biol. 320, 991–1009.
Ghosh G., Duyne G.V., Ghosh S., Sigler P.B. (1995) Structure of NF‑κB p50 homodimer bound to a κB site. Nature. 373, 303–310.
Chen Y.-Q., Ghosh S., Ghosh G. (1998) A novel DNA recognition mode by NF-κB p65 homodimer. Nat. Struct. Biol. 5, 67–73.
Escalante C.R., Shen L., Thana D., Aggaraiwal A.K. (2002) Structure of the NF-κB p50/p65 heterodimer bound to the PRDII DNA element from the interferon-β promoter. Structure. 10, 383–391.
Романенков А.С., Устюгов А.А., Зацепин Т.С., Никулова А.А., Колесников И.В., Метелев В.Г., Орецкая Т.С., Кубарева Е.А. (2005) Анализ белково-нуклеиновых контактов в комплексах фактора транскрипции NF-κB c ДНК. Биохимия. 70, 1474–1487.
Kim Y., Ho S.O., Gassman N.R., Korlann Y., Lan-dorf E.V., Collart F.R., Weiss S. (2008) Efficient site-specific labeling of proteins via cysteines. Bioconjug. Chem. 19, 786–791.
Kapanidis A.N., Weiss S. (2002) Fluorescent probes and bioconjugation chemistries for single-molecule fluorescence analysis of biomolecules. J. Chem. Phys. 117, 10953–10964.
Reck-Peterson S.L., Derr N.D., Stuurman N. (2010) Imaging single molecules using total internal reflection fluorescence microscopy (TIRFM). Cold Spring Harb. Protoc. 3, pdb.top73.
Liu J., Hanne J., Britton B.M., Bennett J., Kim D., Lee J.B., Fishel R. (2016) Cascading MutS and MutL sliding clamps control DNA diffusion to activate mismatch repair. Nature. 539, 583–587.
Рогулин Е.А., Перевязова Т.А., Железная Л.А., Матвиенко Н.И. (2004) Плазмида pRARE в качестве вектора для клонирования при получении суперпродуцента сайт-специфической никазы N.BspD6I. Биохимия. 69, 1381−1386.
Laemmli U.K. (1970) Cleavage of structural proteins during the assembly of the head of the bacteriophage T4. Nature. 227, 680–685.
Монахова М.В., Кубарева Е.А., Романова Е.А., Семкина А.С., Набережнов Д.С., Рао Д.Н., Зацепин Т.С., Орецкая Т.С. (2019) Синтез фрагментов ДНК, содержащих β-дикетогруппу для аффинной модификации белков. Биоорган. химия. 45, 303‒314.
Pingoud A., Wilson G.G., Wende W. (2014) Type II restriction endonucleases – a historical perspective and more. Nucleic Acids Res. 42, 7489–527.
Bitinaite J., Wah D.A., Aggarwal A.K., Schildkraut I. (1998) FokI dimerization is required for DNA cleavage. Proc. Natl. Acad. Sci. USA. 95, 10570–10575.
Friedhoff P., Manelyte L., Giron-Monzon L., Winkler I., Groothuizen F.S., Sixma T.K. (2017) Use of single-cysteine variants for trapping transient states in DNA mismatch repair. Methods Enzymol. 592, 77–101.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология