

Молекулярная биология, 2020, T. 54, № 5, стр. 826-836
Некодирующий полиморфизм rs6832151 ‒ перспективный кандидат для геномного редактирования с целью поиска новых механизмов развития аутоиммунных заболеваний
А. С. Устюгова a, *, М. А. Афанасьева a
a Институт молекулярной биологии им. В. А. Энгельгардта Российской академии наук
119991 Москва, Россия
* E-mail: ustugovaalina@yandex.ru
Поступила в редакцию 02.03.2020
После доработки 18.03.2020
Принята к публикации 18.03.2020
Аннотация
В настоящее время в качестве мишеней для фармакологических препаратов используется лишь малая доля закодированных в геноме белков. Полногеномный поиск ассоциаций считается мощным инструментом выявления локусов, задействованных в развитии комплексных заболеваний, таких как аутоиммунные расстройства. Однако требуется дальнейшая работа по идентификации функционально связанных с патогенезом однонуклеотидных полиморфизмов (SNP) в ассоциированных локусах и определению их генов-мишеней. Дело в том, что в большинстве случаев SNP являются некодирующими, а их гены-мишени могут располагаться на расстоянии до миллионов пар оснований от полиморфного участка. Используя биоинформатические инструменты и базы данных, мы обнаружили пять межгенных, ассоциированных с аутоиммунными заболеваниями полиморфизмов с высокой вероятностью каузативности, для которых не известны гены-мишени. Проанализировано влияние этих SNP на активность окружающих энхансеров в составе плазмиды, кодирующей ген-репортер люциферазы. Риск-аллель полиморфизма rs6832151(G) увеличивал экспрессию репортерного гена при высокой активности соответствующего энхансера в Т-клеточной линии СЕМ. Гены-мишени этого полиморфизма могут быть в дальнейшем установлены путем внесения точечных мутаций в геном и сравнения транскриптомов полученных сублиний, несущих альтернативные аллели rs6832151.
ВВЕДЕНИЕ
В настоящее время многие аутоиммунные заболевания не поддаются полному терапевтическому контролю и приводят к инвалидности, снижению качества жизни и ранней смерти больных. Суммарная распространенность этих заболеваний составляет 7.6–9.4% в мировой популяции [1]. Аутоиммунные заболевания возникают из-за дефектов в механизмах иммунологической толерантности. Патологический процесс, вероятно, начинается с активации врожденного иммунитета после распознавания собственных или чужеродных молекул, с последующим воспалением и активацией аутореактивных Т- и В-клеток, которые в норме присутствуют у каждого человека [2]. Наряду с факторами внешней среды и эпигенетическими особенностями, на развитие этих заболеваний существенно влияет генетический компонент [3]. Полногеномные исследования ассоциаций (Genome-wide association studies, GWAS) призваны выявить новые локусы, участвующие в патогенезе комплексных заболеваний и таким образом расширить наше понимание их механизмов и возможности для терапии. С помощью масштабных GWAS были найдены сотни однонуклеотидных полиморфизмов (single nucleotide polymorphisms, SNP), ассоциированных с аутоиммунными патологиями. Большинство из этих SNP находятся в некодирующих областях [4 ] , что не позволяет напрямую установить механизм их влияния на патологический процесс.
Некодирующие SNP могут влиять на сплайсинг РНК и ее стабильность, а также на активность промоторов и энхансеров, причем последнее наиболее распространено и около 60% некодирующих полиморфизмов, ассоциированных с аутоиммунными заболеваниями, расположены в энхансерах, активных в тех или иных клетках иммунной системы [5]. Определение генов-мишеней таких полиморфизмов ‒ сложная задача. Широко применявшаяся практика отнесения SNP к ближайшему гену оказалась необоснованной, так как было показано, что полиморфизмы могут влиять на развитие болезни, регулируя экспрессию одного или нескольких генов на удалении до миллионов пар оснований или на другой хромосоме, даже будучи расположенными в интроне [6].
В настоящее время для определения генов-мишеней доступны такие инструменты, как поиск eQTL (expression quantitative trait loci – SNP, влияющих на уровень экспрессии каких-либо генов), путем анализа транскриптомов, геномное редактирование и дополняющие их методы изучения конформации хромосом (3С, 4С и т.п.) [6, 7]. При поиске eQTL анализируют транскриптомы клеток и тканей доноров, генотипированных по обширной панели SNP, и выявляют связь между уровнями экспрессии различных РНК и генотипами. Этот метод позволяет обнаружить влияние SNP на экспрессию in vivo, но в силу высокой вариабельности в работе энхансеров в зависимости от конкретного типа клеток или даже их функционального состояния [8] это удается далеко не всегда, хотя перечень исследованных субпопуляций лимфоцитов продолжает расти и это ограничение, возможно, будет преодолено в будущем.
Нас заинтересовали полиморфизмы с сильными признаками каузативности, для которых пока неизвестны eQTL, поскольку в дальнейшем мы планируем выявить их гены-мишени, используя систему геномного редактирования CRISPR/Cas. Получение сублиний релевантных клеточных линий, несущих альтернативные аллели таких SNP, путем точного геномного редактирования и сравнение их транскриптомов позволят обнаружить новых неочевидных участников патогенеза ассоциированных заболеваний. Данная процедура очень трудоемка, поэтому кандидатные полиморфизмы должны быть тщательно подобраны с помощью методов точного картирования [9]. Также необходима предварительная оценка их функциональности в релевантных клеточных линиях, чтобы отобрать наиболее перспективные для редактирования полиморфизмы и подходящую клеточную модель.
Данная работа посвящена поиску межгенных SNP, ассоциированных с аутоиммунными заболеваниями, для которых в настоящее время неизвестны гены-мишени, и выбору из них наиболее перспективных кандидатов для геномного редактирования. Путем комплексного биоинформатического анализа мы отобрали пять полиморфизмов с наибольшей вероятностью каузативности и изучили их влияние на экспрессию гена-репортера в ряде лейкоцитарных клеточных линий человека.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Отбор однонуклеотидных полиморфизмов с высокой вероятностью каузативности. При выборе кандидатных SNP нами были использованы данные GWAS, полученные с применением чипа с плотным покрытием аутоиммунных локусов и локусов, задействованных в работе иммунной системы, ‒ Immunochip [5]. Для отбора потенциально функциональных SNP использован алгоритм PICS (Probabilistic Identification of Causal SNPs, https://pubs.broadinstitute.org/pubs/finemapping/), разработанный Farh с соавторами. Этот алгоритм позволяет вычислить вероятность каждого полиморфизма быть каузативным в каждой группе SNP, сцепленных с индексным (при эмпирическом пороге алгоритма r2 > 0.5), учитывая структуру гаплотипов и картину ассоциаций в локусе.
Выбор полиморфизмов, лежащих в энхансерах, характерных для иммунных клеток, проводили с помощью сервиса USCS Genome Browser (сборка ‒ февраль 2009 года, GRCh37/hg19) на основе эпигенетических данных проекта ENCODE (https://www.encodeproject.org/, [10]) по ацетилированию Lys27 гистона H3 (H3K27Ac) для первичных иммуноцитов [11]. Для определения границ энхансеров были учтены также данные по чувствительности к ДНКазе I и кластеризации факторов транскрипции (ТФ) на ДНК. Контрольная последовательность (имеющая схожий размер, но не несущая эпигенетических признаков активного энхансера или репрессора в лейкоцитах) подбиралась на неактивных участках генома согласно комплексным эпигенетическим характеристикам проекта NIH Epigenomics Roadmap Consortium [12].
Координаты последовательностей представлены в табл. 1. Эпигенетические данные представлены на рис. 1S‒5S Приложения.
Таблица 1.
Координаты SNP и соответствующих энхансеров, клонированных в репортерные векторы (сборка генома человека от февраля 2009 г., GRCh37/hg19)
| SNP | Координаты SNP | Координаты последовательностей энхансеров |
|---|---|---|
| rs6832151 | chr4:40303633 | chr4:40302685-40304960 |
| rs4772201 | chr13:100086259 | chr13:100085858-100087493 |
| rs2163226 | chr2:43361256 | chr2:43360220-43361931 |
| rs61972489 | chr13:100085274 | chr13:100083789-100086058 |
| rs2387397 | chr10:6390192 | chr10:6388584-6391070 |
| Контрольная последовательность | – | chr17:38087831-38089396 |
Рис. 1.
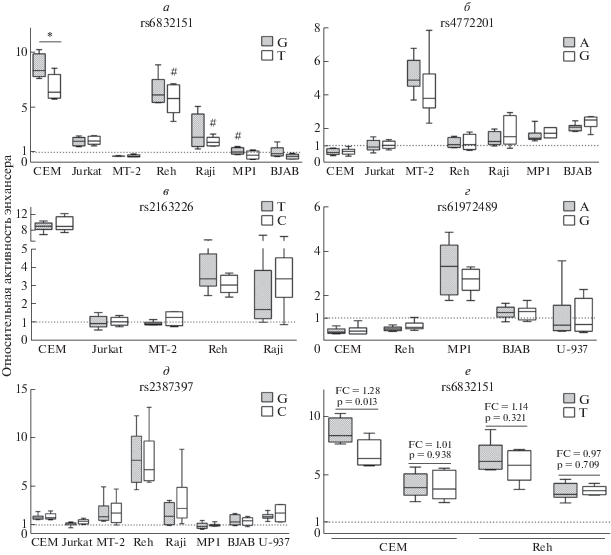
Влияние анализируемых полиморфизмов на активность соответствующих энхансеров в люциферазном тесте. Относительный сигнал люциферазы Lampyridae к сигналу люциферазы Renilla нормировали на сигнал, полученный от клеток, трансфицированных плазмидой с контрольной последовательностью (а‒д). Результаты двух независимых люциферазных тестов для полиморфизма rs6832151 на клеточных линиях СЕМ и Reh (е). Риск-аллели полиморфизмов выделены штриховкой. Пунктирная линия обозначает базовую активность репортерной конструкции, содержащей неактивный контрольный фрагмент вместо энхансера. Уровень значимости посчитан с помощью двустороннего критерия Стьюдента для непарных выборок с неравными дисперсиями. “Усы” показывают минимальное и максимальное значение выборки. * p < 0.05, # n = 5, p ‒ p-значение, FC (fold change) ‒ отношение активности энхансера, содержащего rs6832151(G) аллель, к активности энхансера, содержащего rs6832151(Т). Число технических повторов n = 6.
Оценка влияния альтернативных вариантов SNP на вероятность связывания релевантных ТФ проводилась с помощью сервиса PERFECTOS-APE [13] с использованием баз данных HOCOMOCO11, JASPAR и SELEX [14‒16]. Рассматривали белковые факторы, для которых различие в аллельном варианте SNP давало наибольшее теоретическое изменение силы связывания ТФ (fold change > 5.0) и наименьшее p-значение для мотива связывания (p‑value < 0.0005).
Получение репортерных конструкций. Последовательности энхансеров, содержащие отобранные полиморфизмы, и контрольная последовательность были получены методом полимеразной цепной реакции (ПЦР) с использованием коммерческой очищенной геномной ДНК человека (“Promega”, США). Для получения конструкций использовали праймеры, содержащие сайты рестрикции (представлены строчными буквами в табл. 2S Приложения) для рестриктазы BamHI (кроме rs61972489 и rs2387397 – BglII) в прямых праймерах и для SalI в обратных праймерах.
Таблица 2.
Условия электропорации для каждой клеточной линии
| Клеточная линия | Напряжение, В | Длительность импульса, мс | Количество импульсов |
|---|---|---|---|
| CEM | 1400 | 20 | 1 |
| Jurkat | 1350 | 10 | 3 |
| MT-2 | 1400 | 30 | 1 |
| Reh | 1300 | 10 | 3 |
| Raji | 1300 | 30 | 1 |
| MP1 | 1300 | 10 | 3 |
| BJAB | 850 | 30 | 2 |
| U-937 | 1400 | 30 | 1 |
Полученные энхансеры и контрольная последовательность были клонированы в вектор pGL3-basic (“Promega”) после гена люциферазы Lampyridae с помощью рестриктаз BamHI(BglII)/SalI. Синтетический промотор из вектора pGL4.24 (“Promega”) встраивали в плазмиду перед геном люциферазы с использованием сайтов рестрикции HindIII и NcoI. Препараты плазмид получали с помощью наборов Plasmid Midiprep (“Евроген”, Россия) и NucleoBond Xtra Midi EF (“Macherey-Nagel”, Германия). Концентрации плазмид измеряли на спектрофотометре NanoDrop™ 1000 (“Thermo Fisher Scientific”, США) и выравнивали для сравниваемых конструкций.
Получение альтернативных вариантов полиморфизмов. Для получения последовательностей энхансеров с альтернативными аллельными вариантами SNP использовали ПЦР-мутагенез [17]. Однонуклеотидные мутации были внесены праймерами с точечной мутацией (табл. 3S Приложения), соответствующей альтернативному аллелю полиморфизма, как описано ранее [18].
Таблица 3.
Отсортированный по величине отношения рисков (OR – Odds Ratio) список отобранных для исследования полиморфизмов с высокой вероятностью каузативности и неизвестными генами-мишенями
| SNP-риск- аллель |
ORa | Заболевание | Вероятность PICS | Клетки с наилучшими признаками энхансера в области SNPb |
|---|---|---|---|---|
| rs6832151-G | 1.24 | Аутоиммунные заболевания щитовидной железы | 0.5257 | Центробласты |
| rs2387397-G | 1.14 | Целиакия | 0.315 | Центробласты |
| rs2163226-T | 1.12 | Рассеянный склероз | 0.3613 | Th0 |
| rs4772201-A | 1.10 | Рассеянный склероз | 0.5382 | Активированные CD25–IL-17– Th |
| rs61972489-A | 1.10 | Рассеянный склероз | 0.1485 | CD14+ клетки |
a Значения OR приведены согласно GWAS Catalog (https://www.ebi.ac.uk/gwas), для rs61972489 данные отсутствовали, поэтому использована величина OR для соответствующего индексного SNP (rs4772201). b Th ‒ Т-хелперы.
Клеточные линии. Линия MT-2 была получена в рамках программы NIH AIDS Research and Reference Reagent Program. Клеточные линии U-937 и Raji были любезно предоставлены д.б.н. Прасоловым В.С. (Институт молекулярной биологии им. В.А. Энгельгардта РАН, Москва, Россия) и к.б.н. Дмитриевым С.Е. (НИИ физико-химической биологии им. А.Н. Белозерского, Москва, Россия) соответственно. Клетки MP1, Reh и BJAB были любезно предоставлены доктором Clark E.A. (University of Washington, США), CEM и Jurkat были предоставлены д.б.н. Филатовым А.В. (ФГБУ “ГНЦ институт иммунологии” ФМБА России, Москва, Россия). Все клетки культивировали в среде RPMI-1640 (“ПанЭко”, Россия) с добавлением 10% FBS (“Biosera”, Франция), 2 мМ L-глютамина (“Thermo Fisher Scientific”, США), 100 ед./мл пенициллина, 100 мкг/мл стрептомицина (“Thermo Fisher Scientific”, США) при 37°C и 5% CO2. Активацию моноцитарной линии U-937 проводили как описано ранее [19].
Люциферазный тест. Трансфекцию клеток репортерными конструкциями проводили методом электропорации с использованием Neon™ Transfection System (“Thermo Fisher Scientific”, США) [20]. Использовали носики объемом 100 мкл, 1.5 млн клеток на экспериментальную точку. Для каждой линии предварительно проводили подбор условий электропорации с плазмидой peGFP. Оптимальными считались условия, при которых число живых флуоресцирующих клеток было максимальным (табл. 2).
Все клетки для одного эксперимента ресуспендировали в необходимом количестве буфера R (из расчета 110 мкл на точку), который содержал контрольную плазмиду, кодирующую ген люциферазы Renilla pRL-CMV (“Promega”, США), в расчете 0.5 мкг на точку. Сигнал от контрольной плазмиды использовали для нормализации на внешние вариабельные параметры, такие как эффективность трансфекции, эффективность лизиса клеток перед измерением люминесцентного сигнала и т. п. Далее суспензию клеток делили по экспериментальным точкам и на каждую независимо добавляли 5 мкг плазмиды, содержащей последовательности исследуемых энхансеров. Через 24 ч клетки лизировали и измеряли сигналы люцифераз с использованием набора Dual-Luciferase Reporter Assay System (“Promega”, США) и планшетного люминометра Hidex Bioscan Plate Chameleon Luminometer с программным обеспечением MicroWin 2 000 software. При проведении люциферазного теста использовали минимум два препарата плазмид, чтобы учесть вклад возможной вариабельности чистоты препаратов.
Статистическая обработка результатов. Статистическую значимость различия в сигнале люциферазы между альтернативными аллелями SNP определяли с использованием двустороннего критерия Стьюдента для непарных выборок с неравными дисперсиями [21]. Расчет выполняли в MS Excel 2016 (“Microsoft”, США).
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
В результате комплексного биоинформатического анализа найдено пять межгенных SNP с высокой вероятностью каузативности и неизвестными мишенями
Из данных GWAS, которые были получены с помощью Immunochip и проанализированы Farh с соавторами с помощью математического алгоритма точного картирования PICS, отобрали 77 полиморфизмов по нескольким критериям:
● относительная вероятность каузативности (PP, PICS Probability) более 0.1;
● пересечение с энхансерной областью хотя бы в одном релевантном типе клеток;
● расположение в межгенных участках (SNP должен находиться вне тела гена и выше 100 тыс. п.н. от старта транскрипции);
● не является eQTL, то есть для полиморфизма не должны быть известны гены-мишени.
Отсортированный по величине отношения рисков (Odds Ratio, OR) список представлен в табл. 1S Приложения (полиморфизмы, отобранные для функционального анализа, представлены в табл. 3).
Далее индивидуально для каждого из 77 отобранных полиморфизмов по данным ENCODE проанализировали расположение относительно областей посадки ТФ, гиперчувствительности к ДНКазе I и ChIP-seq-пиков H3K27Ac в нескольких субпопуляциях первичных лейкоцитов: В‑клетках, Т-клетках, наивных CD4+ αβТ-клетках, CD4+ αβТ-клетках памяти, CD8+ αβТ-клетках, CD4+CD25+ αβТ-регуляторных клетках, NK-клетках, нейтрофилах и CD14+ моноцитах. В результате найдено пять полиморфизмов, находящихся в областях с признаками активных лейкоцитарных энхансеров и, соответственно, имеющих большую вероятность влиять на экспрессию генов (рис. 1S‒5S Приложения): rs6832151, rs4772201, rs61972489, rs2163226 и rs2387397.
В базе данных PubMed мы не нашли работ по исследованию генов-мишеней выбранных полиморфизмов. В базах данных GTEx и DICE также нет данных о связи этих SNP с уровнем экспрессии какой-либо мРНК. Но имеются данные о связи rs2163226 с длинной некодирующей РНК (lncРНК). Согласно GTEx, от генотипа по данному полиморфизму зависит уровень экспрессии lncРНК AC093609.1 в слизистой оболочке пищевода. Однако, поскольку rs2163226 ассоциирован с рассеянным склерозом, это не проливает свет на механизм повышения риска, и гены-мишени в клетках, релевантных данному заболеванию, остаются неизвестными [22].
Высокая вероятность каузативности отобранных полиморфизмов подтверждена анализом потенциального влияния их риск-аллелей на связывание транскрипционных факторов
Один из подходов к точному картированию заключается в поиске мотивов связывания ТФ, которые разрушаются или образуются альтернативными аллелями SNP. Для каждого из пяти отобранных полиморфизмов с помощью ресурса PERFECTOS-APE и коллекций мотивов связывания ТФ HOCOMOCO, Jaspar и SELEX удалось предсказать белковые факторы, сайты посадки которых создаются или разрушаются их риск-аллелями. Кроме того, с использованием баз данных DICE [23] и GTEx [24] проанализировано наличие мРНК этих ТФ в релевантных типах клеток (табл. 4).
Таблица 4.
Анализ предсказанного влияния риск-аллелей полиморфизмов на изменение связывания транскрипционных факторов, экспрессирующихся в различных клетках иммунной системы
| Риск- аллель |
ТФ | Коллекция мотивов связывания ТФ | Модификация сайта посадки ТФ | Fold changea | Клетки, экспрессирующие ТФ |
|---|---|---|---|---|---|
| rs6832151 | |||||
| G | NEUROG2 | HOCOMOCO | Разрушает | 17.22 | Лимфобластоидные линии |
| GFI1 | Jaspar, HOCOMOCO | Разрушает | 6.93 | Т-клетки и моноциты, цельная кровь, селезенка и лимфобластоидные линии | |
| GFI1B | HOCOMOCO | Разрушает | 37.3 | CD14lowCD16+ моноциты, цельная кровь и селезенка | |
| rs4772201 | |||||
| А | CXXC1 | HOCOMOCO | Разрушает | 271.8 | Клетки иммунной системы |
| rs61972489 | |||||
| A | HES1 | HOCOMOCO | Разрушает | 92.65 | В-клетки и моноциты |
| rs2163226 | |||||
| Т | MTF1 | HOCOMOCO | Разрушает | 7.61 | Клетки иммунной системы |
| IRF5 | SELEX | Cоздает | 9.44 | Клетки иммунной системы | |
| rs2387397 | |||||
| G | ARNTb | HOCOMOCO | Cоздает | 65.11 | Повсеместно |
| KLF8 | HOCOMOCO | Cоздает | 26.19 | Т-, В-, NK-клетки и клетки пищеварительной системы | |
| BATF3b | HOCOMOCO | Cоздает | 13.03 | Моноциты и активированные Т-клетки | |
| TFCP2 | HOCOMOCO | Разрушает | 21.43 | Повсеместно | |
a Теоретическое изменение силы связывания ТФ (во сколько раз повышается или понижается вероятность связывания ТФ в зависимости от аллеля SNP). b Есть область связывания по данным ChIP-Seq в лимфоцитах человека согласно ресурсу GTRD (Gene Transcription Regulation Database) [25].
Риск-аллель полиморфизма rs6832151(G) может разрушать сайт посадки NEUROG2, экспрессирующегося в лимфобластоидных линиях, но не в первичных лейкоцитах. Вероятность участия NEUROG2 в реализации потенциального эффекта rs6832151 очень мала, так как экспрессионный профиль В-клеток меняется после заражения вирусом Эпштейна-Барр. Однако риск-аллель G также разрушает сайты посадки транскрипционных репрессоров GFI1 и GFI1B, экспрессирующихся в первичных клетках, поэтому они могут быть более вероятными участниками реализации функционального эффекта rs6832151.
Риск-аллель полиморфизма rs61972489(A) может разрушать сайт связывания ТФ HES1, экспрессируемого В-клетками и моноцитами. Интересно, что именно в моноцитах наблюдается самый высокий пик H3K27Ac (рис. 4S Приложения).
Риск-аллель rs2387397(G) может создавать сайты связывания нескольких ТФ, которые экспрессируются в клетках, способных участвовать в патогенезе целиакии, в том числе сайт связывания белкового фактора BATF3. Показано, что этот ТФ участвует в контроле за дифференцировкой CD8+ дендритных клеток тимуса [26].
Использование коллекций мотивов связывания ТФ, построенных на основании ChIP-seq данных, не позволило с помощью PERFECTOS-APE получить надежные предсказания для полиморфизма rs2163226. Однако, согласно коллекции мотивов SELEX, этот полиморфизм может затрагивать сайт связывания IRF5, который в последнее время активно изучают в контексте аутоиммунных заболеваний как члена IRF-семейства [27].
В люциферазном тесте полиморфизм rs6832151 влиял на регуляцию экспрессии репортерного гена при сильной работе соответствующего энхансера
Участки генома с признаками регуляторных элементов, содержащие пять отобранных полиморфизмов с неизвестными генами-мишенями (отмечены серым на рис. 1S‒5S Приложения), были клонированы в видоизмененную плазмиду pGL3-Basic после гена люциферазы Lampyridae под минимальным промотором из вектора pGL4.24. После секвенирования полученных векторов с помощью ПЦР-мутагенеза для каждой конструкции создали вариант, несущий альтернативный аллель соответствующего SNP.
Способность отобранных полиморфизмов влиять на работу соответствующего энхансера проанализирована на широкой панели клеточных линий, которые являются моделями иммуноцитов: Т-хелперов (CEM и Jurkat), Т-регуляторных клеток (МТ-2), предшественников В-клеток (Reh), зрелых В-лимфоцитов (MP1, Raji, BJAB) и моноцитов (U-937) (табл. 4S Приложения). Активность люциферазы, наблюдаемая в трансфицированной репортерными плазмидами моноцитарной клеточной линии U-937, была на уровне фона, поэтому для дополнительного стимулирования клеток мы провели активацию с использованием форбол-12-миристат-13-ацетата и липополисахарида.
Данные люциферазных тестов представлены на рис. 1. Активность энхансерных областей полиморфизмов rs4772201 и rs61972489 проявилась только в одной клеточной линии: МТ-2 (модель Т-регуляторных клеток) (рис. 1б) и МР1 (модель зрелых В-клеток) (рис. 1г) соответственно. Энхансер, содержащий полиморфизм rs2163226, проявил сильную активность в клеточной линии СЕМ и умеренную активность в модели предшественников (Reh) и зрелых (Raji) В-лимфоцитов (рис. 1в). Энхансер, содержащий полиморфизм rs2387397, был активен в нескольких клеточных линиях (рис. 1д). Энхансерная область полиморфизма rs6832151 проявила сильную активность в клеточных линиях CEM и Reh (рис. 1а). Статистически значимая разница во влиянии полиморфизма на экспрессию гена люциферазы в зависимости от аллеля была выявлена только для полиморфизма rs6832151 на клеточной линии СЕМ, однако в последующих экспериментах энхансер работал слабо, и разница во влиянии аллелей стала недетектируемой (рис. 1е).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Для каждого сигнала ассоциации, обнаруживаемого в ходе GWAS (так называемого индексного SNP), существует множество сцепленных с ним полиморфизмов, каждый из которых может быть каузативным, то есть лежать в основе молекулярного механизма, который опосредует риск развития аутоиммунного заболевания. С целью сузить список возможных каузативных полиморфизмов разработаны различные методы точного картирования ассоциаций, имеющие свои сильные и слабые стороны [9]. В исследовании мы использовали алгоритм точного картирования, разработанный Farh с соавт. [5]. Преимущество этого подхода заключается в сочетании статистической модели и обширных эпигенетических данных по модификации гистонов, полученных на узких популяциях первичных лейкоцитов человека. Мы ограничили входящие данные результатами GWAS, полученными с помощью технологии Immunochip. В отличие от остальных исследований, где доноров генотипируют по универсальной панели SNP и каждый кластер сцепленных полиморфизмов обычно представлен лишь одним маркерным полиморфизмом, в Immunochip области хромосом, связанные с работой иммунной системы или ранее выявленные как ассоциированные с аутоиммунными и воспалительными заболеваниями, представлены всеми известными на момент создания этого чипа SNP [28]. Таким образом, для каждого полиморфизма из группы сцепления имеется независимая оценка значимости ассоциации. Кроме того, мы использовали данные ENCODE для определения расположения SNP относительно пика H3K27Ac, участков гиперчувствительности к ДНКазе I и плотных кластеров посадки транскрипционных факторов в ряде первичных лейкоцитов человека и клеточных линиях.
Для грубой оценки функциональности выбранных полиморфизмов и подбора подходящей клеточной модели для геномного редактирования использован люциферазный тест. Этот метод имеет ряд ограничений [7]. Во-первых, сигнал, полученный в люциферазных тестах, зависит от состояния клеток в момент проведения эксперимента, прежде всего количества и соотношения ТФ, поэтому они не всегда воспроизводимы. Во-вторых, при таком способе изучения функциональности SNP можно исследовать только один полиморфизм, тогда как на экспрессию гена могут оказывать влияние несколько сцепленных SNP [29]. И наконец, что наиболее важно, модель репортерного вектора не отражает работу энхансера в контексте нативного генома. Структура хроматина очень сложна и на транскрипцию влияет множество факторов в совокупности: взаимодействие между ДНК, гистонами и некодирующими РНК. Кроме того, часто инициация транскрипции запускается через взаимодействие элементов, расположенных на больших расстояниях друг от друга [30].
Тем не менее у люциферазного теста есть важные преимущества ‒ это простота и относительная доступность. Используя его, можно проанализировать группу полиморфизмов на большом числе разнообразных типов клеток за достаточно короткий промежуток времени, что затруднительно сделать с помощью других методов. Кроме того, люциферазный тест позволяет анализировать полиморфизмы, гены-мишени которых неизвестны, и дает ценную информацию о типах клеток, в которых активен исследуемый энхансер.
Результаты люциферазных тестов позволили нам выделить полиморфизм rs6832151 как наиболее перспективный кандидат для редактирования и выбрать линии CEM и Reh как подходящие для этого клеточные модели. Риск-аллель rs6832151(G)-статистически значимо повышает активность соответствующего энхансера в клеточной линии СЕМ при его активной работе (тенденция наблюдается и в линии Reh). Ряд других характеристик также выделяет rs6832151 среди пяти отобранных полиморфизмов. Он ассоциирован с развитием болезни Грейвса (p = 1.0 × 10–13) [31] с наибольшим соотношением рисков из всех пяти кандидатов (OR = 1.24). Полиморфизм rs6832151 также имеет исключительно высокую вероятность каузативности (PP = 0.5257) и находится в энхансерных областях генома Т- и В-клеток. Кроме того, ассоциация полиморфизма rs6832151 с болезнью Грейвса показана на популяциях разного этнического происхождения, что считается одним из общепризнанных критериев каузативности, так как в разных этнических группах существенно отличается структура сцепленности SNP [31‒33].
По предсказанию PERFECTOS-APE, риск-аллель rs6832151(G) разрушает сайт посадки транскрипционного репрессора GFI1. Роль этого ТФ в клетках иммунной системы сейчас активно исследуют. Показано, что он участвует в регуляции дифференцировки Т- и В-лимфоцитов [34]. Согласно данным CCLE (Cancer Cell Line Encyclopedia) [35], экспрессия GFI1 очень низка в клеточных линиях СЕМ и Reh. Это может объяснять нестабильность эффекта rs6832151 в люциферазном тесте.
Таким образом, по нескольким независимым критериям: величине отношения рисков возникновения ассоциированного заболевания между альтернативными аллелями, признакам функциональности in vitro, статистическому (вероятность PICS), эпигенетическому и транс-этническому точному картированию, а также потенциальному изменению мотива связывания релевантного ТФ, rs6832151 ‒ перспективный кандидат для геномного редактирования с целью подтверждения его функциональности в контексте генома и нахождения его генов-мишеней в клеточной линии СЕМ ‒ модели Т-лимфоцитов человека.
Авторы выражают особую благодарность И.Е. Воронцову за помощь в редактировании и оформлении работы. Авторы также благодарят Центр высокоточного редактирования и генетических технологий для биомедицины ИМБ РАН за методическую помощь.
Исследование выполнено при финансовой поддержке Российского научного фонда в рамках научного проекта № 18-75-00072.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Дополнительные материалы к этой статье доступны по https://doi.org/10.31857/S0026898420040175 для авторизованных пользователей.
Список литературы
Gutierrez-Arcelus M., Rich S.S., Raychaudhuri S. (2016) Autoimmune diseases – connecting risk alleles with molecular traits of the immune system. Nat. Rev. Genetics. 17(3), 160.
Theofilopoulos A.N., Kono D.H., Baccala R. (2017) The multiple pathways to autoimmunity. Nat. Immunol. 18(7), 716.
Costenbade K.H., Gay S., Alarcón-Riquelme M.E., Iaccarino L., Doria A. (2012) Genes, epigenetic regulation and environmental factors: which is the most relevant in developing autoimmune diseases? Autoimmun. Rev. 11(8), 604–609.
Wellcome Trust Case Control Consortium, Australo-Anglo-American Spondylitis Consortium (TASC), Burton P.R., Clayton D.G., Cardon L.R., Craddock N., Deloukas P., Duncanson A., Kwiatkowski D.P., McCarthy M.I., Ouwehand W.H., Samani N.J., Todd J.A., Donnelly P., Barrett J.C., Davison D., Easton D., Evans D.M., Leung H., Marchini J.L., Morris A.P., Spencer C.C.A., Tobin M.D., Attwood A.P., Boorman J.P., Cant B., Everson U., Hussey J.M., Jolley J.D., Knight A.S., Koch K., Meech E., Nutland S., Prowse C.V., Stevens H.E., Taylor N.C., Walters G.R., Walker N.M., Watkins N.A., Winzer T., Jones R.W., McArdle W.L., Ring S.M., Strachan D.P., Pembrey M., Breen G., Clair D.St., Caesar S., Gordon-Smith K., Jones L., Fraser C., Green E.K., Grozeva D., Hamshere M.L., Holmans P.A., Jones I.R., Kirov G., Moskivina V., Nikolov I., O’Donovan M.C., Owen M.J., Collier D.A., Elkin A., Farmer A., Williamson R., McGuffin P., Young A.H., Ferrier I.N., Ball S.G., Balmforth A.J., Barrett J.H., Bishop T.D., Iles M.M., Maqbool A., Yuldasheva N., Hall A.S., Braund P.S., Dixon R.J., Mangino M., Stevens S., Thompson J.R., Bredin F., Tremelling M., Parkes M., Drummond H., Lees C.W., Nimmo E.R., Satsangi J., Fisher S.A., Forbes A., Le-wis C.M., Onnie C.M., Prescott N.J., Sanderson J., Matthew C.G., Barbour J., Mohiuddin M.K., Todhunter C.E., Mansfield J.C., Ahmad T., Cummings F.R., Jewell D.P., Webster J., Brown M.J., Lathrop M.G., Connell J., Dominiczak A., Marcano C.A.B., Burke B., Dobson R., Gungadoo J., Lee K.L., Munroe P.B., Newhouse S.J., Onipinla A., Wallace C., Xue M., Caulfield M., Farrall M., Barton A., Biologics in RA Genetics and Genomics Study Syndicate (BRAGGS) Steering Committee, Bruce I.N., Donovan H., Eyre S., Gilbert P.D., Hilder S.L., Hinks A.M., John S.L., Potter C., Silman A.J., Symmons D.P.M., Thomson W., Worthington J., Dunger D.B., Widmer B., Frayling T.M., Freathy R.M., Lango H., Perry J.R.B., Shields B.M., Weedon M.N., Hattersley A.T., Hitman G.A., Walker M., Elliott K.S., Groves C.J., Lindgren C.M., Rayner N.W., Timpson N.J., Zeggini E., Newport M., Sirugo G., Lyons E., Vannberg F., Hill A.V.S., Bradbury L.A., Farrar C., Pointon J.J., Wordsworth P., Brown M.A., Franklyn J.A., Heward J.M., Simmonds M.J., Gough S.C.L., Seal S., Breast Cancer Susceptibility Collaboration (UK), Stratton M.R., Rahman N., Ban M., Goris A., Sawcer S.J., Compston A., Conway D., Jallow M., Newport M., Sirugo G., Rockett K.A., Bumpstead S.J., Chaney A., Downes K., Ghori M.J.R., Gwilliam R., Hunt S.E., Inouye M., Keniry A., King E., McGinnis R., Potter S., Ravindrarajah R., Whittaker P., Widden C., Withers D., Cardin N.J., Davison D., Ferreira T., Pereira-Gale J., Hallgrimsdóttir I.B., Howie B.N., Su Z., Teo Y.Y., Vukcevic D., Bentley D., Brown M.A., Compston A., Farrall M., Hall A.S., Hattersley A.T., Hill A.V.S., Parkes M., Pembrey M., Stratton M.R., Mitchell S.L., Newby P.R., Brand O.J., Carr-Smith J., Pearce S.H.S., McGinnis R., Keniry A., Deloukas P., Reveille J.D., Zhou X., Sims A.-M., Dowling A., Taylor J., Doan T., Davis J.C., Savage L., Ward M.M., Learch T.L., Weisman M.H., Brown M. (2007) Association scan of 14,500 nonsynonymous SNPs in four diseases identifies autoimmunity variants. Nat. Genet. 39(11), 1329.
Farh K.K.H., Marson A., Zhu J., Kleinewietfeld M., Housley W.J., Beik S., Shoresh N., Whitton H., Ryan R.J.H., Shishkin A.A., Hatan M., Carrasco-Alfonso M.J., Mayer D., Luckey C.J., Patsopoulos N.A., Jager P.L.D., Kuchroo V.K., Epstein C.B., Daly M.J., Hafler D.A., Bernstein B.E. (2015) Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature. 518(7539), 337–343.
Zhu Y., Tazearslan C., Suh Y. (2017) Challenges and progress in interpretation of non-coding genetic variants associated with human disease. Exp. Biol. Med. 242(13), 1325–1334.
Gallagher M.D., Chen-Plotkin A.S. (2018) The post-GWAS era: from association to function. Am. J. Hum. Genet. 102(5), 717–730.
Murphy A., Chu J.H., Xu M., Carey V.J., Lazarus R., Liu A., Szefler S.J., Strunk R., Demuth K., Castro M., Hansel N.N., Diette G.B., Vonakis B.M., Adkinson N.F.J., Klanderman B.J., Senter-Sylvia J., Ziniti J., Lange C., Pastinen T., Raby B.A. (2010) Mapping of numerous disease-associated expression polymorphisms in primary peripheral blood CD4+ lymphocytes. Hum. Mol. Genet. 19(23), 4745–4757.
Spain S.L., Barrett J.C. (2015) Strategies for fine-mapping complex traits. Hum. Mol. Genet. 24(R1), R111–R119.
ENCODE Project Consortium. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature. 489(7414), 57–74.
Gorbacheva A.M., Korneev K.V., Kuprash D.V., Mitkin N.A. (2018) The Risk G allele of the single-nucleotide polymorphism rs928413 creates a CREB1-binding site that activates IL33 Promoter in lung epithelial cells. Int. J. Mol. Sci. 19(10), 2911.
Bernstein B.E., Stamatoyannopoulos J.A., Costello J.F., Ren B., Milosavljevic A., Meissner A., Kellis M., Marra M.A., Beaudet A.L., Ecker J.R., Farnham P.J., Hirst M., Lander E.S., Mikkelsen T.S., Thomson J.A. (2010) The NIH roadmap epigenomics mapping consortium. Nat. Biotechnol. 28(10), 1045–1048.
Vorontsov I.E., Kulakovskiy I.V., Khimulya G., Nikolaeva D.D., Makeev V.J. (2015) PERFECTOS-APE ‒ Predicting regulatory functional effect of SNPs by approximate P-value estimation. In: International Conference on Bioinformatics Models, Methods and Algorithms, vol. 2. SCITEPRESS, pp. 102–108.
Kulakovskiy I.V., Vorontsov I.E., Yevshin I.S., Sharipov R.N., Fedorova A.D., Rumynskiy E.I., Medvedeva Y.A., Magana-Mora A., Bajic V.B., Papatsenko D.A., Kolpakov F.A., Makeev V.J. (2018) HOCOMOCO: towards a complete collection of transcription factor binding models for human and mouse via large-scale ChIP-Seq analysis. Nucleic Acids Res. 46(D1), D252–D259.
Sandelin A., Alkema W., Engström P., Wasserman W.W., Lenhard B. (2004) JASPAR: an open-access database for eukaryotic transcription factor binding profiles. Nucleic Acids Res. 32(Database issue), D91–D94.
Jolma A., Kivioja T., Toivonen J., Cheng L., Wei G., Enge M., Taipale M., Vaquerizas J.M., Yan J., Sillanpää M.J., Bonke M., Palin K., Talukder S., Hughes T.R., Luscombe N.M., Ukkonen E., Taipale J. (2010) Multiplexed massively parallel SELEX for characterization of human transcription factor binding specificities. Genome Res. 20(6), 861–873.
Pogulis R.J., Vallejo A.N., Pease L.R. (1996) In vitro recombination and mutagenesis by overlap extension PCR. In: In vitro mutagenesis protocols. Humana Press, pp. 167–176
Ustiugova A.S., Korneev K.V., Kuprash D.V., Afanasyeva M.A. (2019) Functional SNPs in the human autoimmunity-associated locus 17q12-21. Genes (Basel). 10(2), 77.
Korneev K.V., Sviriaeva E.N., Mitkin N.A., Gorbacheva A.M., Uvarova A.N., Ustiugova A.S., Polanovsky O.L., Kulakovskiy I.V., Afanasyeva M.A., Schwartz A.M., Kuprash D.V. (2020) Minor C allele of the SNP rs7873784 associated with rheumatoid arthritis and type-2 diabetes mellitus binds PU.1 and enhances TLR4 expression. Biochim. Biophys. Acta Mol. Basis Dis. 1866(3), 165626.
Mitkin N.A., Muratova A.M., Korneev K.V., Pavshintsev V.V., Rumyantsev K.A., Vagida M.S., Uvarova A.N., Afanasyeva M.A., Schwartz A.M., Kuprash D.V. (2018) Protective C allele of the single-nucleotide polymorphism rs1335532 is associated with strong binding of Ascl2 transcription factor and elevated CD58 expression in B-cells. Biochim. Biophys. Acta Mol. Basis Dis. 1864(10), 3211–3220.
Buckley M., Gjyshi A., Mendoza-Fandiño G., Baskin R., Carvalho R.S., Carvalho M.A., Woods N.T., Monteiro A.N. (2016) Enhancer scanning to locate regulatory regions in genomic loci. Nat. Protoc. 11(1), 46.
Kotelnikova E., Kiani N.A., Messinis D., Pertsovskaya I., Pliaka V., Bernardo-Faura M., Rinas M., Vila G., Zubizarreta I., Pulido-Valdeolivas I., Sakellaropoulos T., Faigle W., Silberberg G., Masso M., Stridh P., Behrens J., Olsson T., Martin R., Paul F., Alexopoulos L.G., Saez-Rodriguez J., Tegner J., Villoslada P. (2019) MAPK pathway and B cells overactivation in multiple sclerosis revealed by phosphoproteomics and genomic analysis. Proc. Natl. Acad. Sci. USA. 116(19), 9671–9676.
Schmiedel B.J., Singh D., Madrigal A., Valdovino-Gonzalez A.G., White B.M., Zapardiel-Gonzalo J., Ha B., Altay G., Greenbaum J.A., McVicker G., Seumois G., Rao A., Kronenberg M., Peters B., Vijayanand P. (2018) Impact of genetic polymorphisms on human immune cell gene expression. Cell. 175(6), 1701–1715.
GTEx Consortium. (2013) The genotype-tissue expression (GTEx) project. Nat. Genet. 45(6), 580.
Yevshin I., Sharipov R., Kolmykov S., Kondrakhin Y., Kolpakov F. (2019) GTRD: a database on gene transcription regulation ‒ 2019 update. Nucleic Acids Res. 47(D1), D100–D105.
Chandra J., Kuo P.T., Hahn A.M., Belz G.T., Frazer I.H. (2017) Batf3 selectively determines acquisition of CD8+ dendritic cell phenotype and function. Immunol. Cell Biol. 95(2), 215–223.
Almuttaqi H., Udalova I.A. (2019) Advances and challenges in targeting IRF5, a key regulator of inflammation. FEBS J. 286(9), 1624–1637.
Cortes A., Brown M.A. (2011) Promise and pitfalls of the Immunochip. Arthritis Res. Ther. 13(1), 101.
Corradin O., Saiakhova A., Akhtar-Zaidi B., Myeroff L., Willis J., Cowper-Sal R., Lupien M., Markowitz S., Scacheri P.C. (2014) Combinatorial effects of multiple enhancer variants in linkage disequilibrium dictate levels of gene expression to confer susceptibility to common traits. Genome Res. 24(1), 1–13.
Sanyal A., Lajoie B.R., Jain G., Dekker J. (2012) The long-range interaction landscape of gene promoters. Nature. 489(7414), 109–113.
Szymański K., Bednarczuk T., Krajewski P., Płoski R. (2012) The replication of the association of the rs6832151 within chromosomal band 4p14 with Graves’ disease in a Polish Caucasian population. Tissue Antigens. 79(5), 380–383.
Chu X., Pan C.M., Zhao S.X., Liang J., Gao G.Q., Zhang X.M., Yuan G.Y., Li C.G., Xue L.Q., Shen M., Liu W., Xie F., Yang S.Y., Wang H.F., Shi J.Y., Sun W.W., Du W.H., Zuo C.L., Shi J.X., Liu B.L., Guo C.C., Zhan M., Gu Z.H., Zhang X.N., Sun F., Wang Z.Q., Song Z.Y., Zou C.Y., Sun W.H., Guo T., Cao H.M., Ma J.H., Han B., Li P., Jiang H., Huang Q.H., Liang L., Liu L.B., Chen G., Su Q., Peng Y.D., Zhao J.J., Ning G., Chen Z., Chen J.L., Chen S.J., Huang W., Song H.D., China Consortium for Genetics of Autoimmune Thyroid Disease. (2011) A genome-wide association study identifies two new risk loci for Graves’ disease. Nat. Genet. 43(9), 897.
Cooper J.D., Simmonds M.J., Walker N.M., Burren O., Brand O.J., Guo H., Wallace C., Stevens H., Coleman G., Wellcome Trust Case Control Consortium, Franklyn J.A., Todd J.A., Gough S.C.L. (2012) Seven newly identified loci for autoimmune thyroid disease. Hum. Mol. Genet. 21(23), 5202–5208.
Van der Meer L.T., Jansen J.H., Van Der Reijden B.A. (2010) Gfi1 and Gfi1b: key regulators of hematopoiesis. Leukemia. 24(11), 1834.
Ghandi M., Huang F.W., Jané-Valbuena J., Kryukov G.V., Lo C.C., McDonald E.R., Barretina J., Gelfand E.T., Bielski C.M., Li H., Hu K., Andreev-Drakhlin A.Y., Kim J., Hess J.M., Haas B.J., Aguet F., Weir B.A., Rothberg M.V., Paolella B.R., Lawrence M.S., Akbani R., Lu Y., Tiv H.L., Gokhale P.C., de Weck A., Mansour A.A., Oh C., Shih J., Hadi K., Rosen Y., Bistline J., Venkatesan K., Reddy A., Sonkin D., Liu M., Lehar J., Korn J.M., Porter D.A., Jones M.D., Golji J., Caponigro G., Taylor J.E., Dunning C.M., Creech A.L., Warren A.C., McFarland J.M., Zamanighomi M., Kauffmann A., Stransky N., Imielinski M., Maruvka Y.E., Cherniack A.D., Tsherniak A., Vazquez F., Jaffe J.D., Lane A.A., Weinstock D.M., Johannessen C.M., Morrissey M.P., Stegmeier F., Schlegel R., Hahn W.C., Getz G., Mills G.B., Boehm J.S., Golub T.R., Garraway L.A., Sellers W.R. (2019). Next-generation characterization of the cancer cell line encyclopedia. Nature. 569(7757), 503–508.
Foley G.E., Lazarus H., Farber S., Uzman B.G., Boone B.A., McCarthy R.E. (1965) Continuous culture of human lymphoblasts from peripheral blood of a child with acute leukemia. Cancer. 18(4), 522–529.
Schneider U., Schwenk H.U., Bornkamm G. (1977) Characterization of EBV-genome negative “null” and “T” cell lines derived from children with acute lymphoblastic leukemia and leukemic transformed non-Hodgkin lymphoma. Int. J. Cancer. 19(5), 621–626.
Hamano R., Wu X., Wang Y., Oppenheim J.J., Chen X. (2015) Characterization of MT-2 cells as a human regulatory T cell-like cell line. Cell. Mol. Immunol. 12(6), 780–782.
Matsuo Y., Drexler H.G. (1998) Establishment and characterization of human B cell precursor-leukemia cell lines. Leuk. Res. 22(7), 567–579.
Pistillo M.P., Tanigaki N., Mazzoleni O., Ciccone E., Hämmerling U., Park M., Terasaki P.I., Ferrara, G.B. (1987) Human lymphoblastoid cell lines secreting antibodies with restricted HLA specificity. Immunogenetics. 25(3), 145–151.
Karpova M.B., Schoumans J., Ernberg I., Henter J.I., Nordenskjöld M., Fadeel B. (2005) Raji revisited: cytogenetics of the original Burkitt’s lymphoma cell line. Leukemia. 19(1), 159–161.
Klein G., Lindahl T., Jondal M., Leibold W., Menézes J., Nilsson K., Sundström C. (1974) Continuous lymphoid cell lines with characteristics of B cells (bone-marrow-derived), lacking the Epstein–Barr virus genome and derived from three human lymphomas. Proc. Natl. Acad. Sci. USA. 71(8), 3283–3286.
Sundström C., Nilsson K. (1976) Establishment and characterization of a human histiocytic lymphoma cell line (U-937). Int. J. Cancer. 17(5), 565–577.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология