

Молекулярная биология, 2021, T. 55, № 1, стр. 42-53
Причины и последствия геномной нестабильности при психических и нейродегенеративных заболеваниях
И. Ю. Юров a, b, *, С. Г. Ворсанова a, b, О. С. Куринная a, b, М. А. Зеленова a, b, К. С. Васин a, b, Ю. Б. Юров a, b
a Научный центр психического здоровья
115522 Москва, Россия
b Научно-исследовательский клинический институт педиатрии им. акад. Ю.Е. Вельтищева
Российского национального медицинского университета им. Н.И. Пирогова
125412 Москва, Россия
* E-mail: ivan.iourov@gmail.com
Поступила в редакцию 29.06.2020
После доработки 11.07.2020
Принята к публикации 20.07.2020
Аннотация
Известно, что каждый нейрон имеет 100–10 000 связей (синапсов) с другими нервными клетками, а геномные аномалии, затрагивающие даже небольшое количество клеток головного мозга, могут приводить к функциональным изменениям центральной нервной системы (ЦНС). Так, при нейродегенеративных заболеваниях (болезнь Альцгеймера и атаксия-телеангиэктазия) в клетках головного мозга наблюдается геномная и хромосомная нестабильность. При различных формах нарушения психики (шизофрения, аутистические расстройства, умственная отсталость, эпилепсия) наблюдается соматический мозаицизм, специфичный для ЦНС. Изучение генетических процессов в нервных клетках позволяет выделить определенное количество генных сетей и процессов-кандидатов, изменение которых приводит к нарушению стабильности генома. Соматические мутации возникают в головном мозге преимущественно на ранних этапах онтогенеза. Следовательно, наиболее вероятно, что вариабельность генома и соматический мозаицизм в различных областях головного мозга обусловлены нарушением таких процессов, как регуляция клеточного цикла, репарация/репликация ДНК и программируемая клеточная гибель. Помимо этого, при нейродегенеративных заболеваниях в клетках ЦНС происходят такие процессы, как эндомитоз/эндоредупликация и вхождение в клеточный цикл, аномальное для постмитотических нейронов. Принимая во внимание эти данные, предполагается, что нестабильность генома представляет собой один из ключевых элементов патогенетического каскада при болезнях мозга. В представленном обзоре рассмотрены соматические вариации генома в головном мозге при нейродегенеративных и психических заболеваниях, а также исследования, посвященные возможным причинам возникновения и последствиям геномной нестабильности в клетках ЦНС.
ВВЕДЕНИЕ
За последние два десятилетия было неоднократно показано, что вариации генома могут быть причиной болезней центральной нервной системы (ЦНС). Подробно описан широкий спектр генных и хромосомных мутаций, а также вариаций числа копий последовательностей ДНК (copy number variations — CNV), способных приводить к развитию нейродегенеративных и психических заболеваний, представляющих значимую медицинскую и социальную проблему [1–5]. Особый интерес представляет анализ геномных вариаций непосредственно в клетках ЦНС, поскольку при соответствующих заболеваниях патологические процессы происходят именно в головном мозге. Примечательно, что геномная вариабельность может быть специфичной для популяции нервных клеток (тканеспецифический соматический мозаицизм) [6, 7]. В зависимости от количества аномальных клеток этот феномен может быть причиной как межклеточного функционального разнообразия (фоновый/минимальный уровень соматических мутаций), так и нарушения гомеостаза клеток ЦНС при нервно-психических болезнях (увеличения уровня соматических мутаций) [8–11]. Если процесс накопления соматических мутаций носит прогрессирующий характер, то наблюдается геномная и/или хромосомная нестабильность (genome instability — GIN; chromosome instability — CIN), которая, как правило, является одним из ключевых элементов патогенетического каскада при ряде нейродегенеративных заболеваний [12, 13]. Таким образом, последствия GIN/CIN и соматического мозаицизма в клетках головного мозга могут различаться в зависимости от размера пораженной клеточной популяции.
Несмотря на то, что механизмы межклеточной вариабельности генома, включая мозаицизм и GIN/CIN, до конца не изучены, существует ряд убедительных моделей, описывающих молекулярные и клеточные процессы, приводящие к геномным вариациям в соматических клетках. В частности, известно, что соматические мутации аккумулируются, в основном, на самых ранних и поздних стадиях онтогенеза [14, 15]. Помимо этого, регулярные изменения генома (генные мутации и CNV), затрагивающие гены, которые участвуют в регуляции клеточного цикла, поддержании стабильности генома и запрограммированной клеточной гибели, могут быть причиной GIN или CIN [16–18]. Учитывая постмитотическую природу нейронов, особое внимание уделяется их аномальному вхождению в клеточный цикл, которое способно привести к хромосомному дисбалансу и клеточной гибели [19, 20]. Обосновано предположение о том, что нарушение стабильности генома клеток головного мозга обусловлено взаимодействием генетических и средовых факторов, инициирующим каскад аномальных внутриклеточных процессов, в результате которых формируется геномная (хромосомная) патология [21]. Механизм возникновения соматического мозаицизма и GIN/CIN, по-видимому, определяет возможное воздействие этих форм геномной вариабельности на функционирование головного мозга в норме и при нервных и психических заболеваниях.
В настоящей работе рассмотрена вариабельность генома в клетках головного мозга при нейродегенеративных и психических заболеваниях. Особое внимание уделено возможным причинам возникновения соматического мозаицизма и GIN/CIN в нейронах ЦНС. На основе анализа данных о геномных и хромосомных аномалиях в нервных клетках при болезнях мозга выделены генные сети и процессы-кандидаты, изменения которых могут приводить к GIN и CIN и, как следствие, к нарушению функционирования ЦНС.
Рис. 1.
Схематическое изображение механизма нейродегенеративных и психических болезней, связанного с соматическим мозаицизмом и GIN/CIN (подробное описание в тексте). Использованы части иллюстраций, показывающие результаты анализа анеуплоидии методом FISH, взятые из работ Yurov и соавт. 2007, 2011 и 2014 [25, 61] (Creative Commons Attribution License 2.0).
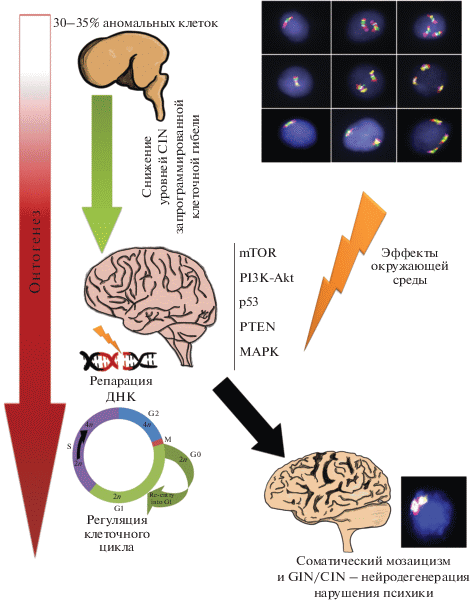
ОНТОГЕНЕТИЧЕСКИЕ ВАРИАЦИИ ГЕНОМА В КЛЕТКАХ ЦНС
Считается, что такие основные параметры ЦНС, как жизнеспособность клеточных популяций, предрасположенность к клеточной гибели и функциональные особенности, закладываются в ходе раннего онтогенеза (в основном, в первом и частично во втором триместре внутриутробного развития). В этот период также происходит беспрецедентное увеличение числа клеток эмбрионального мозга (со скоростью примерно 300000 клеток в минуту) [22, 23]. Изучение генетических особенностей этих клеточных популяций показало, что 30–35% клеток эмбрионального мозга являются анеуплоидными (увеличение или уменьшение количества хромосом в клетке, некратное гаплоидному набору (23 хромосомы) у человека). Фактически, в эмбриональном мозге человека наблюдается хромосомный мозаицизм, ограниченный тканями ЦНС, а также CIN, крайне похожая на CIN в раковых клетках [24, 25]. Примечательно, что помимо анеуплоидии клетки эмбрионального мозга также содержат множественные CNV, размер которых составляет менее 1 млн.п.н. [26, 27]. Таким образом, обоснован вывод о том, что в клеточных популяциях эмбрионального мозга человека наблюдается GIN на хромосомном (анеуплоидия) и субхромосомном/надмолекулярном (CNV) уровнях. Следует отметить, что уровни GIN/CIN максимальны именно в тот момент, когда максимальна скорость увеличения числа клеток в эмбриональном мозге. По-видимому, столь стремительный рост числа клеток вносит свой вклад в образование GIN и CIN [28]. На генном уровне (вариации нуклеотидной последовательности ДНК в виде генных мутаций и/или однонуклеотидных полиморфных замен, или SNP) подобные изменения отсутствуют, а число SNP и других форм изменений ДНК (мутаций) на генном уровне соответствует изменениям в половых клетках человека и других тканях [27–31]. На поздних стадиях внутриутробного развития общее число клеток в головном мозге плода снижается на 30–70% [32]. Молекулярно-генетические (молекулярно-цитогенетические) исследования головного мозга в постнатальном периоде свидетельствуют о том, что число анеуплоидных клеток, как и клеток с множественными CNV, снижается до 10% и меньше [10, 24, 26, 33]. Подобная корреляция уменьшения общего числа клеток и уровней GIN в ходе онтогенеза головного мозга объясняется тем, что хромосомные аномалии и CIN – это онтогенетический механизм регуляции размера клеточных популяций в ЦНС. Предполагается, что GIN/CIN представляют собой элемент каскада процессов запрограммированной клеточной гибели (например, посредством “митотической катастрофы”), которая считается одним из ключевых способов межклеточной селекции для достижения функционального и структурного разнообразия нервных клеток у человека [7, 21, 27–29, 32]. Необходимо отметить, что подобные процессы наблюдаются в эмбриональном мозге и других позвоночных (мышей и рыб), но в постнатальном периоде число клеток, пораженных GIN/CIN, снижается в значительно меньшей степени [34–36]. Все это позволило предположить, что онтогенетические вариации генома в мозге плода – это специфичный для человека феномен, за счет которого достигается уникальная сложность и пластичность ЦНС [22, 27, 29]. Нарушение запрограммированной клеточной гибели может приводить к тому, что в постнатальном мозге будут присутствовать клетки с геномными и/или хромосомными аномалиями, которые образуются на ранних стадиях внутриутробного развития.
Аккумуляция соматических мутаций (хромосомные и генные мутации) рассматривается как один из наиболее важных генетических механизмов старения. Соматический мозаицизм, а также GIN/CIN наносят значительный ущерб клеточным популяциям, вызывая дегенерацию тканей, включая головной мозг [37–39]. Тем не менее, отсутствуют однозначные данные об увеличении уровней мозаицизма и GIN/CIN на поздних стадиях онтогенеза мозга человека. В ряде работ отмечено, что при старении возрастает уровень спорадической анеуплоидии в клетках ЦНС [37, 38, 40], в то время как в других публикациях сообщается об отсутствии существенного роста числа клеток с хромосомными/генными мутациями или CNV [41, 42]. По разным данным уровень соматического мозаицизма и GIN в клетках головного мозга составляет от 0.7 до 17% у индивидов старше 50 лет, а среди лиц моложе 50 лет — от 4.8 до 12%. Наименьший уровень определен в работах, выполненных с использованием секвенирования генома индивидуальных клеток, которое позволяет выявлять вариации генома с наивысшим разрешением, но обладает существенным ограничением по количеству исследуемых клеток (максимум 100 клеток). Наивысший уровень выявлен с помощью молекулярно-цитогенетических методов, основанных на флуоресцентной гибридизации in situ (FISH), которые позволяют детектировать отдельные формы хромосомных и субхромосомных аномалий. С помощью этих методов можно изучать крупные клеточные популяции (1000 клеток и более) [28, 43–45]. Несмотря на отсутствие однозначных данных об увеличении уровня соматического мозаицизма и GIN/CIN в клетках ЦНС в зависимости от возраста, аккумуляция соматических мутаций рассматривается в настоящее время в качестве одного из наиболее вероятных механизмов нарушений функционирования головного мозга в позднем онтогенезе [14, 38]. Подобное утверждение справедливо также для нейродегенеративных заболеваний [10, 46, 47] и психических болезней [21, 48, 49], проявляющихся, в основном, у пожилых индивидов. Таким образом, следует с высокой вероятностью утверждать, что повышение уровня соматических мутаций и GIN может быть механизмом, приводящим к дисфункции ЦНС.
ГЕНОМНАЯ НЕСТАБИЛЬНОСТЬ ПРИ НЕЙРОДЕГЕНЕРАЦИИ
В настоящее время существует множество теорий относительно молекулярных причин нейродегенерации [50]. Среди них особое место занимают соматические мутации и GIN в клетках головного мозга [51, 52]. Более того, опубликовано много сообщений, согласно которым патологические процессы, наблюдаемые при нейродегенеративных заболеваниях, приводят к образованию GIN/CIN. В число этих процессов входят нарушение репарации ДНК [53, 54], аномальное вхождение нейронов в клеточный цикл [9, 20], репликативный стресс [55], нарушение процессов регуляции сегрегации хромосом в митозе [56, 57]. В частности, при болезни Альцгеймера (одной из самых распространенных болезней, ассоциированной с нейродегенерацией и поздней манифестацией) наблюдаются процессы, подобные малигнизации [58]. Эти процессы приводят к тому, что нейроны либо погибают, либо находятся в “состарившемся” состоянии (senescence-like state) [59, 60]. Примечательно, что при болезни Альцгеймера более чем в 2 раза увеличивается число клеток головного мозга с анеуплоидией (моносомией или потерей) хромосомы Х, цитогенетическим маркером процесса старения организма [61]. Необходимо отметить, что на протяжении нескольких последних десятилетий обсуждается возможность ассоциации анеуплоидии хромосомы 21 (трисомия, или наличие дополнительной хромосомы) с болезнью Альцгеймера, поскольку при синдроме Дауна (трисомия хромосомы 21) наблюдается деменция по “альцгеймеровскому типу” после 35 лет. Помимо этого, на хромосоме 21 в участке 21q21.3 расположен ген APP, кодирующий трансмембранный белок, предшественник бета-амилоида – компонента амилоидных бляшек при болезни Альцгеймера (маркер заболевания) [47]. Мутации в этом гене ассоциированы с нарушением сегрегации хромосом и хромосомным мозаицизмом (CIN), включая трисомию хромосомы 21 [62]. Изучение вариаций последовательностей гена APP в клетках ЦНС при болезни Альцгеймера показало, что среди механизмов этого заболевания могут быть CNV с участием этого гена (увеличение числа копий) и повышенный уровень его соматической рекомбинации [63]. Изучение хромосомного набора в постмортальных образцах головного мозга индивидов с болезнью Альцгеймера выявило в 6–15% нервных клеток CIN, преимущественно вовлекающую в нестабильность хромосому 21, в виде анеуплоидии (моносомии и трисомии) [10, 64]. Анализ последствий анеуплоидии в клетках ЦНС при данном заболевании показал, что она приводит к клеточной гибели [13]. Гены, мутации которых связаны с редкими семейными формами болезни Альцгеймера (пресенилины 1 и 2 /PS1 и PS2/ и MAPT), участвуют в процессах регуляции сегрегации хромосом в митозе и ассоциированы с образованием CIN [47]. Описаны также случаи заболевания, при которых в клетках головного мозга обнаружен соматический мозаицизм по мутациям генов PS1, PS2, MAPT и APP [65]. Таким образом, можно утверждать, что нейродегенеративные процессы при болезни Альцгеймера обусловлены CIN (вовлекает преимущественно хромосому 21), которая, по-видимому, возникает за счет нарушения таких генных сетей (pathways), как регуляция клеточного цикла, сегрегация хромосом в митозе, репарация ДНК и запрограммированная клеточная гибель.
CIN в виде анеуплоидии и разрывов интерфазных хромосом, ограниченная клетками мозжечка, ассоциирована с механизмом нейродегенерации при атаксии-телеангиэктазии — болезни, вызываемой мутациями в гене АТМ, кодирующем белок, необходимый для поддержания стабильности генома [12]. Мутации гена MAPT, вызывающие лобно-височную (фронто-темпоральную) дегенерацию, приводят к CIN в виде анеуплоидии (трисомии) хромосом 12 и 21 в клетках ЦНС индивидов с этим заболеванием [66]. При деменции с тельцами Леви наблюдается повышенный уровень анеуплоидии, которая, вероятно, приводит к характерной гистопатологической картине [67]. Аккумуляция трисомных клеток в ЦНС также выявляется и при болезни Нимана–Пика (тип С1), связанной с мутациями в гене NPC1 [56]. Исследования вариабельности генома в клетках головного мозга при болезни Паркинсона выявили локальную нестабильность в виде CNV (увеличение числа копий) гена SNCA в дофаминергических нейронах; мутации этого гена считаются одной из причин семейных случаев этого заболевания [68]. Как можно заметить, между нейродегенеративными процессами и малигнизацией клеток имеется много параллелей [21, 54, 57, 58, 64]. Возникает вопрос относительного различия между GIN/CIN при онкологических заболеваниях и при нейродегенерации [69]. Детальное исследование этой проблемы позволило предложить модель, объясняющую это различие. Так, клеточные популяции, вызывающие онкологические заболевания, характеризуются GIN/CIN, которые возникают за счет соматических мутаций в отдельных клетках и клональной эволюции, тогда как при нейродегенерации GIN и CIN (как правило, специфического типа) накапливаются в ходе онтогенеза и после определенного взаимодействия с окружающей средой в клетках с GIN/CIN инициируются процессы запрограммированной гибели [70]. Суммируя результаты изучения причин и последствий геномной патологии при нейродегенеративных заболеваниях, можно сделать следующие выводы: 1) GIN/CIN (преимущественно в виде анеуплоидии) – это элемент каскада нейродегенеративных процессов; 2) патология ЦНС при нейродегенеративных заболеваниях обуславливается нарушением таких процессов, как регуляция клеточного цикла, репарация и репликация ДНК, запрограммированная клеточная гибель; 3) очень вероятно, что изменение генных сетей, обеспечивающих отсутствие “сбоев” в осуществлении этих процессов, может быть причиной нейродегенерации.
ГЕНОМНАЯ НЕСТАБИЛЬНОСТЬ ПРИ ПСИХИЧЕСКИХ ЗАБОЛЕВАНИЯХ
В последние годы идея о том, что нервные и психические заболевания могут быть вызваны геномными вариациями непосредственно в клетках головного мозга, стала общепринятой [71, 72]. Первая работа в области изучения геномной (хромосомной) патологии при психических болезнях посвящена анализу клеток ЦНС при шизофрении (самом распространенном психическом заболевании). В результате обнаружили, что отдельные случаи шизофрении могут быть связаны с анеуплоидией хромосом 18 и Х [73]. Дальнейшие молекулярно-цитогенетические исследования хромосомного мозаицизма в постмортальных образцах головного мозга больных шизофренией (n = 22) позволили обнаружить CIN в виде анеуплоидии хромосомы 1 [74] и половых хромосом [75]. Ассоциация этих вариаций генома клеток головного мозга с шизофренией установлена с помощью непараметрических статистических методов (в частности, U-критерий Манна–Уитни). Изучение GIN с помощью секвенирования выявило SNP, специфичные для популяций клеток ЦНС пациентов с шизофренией [76]. Описаны также случаи шизофрении, при которых в клетках головного мозга находили неспецифические CNV [77] и делеции в участках 2q31.2 (ген PRKRA), 5q35.2 (ген BOD1) и 7p15.2 (ген CBX3) [78], не выявленные в контрольных образцах. GIN и CIN также неоднократно обнаруживали в различных типах клеток (клетки крови и биопсии кожи) больных шизофренией [79]. Эти данные позволили сделать обоснованный вывод о том, что GIN/CIN представляет собой один из механизмов этого тяжелого и распространенного заболевания [80]. Важно отметить, что подходы к поиску причин соматической анеуплоидии в различных клеточных популяциях (например, в раковых клетках), основанные на принципах системной биологии, позволили выделить ряд процессов-кандидатов [81], также изменяемых при психических заболеваниях [21, 48, 71].
Среди психических заболеваний, обладающих ярко выраженной генетической компонентой, выделяют аутистические расстройства, или расстройства аутистического спектра (РАС) [82, 83]. При данной форме нарушения психики систематически выявляют соматический мозаицизм в виде мозаичной анеуплоидии, структурных хромосомных аномалий, CNV и генных мутаций [84, 85]. Изучение постмортальных клеток головного мозга индивидов с РАС выявило специфические соматические мутации в генах SCN1A, SCN2A, SETD2, ARID1B [86]. К сожалению, до настоящего времени не опубликованы результаты анализа GIN/CIN в клетках ЦНС при РАС. При других формах нарушения развития ЦНС (умственная отсталость и эпилепсия) в клетках головного мозга неоднократно находили соматические мутации в различных генах, среди которых особое место занимают гены AKT1, AKT3, MTOR, PIK3CA, TSC1 и TSC2, поскольку кодируемые ими белки участвуют во множестве процессов, необходимых для обеспечения стабильности генома [87–89]. Спектр мутаций не ограничивается перечисленными генами, хотя необходимо отметить, что в нескольких работах описаны нарушения именно этих генов [72, 87, 89]. GIN/CIN ассоциированы с поведенческими нарушениями (в частности, посттравматический синдром, болезнь “войны в заливе”), а ухудшение состояния пациентов коррелирует, как правило, с ростом уровня GIN или CIN [90, 91].
Изучение генов-кандидатов показало, что с шизофренией может быть ассоциирован крупный кластер генов, вовлеченных в сеть клеточного цикла [92, 93]. Нарушения клеточного цикла обнаруживали ранее в экспериментальных моделях этого заболевания [94]. Подобные изменения также найдены и при РАС [95, 96], при которых наблюдаются также нарушения запрограммированной клеточной гибели (апоптоза) [97]. Анализ публикаций по соматическому мозаицизму и GIN/CIN в клетках ЦНС при психических заболеваниях указывает на острую необходимость более активного изучения этих феноменов при РАС, умственной отсталости и эпилепсии. При шизофрении подобных исследований значительно больше, однако в наших знаниях о геномной вариабельности в клетках головного мозга при этом заболевании имеется целый ряд пробелов. Тем не менее, показано, что определенное число случаев психических болезней связано с соматическим мозаицизмом и GIN/CIN в различных областях головного мозга, возможно, обусловленным нарушением процессов регуляции клеточного цикла и запрограммированной клеточной гибели.
ГЕННЫЕ СЕТИ, АССОЦИИРОВАННЫЕ С НАРУШЕНИЕМ СТАБИЛЬНОСТИ ГЕНОМА, В КЛЕТКАХ ЦНС
С помощью методологии системной биологии изучено большое количество данных, полученных в рамках анализа геномных ассоциаций и генетических нарушений (генные мутации, CNV и хромосомные аномалии) при болезнях мозга. Основным результатом этих исследований стало выделение процессов-кандидатов или генных сетей (“молекулярных путей”). Среди них в контексте геномных вариаций и нестабильности в клетках ЦНС можно выделить регуляцию клеточного цикла, включая контроль сегрегации хромосом в митозе и репликацию ДНК, апоптоз, репарацию ДНК, “клеточное старение” (сенесценцию), сигнальный путь MАРК (митоген-активируемые протеинкиназы) [72, 98–101]. Показано, что GIN/CIN и соматический мозаицизм могут быть связаны с мутациями и CNV в генах, кодирующих элементы указанных сетей [16, 39, 102–104]. Имеются сообщения об общих “молекулярных путях” при онкологических и нервно-психических заболеваниях, известных как “Pathways in cancer” [105, 106]. Специфические генные сети, изменение которых ассоциировано с патологией головного мозга при психических заболеваниях и поддержанием стабильности генома, представлены mTOR [88, 100], PI3K-Akt, p53 [72, 89] и PTEN [107]. Как отмечалось ранее, при ряде болезней мозга нарушения этих генных сетей и процессов подтверждены экспериментально. Более того, указанные генные сети рассматривают как мишени для лекарственной терапии [108–113].
Поскольку соматический мозаицизм и GIN/ CIN считаются генетической причиной различных заболеваний (в зависимости от пораженной ткани) [114–116], существует возможность экстраполировать на нервные клетки данные, полученные на других клеточных популяциях. Так, межклеточная геномная вариабельность преимущественно возникает в период внутриутробного развития, а нарушения процессов, инициирующих клеточную гибель, приводят к высокому уровню соматического мозаицизма или GIN/CIN после рождения [14–18, 39]. В митотических клетках соматические хромосомные мутации часто возникают при нарушении сегрегации хромосом в ходе деления клетки и аномальной регуляции клеточного цикла [18, 39]. Однако следует иметь в виду, что клетки ЦНС человека по большой части постмитотические. Это, вероятно, приводит к тому, что нарушения регуляции клеточного цикла, связанные с патологией головного мозга, носят специфический характер и в основном представлены аномальным вхождением в клеточный цикл, эндоредупликацией/эндомитозом и репликативным стрессом [20, 44, 46, 55, 99]. Не исключено, что нейрогенез (образование “новых” нейронов) на поздних стадиях онтогенеза (процесс, который регрессирует на протяжении жизни организма) также может быть причиной возникновения нервных клеток с геномной и/или хромосомной патологией за счет отсутствия канонических форм репарации и нарушения сегрегации хромосом в митозе [7, 22, 99]. Соматические мутации, представляющие связующее звено между генетическими и средовыми компонентами патогенеза болезней мозга, могут быть вызваны отрицательным влиянием окружающей среды [21, 117, 118]. Таким образом, обосновано предположение о том, что нарушение функционирования головного мозга при нейродегенеративных и психических заболеваниях обусловлено сложными взаимодействиями клеточного генома и окружающей среды, а мозаицизм и GIN/CIN можно рассматривать как ключевой элемент каскада патологических процессов, вызванных такими взаимодействиями.
Основываясь на современных представлениях о возможных причинах GIN/CIN и соматического мозаицизма, можно предложить схему патогенеза болезней мозга, связанного с данными феноменами (рис. 1). Высокие уровни GIN/CIN в эмбриональном мозге снижаются за счет запрограммированной клеточной гибели. Нарушение этого процесса, а также отрицательные эффекты окружающей среды, которые, скорее всего, изменяют генные сети mTOR, PI3K-Akt, p53, PTEN и МАРК, могут привести к отклонениям в регуляции клеточного цикла (нерасхождению хромосом в митозе, репликативному стрессу) и репарации ДНК. В результате в клетках головного мозга образуется соматический мозаицизм и/или прогрессирует GIN/CIN. Эти процессы, в свою очередь, нарушают гомеостаз нервных клеток, что критически изменяет функционирование ЦНС как на уровне отдельных клеток, так и определенных областей головного мозга. В зависимости от характера изменений на уровне отдельных нервных клеток, вызванных генетическими причинами, возможна потеря критической клеточной массы (нейродегенерация) или нарушение функциональной активности областей мозга за счет синаптической активности нейронов с аномальным геномом. Следует отметить, что кроме вышеуказанных процессов-кандидатов нарушения функционирования головного мозга при нейродегенеративных и психических заболеваниях, рассматриваются и другие генные сети [4, 50, 119].
ЗАКЛЮЧЕНИЕ
Обзор современных данных о причинах и последствиях вариаций генома в клетках головного мозга при нейродегенеративных и психических заболеваниях показывает, что соматический мозаицизм и GIN/CIN можно рассматривать как механизмы дисфункции ЦНС при болезнях мозга. С определенной долей уверенности можно утверждать, что нарушения генных сетей (“молекулярных путей”) и изменения процессов, необходимых для обеспечения сохранности стабильности генома, регуляции клеточного цикла и запрограммированной клеточной гибели, приводят к соответствующим формам GIN или CIN, которые входят в состав патогенетического каскада при нейродегенерации и других формах дисфункции ЦНС. С высокой вероятностью можно считать, что эти процессы инициируются неблагоприятным влиянием окружающей среды.
Крайне важно отметить, что соматический мозаицизм, GIN или CIN не следует рассматривать как основные (исключительные) механизмы патологии головного мозга. Абсолютизация как описанных патологических процессов, так и других механизмов нарушения функционирования ЦНС может привести к неприемлемо упрощенной картине этиологии болезней мозга, что неминуемо станет преградой на пути к разработке научно обоснованных методов молекулярной диагностики и терапии социально значимых нейродегенеративных и психических заболеваний. Объединение данных, полученных благодаря исследованиям в различных областях молекулярной и клеточной биологии мозга, позволит на основе принципов системной биологии определить обобщенный механизм (general pathway) нарушения функционирования ЦНС за счет соматического мозаицизма и GIN/CIN, что необходимо для успешного лечения болезней мозга [4, 21, 82, 120–123]. Знания, полученные в ходе подобного анализа, будут, несомненно, иметь исключительное значение для таких фундаментальных дисциплин, как геномика человека, нейробиология и молекулярная медицина.
Исследование выполнено при финансовой поддержке Российского фонда фундаментальных исследований в рамках научного проекта № 19-115-50415 (“Экспансия”).
Настоящая статья не содержит каких-либо исследований, выполненных с использованием биологических материалов.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
McGuffin P., Owen M.J., Gottesman I.I. (2002) Psychiatric Genetics and Genomics. Oxford, UK, Oxford University Press.
Lee J.A., Lupski J.R. (2006) Genomic rearrangements and gene copy-number alterations as a cause of nervous system disorders. Neuron. 52, 103–121.
Iourov I.Y., Vorsanova S.G., Yurov Y.B. (2008) Molecular cytogenetics and cytogenomics of brain diseases. Curr. Genomics. 9, 452–465.
Parikshak N.N., Gandal M.J., Geschwind D.H. (2015) Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat. Rev. Genet. 16, 441–458.
Smoller J.W., Andreassen O.A., Edenberg H.J., Faraone S.V., Glatt S.J., Kendler K.S. (2019) Psychiatric genetics and the structure of psychopathology. Mol. Psychiatry. 24, 409–420.
Kingsbury M.A., Yung Y.C., Peterson S.E., Westra J.W., Chun J. (2006). Aneuploidy in the normal and diseased brain. Cell. Mol. Life Sci. 63, 2626–2641.
Iourov I.Y., Vorsanova S.G., Yurov Y.B. (2006) Chromosomal variation in mammalian neuronal cells: known facts and attractive hypotheses. Int. Rev. Cytol. 249, 143–191.
Kingsbury M.A., Friedman B., McConnell M.J., Rehen S.K., Yang A.H., Kaushal D., Chun J. (2005) Aneuploid neurons are functionally active and integrated into brain circuitry. Proc. Natl. Acad. Sci. USA. 102, 6143–6147.
Mosch B., Morawski M., Mittag A., Lenz D., Tarnok A., Arendt T. (2007) Aneuploidy and DNA replication in the normal human brain and Alzheimer’s disease. J. Neurosci. 27, 6859–6867.
Iourov I.Y., Vorsanova S.G., Liehr T., Yurov Y.B. (2009) Aneuploidy in the normal, Alzheimer’s disease and ataxia-telangiectasia brain: differential expression and pathological meaning. Neurobiol. Dis. 34, 212–220.
Knouse K.A., Wu J., Whittaker C.A., Amon A. (2014) Single cell sequencing reveals low levels of aneuploidy across mammalian tissues. Proc. Natl. Acad. Sci. USA. 111, 13409–13414.
Iourov I.Y., Vorsanova S.G., Liehr T., Kolotii A.D., Yurov Y.B. (2009) Increased chromosome instability dramatically disrupts neural genome integrity and mediates cerebellar degeneration in the ataxia-telangiectasia brain. Hum. Mol. Genet. 18, 2656–2669.
Arendt T., Brückner M.K., Mosch B., Lösche A. (2010). Selective cell death of hyperploid neurons in Alzheimer’s disease. Am. J. Pathol. 177, 15–20.
Yurov Y.B., Vorsanova S.G., Iourov I.Y. (2010) Ontogenetic variation of the human genome. Curr. Genomics. 11, 420–425.
Zhang L., Vijg J. (2018) Somatic mutagenesis in mammals and its implications for human disease and aging. Annu. Rev. Genet. 52, 397–419.
Kennedy S.R., Loeb L.A., Herr A.J. (2012) Somatic mutations in aging, cancer and neurodegeneration. Mech. Ageing Dev. 133, 118–126.
Varetti G., Pellman D., Gordon D.J. (2014) Aurea mediocritas: the importance of a balanced genome. Cold Spring Harb. Perspect. Biol. 6, a015842.
Mitrentsi I., Yilmaz D., Soutoglou E. (2020) How to maintain the genome in nuclear space. Curr. Opin. Cell Biol. 64, 58–66.
Herrup K., Yang Y. (2007) Cell cycle regulation in the postmitotic neuron: oxymoron or new biology? Nat. Rev. Neurosci. 8, 368–378.
Arendt T. (2012) Cell cycle activation and aneuploid neurons in Alzheimer’s disease. Mol. Neurobiol. 46, 125–135.
Iourov I.Y., Vorsanova S.G., Yurov Y.B. (2013) Somatic cell genomics of brain disorders: a new opportunity to clarify genetic-environmental interactions. Cytogenet. Genome Res. 139, 181–188.
Muotri A.R., Gage F.H. (2006) Generation of neuronal variability and complexity. Nature. 441, 1087–1093.
Herculano-Houzel S. (2009) The human brain in numbers: a linearly scaled-up primate brain. Front. Hum. Neurosci. 3, 31.
Yurov Y.B., Iourov I.Y., Monakhov V.V., Soloviev I.V., Vostrikov V.M., Vorsanova S.G. (2005) The variation of aneuploidy frequency in the developing and adult human brain revealed by an interphase FISH study. J. Histochem. Cytochem. 53, 385–390.
Yurov Y.B., Iourov I.Y., Vorsanova S.G., Liehr T., Kolotii A.D., Kutsev S.I., Pellestor F., Beresheva A.K., Demidova I.A., Kravets V.S., Monakhov V.V., Soloviev I.V. (2007) Aneuploidy and confined chromosomal mosaicism in the developing human brain. PLoS One. 2, e558.
Rohrback S., April C., Kaper F., Rivera R.R., Liu C.S., Siddoway B., Chun J. (2018) Submegabase copy number variations arise during cerebral cortical neurogenesis as revealed by single-cell whole-genome sequencing. Proc. Natl. Acad. Sci. USA. 115, 10804–10809.
Rohrback S., Siddoway B., Liu C.S., Chun J. (2018) Genomic mosaicism in the developing and adult brain. Dev. Neurobiol. 78, 1026–1048.
Yurov Y.B., Vorsanova S.G., Iourov I.Y. (2018) Human molecular neurocytogenetics. Curr. Genet. Med. Rep. 6, 155–164.
Bushman D.M., Chun J. (2013) The genomically mosaic brain: aneuploidy and more in neural diversity and disease. Semin. Cell. Dev. Biol. 24, 357–369.
Gawad C., Koh W., Quake S.R. (2016) Single-cell genome sequencing: current state of the science. Nat. Rev. Genet. 17, 175–188.
Muyas F., Zapata L., Guigó R., Ossowski S. (2020) The rate and spectrum of mosaic mutations during embryogenesis revealed by RNA sequencing of 49 tissues. Genome Med. 12, 49.
Fricker M., Tolkovsky A.M., Borutaite V., Coleman M., Brown G.C. (2018) Neuronal cell death. Physiol. Rev. 98, 813–880.
McConnell M.J., Lindberg M.R., Brennand K.J., Piper J.C., Voet T., Cowing-Zitron C., Shumilina S., Lasken R.S., Vermeesch J.R., Hall I.M., Gage F.H. (2013) Mosaic copy number variation in human neurons. Science. 342, 632–637.
Kaushal D., Contos J.J., Treuner K., Yang A.H., Kingsbury M.A., Rehen S.K., McConnell M.J., Okabe M., Barlow C., Chun J. (2003) Alteration of gene expression by chromosome loss in the postnatal mouse brain. J. Neurosci. 23, 5599–5606.
Yang A.H., Kaushal D., Rehen S.K., Kriedt K., Kingsbury M.A., McConnell M.J., Chun J. (2003) Chromosome segregation defects contribute to aneuploidy in normal neural progenitor cells. J. Neurosci. 23, 10454–10462.
Zupanc G.K. (2009) Towards brain repair: Insights from teleost fish. Semin. Cell. Dev. Biol. 20, 683–690.
Andriani G.A., Vijg J., Montagna C. (2017) Mechanisms and consequences of aneuploidy and chromosome instability in the aging brain. Mech. Ageing Dev. 161, 19–36.
Vijg J., Dong X., Milholland B., Zhang L. (2017) Genome instability: a conserved mechanism of ageing? Essays Biochem. 61, 305–315.
Iourov I.Y., Vorsanova S.G., Yurov Y.B., Kutsev S.I. (2019) Ontogenetic and pathogenetic views on somatic chromosomal mosaicism. Genes (Basel). 10, E379.
Fischer H.G., Morawski M., Brückner M.K., Mittag A., Tarnok, A., Arendt T. (2012) Changes in neuronal DNA content variation in the human brain during aging. Aging Cell. 11, 628–633.
Van den Bos H., Spierings D.C., Taudt A.S., Bakker B., Porubský D., Falconer E., Novoa C., Halsema N., Kazemier H.G., Hoekstra-Wakker K., Guryev V., den Dunnen W.F., Foijer F., Tatché M.C., Boddeke H.W., Lansdorp P.M. (2016) Single-cell whole genome sequencing reveals no evidence for common aneuploidy in normal and Alzheimer’s disease neurons. Genome Biol. 17, 116.
Chronister W.D., Burbulis I.E., Wierman M.B., Wolpert M.J., Haakenson M.F., Smith A.C.B., Kleinman J.E., Hyde T.M., Weinberger D.R., Bekiranov S., McConnell M.J. (2019) Neurons with complex karyotypes are rare in aged human neocortex. Cell Rep. 26, 825–835.e7.
Iourov I.Y., Liehr T., Vorsanova S.G., Kolotii A.D., Yurov Y.B. (2006) Visualization of interphase chromosomes in postmitotic cells of the human brain by multicolour banding (MCB). Chromosome Res. 14, 223–229.
Faggioli F., Vijg J., Montagna C. (2011) Chromosomal aneuploidy in the aging brain. Mech. Ageing Dev. 132, 429–436.
Andriani G.A., Maggi E., Piqué D., Zimmerman S.E., Lee M., Quispe-Tintaya W., Maslov A., Campisi J., Vijg J., Mar J.C., Montagna C. (2019) A direct comparison of interphase FISH versus low-coverage single cell sequencing to detect aneuploidy reveals respective strengths and weaknesses. Sci. Rep. 9, 10508.
Iourov I.Y., Vorsanova S.G., Yurov Y.B. (2012) Single cell genomics of the brain: focus on neuronal diversity and neuropsychiatric diseases. Curr. Genomics. 13, 477–488.
Potter H., Chial H.J., Caneus J., Elos M., Elder N., Borysov S., Granic A. (2019) Chromosome instability and mosaic aneuploidy in neurodegenerative and neurodevelopmental disorders. Front. Genet. 10, 1092.
Тиганов А.С., Юров Ю.Б., Ворсанова С.Г., Юров И.Ю. (2012) Нестабильность генома головного мозга: этиология, патогенез и новые биологические маркеры психических болезней. Вест. РАМН. 9, 45–53.
Graham E.J., Vermeulen M., Vardarajan B., Bennett D., De Jager P., Pearse R.V. 2nd, Young-Pearse T.L., Mostafavi S. (2019) Somatic mosaicism of sex chromosomes in the blood and brain. Brain Res. 1721, 146345.
Pluvinage J.V., Wyss-Coray T. (2020) Systemic factors as mediators of brain homeostasis, ageing and neurodegeneration. Nat. Rev. Neurosci. 21, 93–102.
Leija-Salazar M., Piette C., Proukakis C. (2018) Somatic mutations in neurodegeneration. Neuropathol. Appl. Neurobiol. 44, 267–285.
Shepherd C.E., Yang Y., Halliday G.M. (2018) Region- and cell-specific aneuploidy in brain aging and neurodegeneration. Neuroscience. 374, 326–334.
Coppedè F., Migliore L. (2015) DNA damage in neurodegenerative diseases. Mutat. Res. 776, 84–97.
Lin X., Kapoor A., Gu Y., Chow M.J., Peng J., Zhao K., Tang D. (2020) Contributions of DNA damage to Alzheimer’s disease. Int. J. Mol. Sci. 21, 1666.
Yurov Y.B., Vorsanova S.G., Iourov I.Y. (2011) The DNA replication stress hypothesis of Alzheimer’s disease. Sci. World J. 11, 2602–2612.
Granic A., Potter H. (2013) Mitotic spindle defects and chromosome mis-segregation induced by LDL/cholesterol-implications for Niemann-Pick C1, Alzheimer’s disease, and atherosclerosis. PLoS One. 8, e60718.
Bajic V., Spremo-Potparevic B., Zivkovic L., Isenovic E.R., Arendt T. (2015) Cohesion and the aneuploid phenotype in Alzheimer’s disease: A tale of genome instability. Neurosci. Biobehav. Rev. 55, 365–374.
Nudelman K.N.H., McDonald B.C., Lahiri D.K., Saykin A.J. (2019) Biological hallmarks of cancer in Alzheimer’s disease. Mol. Neurobiol. 56, 7173–7187.
Fielder E., von Zglinicki T., Jurk D. (2017) The DNA damage response in neurons: die by apoptosis or survive in a senescence-like state? J. Alzheimers Dis. 60, S107–S131.
Baker D.J., Petersen R.C. (2018) Cellular senescence in brain aging and neurodegenerative diseases: evidence and perspectives. J. Clin. Invest. 128, 1208–1216.
Yurov Y.B., Vorsanova S.G., Liehr T., Kolotii A.D., Iourov I.Y. (2014) X chromosome aneuploidy in the Alzheimer’s disease brain. Mol. Cytogenet. 7, 20.
Granic A., Padmanabhan J., Norden M., Potter H. (2010) Alzheimer Abeta peptide induces chromosome mis-segregation and aneuploidy, including trisomy 21: requirement for tau and APP. Mol. Biol. Cell. 21, 511–520.
Lee M.H., Siddoway B., Kaeser G.E., Segota I., Rivera R., Romanow W.J., Liu C.S., Park C., Kennedy G., Long T., Chun J. (2018) Somatic APP gene recombination in Alzheimer’s disease and normal neurons. Nature. 563, 639–645.
Iourov I.Y., Vorsanova S.G., Yurov Y.B. (2011) Genomic landscape of the Alzheimer’s disease brain: chromosome instability — aneuploidy, but not tetraploidy — mediates neurodegeneration. Neurodegener. Dis. 8, 35–37.
Sala Frigerio C., Lau P., Troakes C., Deramecourt V., Gele P., Van Loo P., Voet T., De Strooper B. (2015) On the identification of low allele frequency mosaic mutations in the brains of Alzheimer’s disease patients. Alzheimer’s Dement. 11, 1265–1276.
Caneus J., Granic A., Rademakers R., Dickson D.W., Coughlan C.M., Chial H.J., Potter H. (2018) Mitotic defects lead to neuronal aneuploidy and apoptosis in frontotemporal lobar degeneration caused by MAPT mutations. Mol. Biol. Cell. 29, 575–586.
Yang Y., Shepherd C., Halliday G. (2015) Aneuploidy in Lewy body diseases. Neurobiol. Aging. 36, 1253–1260.
Mokretar K., Pease D., Taanman J.W., Soenmez A., Ejaz A., Lashley T., Ling H., Gentleman S., Houlden H., Holton J.L., Schapira A.H.V., Nacheva E., Proukakis C. (2018) Somatic copy number gains of α-synuclein (SNCA) in Parkinson’s disease and multiple system atrophy brains. Brain. 141, 2419–2431.
Hou Y., Song H., Croteau D.L., Akbari M., Bohr V.A. (2017) Genome instability in Alzheimer disease. Mech. Ageing Dev. 161, 83–94.
Yurov Y.B., Vorsanova S.G., Iourov I.Y. (2019) Chromosome instability in the neurodegenerating brain. Front. Genet. 10, 892.
McConnell M.J., Moran J.V., Abyzov A., Akbarian S., Bae T., Cortes-Ciriano I., Erwin J.A., Fasching L., Flasch D.A., Freed D., Ganz J., Jaffe A.E., Kwan K.Y., Kwon M., Lodato M.A., Mills R.E., Paquola A.C.M., Rodin R.E., Rosenbluh C., Sestan N., Sherman M.A., Shin J.H., Song S., Straub R.E., Thorpe J., Weinberger D.R., Urban A.E., Zhou B., Gage F.H., Lehner T., Senthil G., Walsh C.A., Chess A., Courchesne E., Gleeson J.G., Kidd J.M., Park P.J., Pevsner J., Vaccarino F.M., Brain Somatic Mosaicism Network (2017) Intersection of diverse neuronal genomes and neuropsychiatric disease: the brain somatic mosaicism network. Science. 356, eaal1641.
D'Gama A.M., Walsh C.A. (2018) Somatic mosaicism and neurodevelopmental disease. Nat. Neurosci. 21, 1504–1514.
Yurov Y.B., Vostrikov V.M., Vorsanova S.G., Monakhov V.V., Iourov I.Y. (2001) Multicolor fluorescent in situ hybridization on post-mortem brain in schizophrenia as an approach for identification of low-level chromosomal aneuploidy in neuropsychiatric diseases. Brain Dev. 23, S186–S190.
Yurov Y.B., Iourov I.Y., Vorsanova S.G., Demidova I.A., Kravetz V.S., Beresheva A.K., Kolotii A.D., Monakchov V.V., Uranova N.A., Vostrikov V.M., Soloviev I.V., Liehr T. (2008) The schizophrenia brain exhibits low-level aneuploidy involving chromosome 1. Schizophr. Res. 98, 139–147.
Yurov Y.B., Vorsanova S.G., Demidova I.A., Kolotii A.D., Soloviev I.V., Iourov I.Y. (2018) Mosaic brain aneuploidy in mental illnesses: an association of low-level post-zygotic aneuploidy with schizophrenia and comorbid psychiatric disorders. Curr. Genomics. 19, 163–172.
Fullard J.F., Charney A.W., Voloudakis G., Uzilov A.V., Haroutunian V., Roussos P. (2019) Assessment of somatic single-nucleotide variation in brain tissue of cases with schizophrenia. Transl. Psychiatry. 9, 21.
Kim J., Shin J.Y., Kim J.I., Seo J.S., Webster M.J., Lee D., Kim S. (2014) Somatic deletions implicated in functional diversity of brain cells of individuals with schizophrenia and unaffected controls. Sci. Rep. 4, 3807.
akai M., Watanabe Y., Someya T., Araki K., Shibuya M., Niizato K., Oshima K., Kunii Y., Yabe H., Matsumoto J., Wada A., Hino M., Hashimoto T., Hishimoto A., Kitamura N., Iritani S., Shirakawa O., Maeda K., Miyashita A., Niwa S.I., Takahashi H., Kakita A., Kuwano R., Nawa H. (2015) Assessment of copy number variations in the brain genome of schizophrenia patients. Mol. Cytogenet. 8, 46
Smith C.L., Bolton A., Nguyen G. (2010) Genomic and epigenomic instability, fragile sites, schizophrenia and autism. Curr Genomics. 11, 447-469.
Юров Ю.Б., Ворсанова С.Г., Демидова И.А., Кравец В.С., Востриков В.М., Соловьев И.В., Уранова Н.А., Юров И.Ю. (2016) Геномная нестабильность в клетках головного мозга: хромосомный мозаицизм при шизофрении. Журн. Неврологии и Психиатрии им. С.С. Корсакова. 116, 86–91.
Ye C.J., Regan S., Liu G., Alemara S., Heng H.H. (2018) Understanding aneuploidy in cancer through the lens of system inheritance, fuzzy inheritance and emergence of new genome systems. Mol. Cytogenet. 11, 31.
Ворсанова С.Г., Юров Ю.Б., Сильванович А.П., Демидова И.А., Юров И.Ю. (2013) Современные представления о молекулярной генетике и геномике аутизма. Фунд. Исслед. 4, 356–367.
Castellani C.A., Arking D.E. (2020) High-risk, high-reward genetics in ASD. Neuron. 105, 407–410.
Yurov Y.B., Vorsanova S.G., Iourov I.Y., Demidova I.A., Beresheva A.K., Kravetz V.S., Monakhov V.V., Kolotii A.D., Voinova-Ulas V.Y., Gorbachevskaya N.L. (2007) Unexplained autism is frequently associated with low-level mosaic aneuploidy. J. Med. Genet. 44, 521–525.
Biesecker L.G., Spinner N.B. (2013) A genomic view of mosaicism and human disease. Nat. Rev. Genet. 14, 307–320.
D'Gama A.M., Pochareddyk S., Li M., Jamuar S.S., Reiff R.E., Lam A.N., Sestan N., Walsh C.A. (2015) Targeted DNA sequencing from autism spectrum disorder brains implicates multiple genetic mechanisms. Neuron. 88, 910–917.
Koh H.Y., Lee J.H. (2018) Brain somatic mutations in epileptic disorders. Mol. Cells. 41, 881–888.
Park S.M., Lim J.S., Ramakrishina S., Kim S.H., Kim W.K., Lee J., Kang H.C., Reiter J.F., Kim D.S., Kim H.H., Lee J.H. (2018) Brain somatic mutations in MTOR disrupt neuronal ciliogenesis, leading to focal cortical dyslamination. Neuron. 99, 83–97.e7.
Ye Z., McQuillan L., Poduri A., Green T.E., Matsumoto N., Mefford H.C., Scheffer I.E., Berkovic S.F., Hildebrand M.S. (2019) Somatic mutation: the hidden genetics of brain malformations and focal epilepsies. Epilepsy Res. 155, 106161.
Liu G., Ye C.J., Chowdhury S.K., Abdallah B.Y., Horne S.D., Nichols D., Heng H.H. (2018) Detecting chromosome condensation defects in gulf war illness patients. Curr. Genomics. 19, 200–206.
Vorsanova S.G., Zelenova M.A., Yurov Y.B., Iourov I.Y. (2018) Behavioral variability and somatic mosaicism: a cytogenomic hypothesis. Curr. Genomics. 19, 158–162.
Okazaki S., Boku S., Otsuka I., Mouri K., Aoyama S., Shiroiwa K., Sora I., Fujita A., Shirai Y., Shirakawa O., Kokai M., Hishimoto A. (2016) The cell cycle-related genes as biomarkers for schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry. 70, 85–91.
Yang J., Yan B., Fan Y., Yang L., Zhao B., Zhu F., Zheng J., Wang W., Bai L., Zhang F., Ma X. (2019) Identification of schizophrenia related biological pathways across eight brain regions. Behav. Brain Res. 360, 1–6.
Fan Y., Abrahamsen G., McGrath J.J., Mackay-Sim A. (2012) Altered cell cycle dynamics in schizophrenia. Biol. Psychiatry. 71, 129–135.
Pramparo T., Lombardo M.V., Campbell K., Bar-nes C.C., Marinero S., Solso S., Young J., Mayo M., Dale A., Ahrens-Barbeau C., Murray S.S., Lopez L., Lewis N., Pierce K., Courchesne E. (2015) Cell cycle networks link gene expression dysregulation, mutation, and brain maldevelopment in autistic toddlers. Mol. Syst. Biol. 11, 841.
Gumusoglu S.B., Hing B.W.Q., Chilukuri A.S.S., Dewitt J.J., Scroggins S.M., Stevens H.E. (2020) Chronic maternal interleukin-17 and autism-related cortical gene expression, neurobiology, and behavior. Neuropsychopharmacology. 45, 1008–1017.
Wei H., Alberts I., Li X. (2014) The apoptotic perspective of autism. Int. J. Dev. Neurosci. 36, 13–18.
Pan L., Penney J., Tsai L.H. (2014) Chromatin regulation of DNA damage repair and genome integrity in the central nervous system. J. Mol. Biol. 426, 3376–3388.
Paquola A.C.M., Erwin J.A., Gage F.H. (2017) Insights into the role of somatic mosaicism in the brain. Curr. Opin. Syst. Biol. 1, 90–94.
LiCausi F., Hartman N.W. (2018) Role of mTOR complexes in neurogenesis. Int. J. Mol. Sci. 19, 1544.
Ziats C.A., Rennert O.M., Ziats M.N. (2019) Toward a pathway-driven clinical-molecular framework for classifying autism spectrum disorders. Pediatr. Neurol. 98, 46–52.
Putnam C.D., Allen-Soltero S.R., Martinez S.L., Chan J.E., Hayes T.K., Kolodner R.D. (2012) Bioinformatic identification of genes suppressing genome instability. Proc. Natl. Acad. Sci. USA. 109, E3251–E3259.
Iourov I.Y., Vorsanova S.G., Zelenova M.A., Korostelev S.A., Yurov Y.B. (2015) Genomic copy number variation affecting genes involved in the cell cycle pathway: implications for somatic mosaicism. Int. J. Genomics. 2015, 757680.
Sugaya K. (2019) Chromosome instability caused by mutations in the genes involved in transcription and splicing. RNA Biol. 16, 1521–1525.
Crawley J.N., Heyer W.D., LaSalle J.M. (2016) Autism and cancer share risk genes, pathways, and drug targets. Trends Genet. 32, 139–146.
arkkanen E., Meyer U., Dianov G.L. (2016) DNA damage and repair in schizophrenia and autism: implications for cancer comorbidity and beyond. Int. J. Mol. Sci. 17, 856
Ho J., Cruise E.S., Dowling R.J.O., Stambolic V. (2020) PTEN nuclear functions. Cold Spring Harb. Perspect Med. 10, a036079.
Erickson R.P. (2010) Somatic gene mutation and human disease other than cancer: an update. Mutat. Res. 705, 96–106.
Iourov I.Y., Vorsanova S.G., Yurov Y.B. (2010) Somatic genome variations in health and disease. Curr. Genomics. 11, 387–396.
Campbell I.M., Shaw C.A., Stankiewicz P., Lupski J.R. (2015) Somatic mosaicism: implications for disease and transmission genetics. Trends Genet. 31, 382–392.
Wang L., Zhou K., Fu Z., Yu D., Huang H., Zang X., Mo X. (2017) Brain development and Akt signaling: the crossroads of signaling pathway and neurodevelopmental diseases. J. Mol. Neurosci. 61, 379–384.
Thadathil N., Hori R., Xiao J., Khan M.M. (2019) DNA double-strand breaks: a potential therapeutic target for neurodegenerative diseases. Chromosome Res. 27, 345–364.
Hussain R., Zubair H., Pursell S., Shahab M. (2018) Neurodegenerative diseases: regenerative mechanisms and novel therapeutic approaches. Brain Sci. 8, 177.
Howell K.R., Law A.J. (2020) Neurodevelopmental concepts of schizophrenia in the genome-wide association era: AKT/mTOR signaling as a pathological mediator of genetic and environmental programming during development. Schizophr. Res. 217, 95–104.
Martínez-Cué C., Rueda N. (2020) Cellular senescence in neurodegenerative diseases. Front. Cell. Neurosci. 14, 16.
Wilhelm T., Said M., Naim V. (2020) DNA replication stress and chromosomal instability: dangerous liaisons. Genes (Basel). 11, E642.
Дюжикова Н.А., Даев Е.В. (2018) Геном и стресс-реакция у животных и человека. Экологическая генетика. 16, 4–26.
Horne S.D., Chowdhury S.K., Heng H.H. (2014) Stress, genomic adaptation, and the evolutionary trade-off. Front. Genet. 5, 92.
Резник А.М., Костюк Г.П., Ханнанова А.Н. (2016) Проблемы предпосылок шизофрении по данным молекулярно-генетических исследований. Социальная и клиническая психиатрия. 26, 101–108.
Пузырев В.П. (2014) Медицинская патогенетика. Вавиловский Журн. Генет. Селекции. 18, 7–21.
Iourov I.Y., Vorsanova S.G., Yurov Y.B. (2019) The variome concept: focus on CNVariome. Mol. Cytogenet. 12, 52.
Vorsanova S.G., Yurov Y.B., Iourov I.Y. (2020) Dynamic nature of somatic chromosomal mosaicism, genetic-environmental interactions and therapeutic opportunities in disease and aging. Mol. Cytogenet. 13, 16.
uerreiro R., Gibbons E., Tábuas-Pereira M., Kun-Rodrigues C., Santo G.C., Bras J. (2020) Genetic architecture of common non-Alzheimer’s disease dementias. Neurobiol. Dis. 142, 104946
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология