

Молекулярная биология, 2021, T. 55, № 2, стр. 318-337
Вовлеченность генов белков BRСA1-ассоциированного комплекса наблюдения за геномом (BASC) в развитие многофакторной патологии
Н. П. Бабушкина a, *, А. Е. Постригань a, А. Н. Кучер a
a Научно-исследовательский институт медицинской генетики, Томский национальный
исследовательский медицинский центр Российской академии наук
634050 Томск, Россия
* E-mail: nad.babushkina@medgenetics.ru
Поступила в редакцию 12.08.2020
После доработки 05.09.2020
Принята к публикации 07.09.2020
Аннотация
Генетическая компонента многофакторных заболеваний интенсивно изучается на протяжении многих лет, но до сих пор в этой области остается очень много вопросов. В последние годы появились исследования в области “менделевского кода” – гипотезы, постулирующей существование связи между менделевскими (моногенными) и многофакторными патологиями: полиморфные варианты в генах менделевских заболеваний могут оказаться значимыми для многофакторных патологий, в которых задействованы те же биохимические пути. В настоящем обзоре через призму гипотезы “менделевского кода” представлен взгляд на группу генов, которые могут оказаться перспективными генами-кандидатами широкого спектра патологических состояний, а именно, генов белков систем репарации ДНК. В качестве примера рассмотрены белки BRСA1-ассоциированного комплекса наблюдения за геномом (BASC). Обсуждаются основные функции белков этого комплекса в процессах репарации двухцепочечных разрывов ДНК (ATM, MRE11, NBN, RAD50, BRCA1, BLM) и мисматч-репарации (MSH2, MSH6, MLH1, PMS2, RF-C, PCNA); вовлеченность этих белков в функционирование митохондрий; участие белков BASC в развитии адаптивного иммунного ответа. В 13 (из 16) генах белков комплекса BASC уже известны мутации, приводящие к моногенным заболеваниям, а 11 ассоциированы с многофакторными заболеваниями или отдельными биологическими процессами. Общность патогенетически значимых признаков у больных с мутациями во многих генах белков комплекса BASC и у больных с тяжелыми комбинированными иммунодефицитами позволяет предполагать участие полиморфных вариантов в генах белков систем репарации ДНК в развитии многофакторных заболеваний, вероятно, из-за вовлечения в процессы иммунного ответа. Плейотропные эффекты данных генов указывают на возможность их участия в развитии различных состояний организма – как в норме, так и при патологии.
“МЕНДЕЛЕВСКИЙ КОД” МНОГОФАКТОРНОЙ ПАТОЛОГИИ
Многофакторные заболевания составляют основную проблему здравоохранения во всем мире, поэтому формирование новых стратегий профилактики и лечения широко распространенных патологий по-прежнему находится в фокусе внимания исследователей. Наследственная компонента таких болезней сложна, поэтому генетические исследования в этой области актуальны и в настоящее время, хотя подходы к ее изучению с течением времени существенно меняются.
В последние годы значительный интерес вызывает гипотеза менделевского кода, согласно которой существует связь между менделевскими и многофакторными патологиями: многие из частых вариантов, вносящих свой вклад в формирование сложно наследуемых заболеваний, локализованы в генах, причинных для моногенных патологий [1]. Фенотипические эффекты мутаций, приводящих к синдромальной либо моногенной патологии, чрезвычайно сильны, в то время как полиморфные варианты привносят слабые эффекты, модифицируемые как факторами внешней среды, так и генетическим фоном. Поэтому полиморфные варианты в генах менделевских заболеваний могут оказаться значимыми для многофакторных патологий, в которых задействованы те же биохимические пути. Эта идея получила подтверждение и развитие в ряде исследований. Так, например, показано, что более 23% генов, мутации в которых приводят к высокопенетрантным менделевским заболеваниям, ассоциированы и с многофакторной патологией, причем значения OR для рисковых вариантов таких генов выше, чем для рисковых вариантов генов, не приводящих к моногенным заболеваниям, но ассоциированных с многофакторными [2]. Известно, что проведение полногеномных ассоциативных исследований (Genome-Wide Association Studies, GWAS) позволяет выявить замены в непосредственной близости от генов менделевских заболеваний, причем фенотипические проявления изучаемых сложно наследуемых патологий частично перекрываются с проявлениями моногенных заболеваний [3].
Взгляд через призму гипотезы менделевского кода может дать второе дыхание “кандидатному” подходу в изучении генетической компоненты многофакторной патологии. Наше внимание привлекла группа генов белков систем репарации ДНК. Нарушение процессов репарации ДНК приводит к возникновению целого ряда моногенных и онкологических заболеваний [4, 5]. Вовлеченность полиморфизма генов систем репарации ДНК в развитие патологических состояний разной этиологии в настоящее время активно исследуется, но может ли оно считаться достаточно изученным? Основное внимание сосредоточено на выявлении роли генов систем репарации ДНК в развитии онкопатологии. Известно, что мутации в некоторых генах белков различных систем репарации ДНК обусловливают развитие ряда наследственных онкозаболеваний. Полиморфные варианты в генах этой системы ассоциированы с онкопатологией, чувствительностью к воздействию химических веществ и радиации. Изучение именно этих феноменов чаще всего включает и вовлеченность генетического полиморфизма данных систем генов [5]. Вместе с тем, в единичных исследованиях (в том числе и в GWAS) показано участие полиморфизма генов систем репарации в формировании предрасположенности к широкому спектру сложно наследуемых патологий, таких как заболевания сердечно-сосудистой системы [6–9], психические [10, 11], метаболические [12], иммунологические [13–15] и другие нарушения.
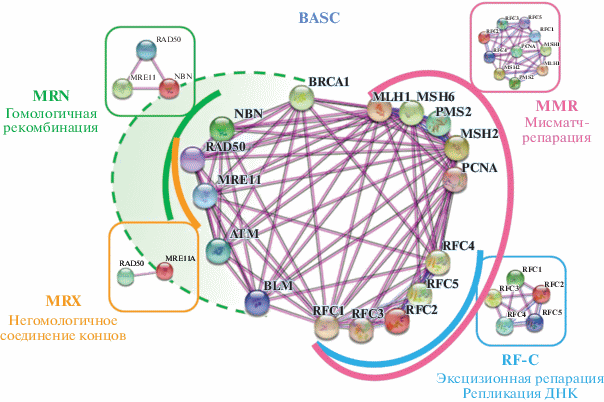
В репарации ДНК участвуют сотни белков. Согласно “GenOntology” в процессы репарации ДНК у человека вовлечены продукты 511 генов (GO: 0006281, фильтры “Homo sapiens”, “experimental evidence”, “UniProt”) [16]. Белки систем репарации образуют, как правило, мультибелковые комплексы, позволяющие исправлять те или иные повреждения ДНК, а также комплексы вне “своих” систем репарации [17, 18]. В настоящем обзоре рассмотрено участие полиморфных вариантов генов белков BRСA1-ассоциированного комплекса наблюдения за геномом (BASC) в формировании предрасположенности к многофакторной патологии. Мультибелковый комплекс BASC содержит опухолевые супрессоры и белки репарации повреждений ДНК – BRCA1, MSH2, MSH6, MLH1, PMS2, RF-C, PCNA, ATM, BLM, MRE11, NBN, RAD50. Комплекс BASC представляет собой динамичную систему, меняющую свой состав как на протяжении клеточного цикла, так и в пределах субклеточных доменов. Белки этого комплекса (рис. 1) участвуют в качестве компонентов в различных системах репарации, а также способны формировать небольшие стабильные субкомплексы, независимые от BRСA1 [19].
Рис. 1.
Комплекс BASC. Сеть построена с помощью оn-line ресурса STRING v:11.0; линии, соединяющие белки в сети, отображают экспериментально доказанные взаимодействия, колокализацию и коэкспрессию. Помимо комплекса BASC указанные белки включены в такие белковые комплексы (все белковые комплексы выделены подчеркиванием), как MRN (MRE11, RAD50, NBN), MRX (MRE11, RAD50), RF-C (RFC1, RFC2, RFC3, RFC4, RFC5), MMR (MLH1, PMS2, MSH2, MSH6, PCNA и RF-C). Наряду с BRCA1, ATM и BLM, белковые комплексы MRN и MRX участвуют в процессах гомологичной рекомбинации; MRX и АТМ – в процессах негомологичного соединения концов. Гетеропентамер RF-C, а также PCNA вовлечены в репликацию ДНК и эксцизионную репарацию. Комплекс MMR осуществляет мисматч-репарацию.
БЕЛКИ КОМПЛЕКСА BASC И РЕПАРАЦИЯ ДВУХЦЕПОЧЕЧНЫХ РАЗРЫВОВ ДНК
Сразу после возникновения двухцепочечного повреждения ДНК близлежащий гистон H2AX очень быстро фосфорилируется (киназой ATM или другими киназами этого семейства), образуя форму gH2AX [20]. Фосфорилирование гистона необходимо для координации сборки репарационного комплекса, а также для дополнительного привлечения необходимых ферментов [21–27]. Нибрин (NBN) в составе комплекса MRN (включающего белки MRE11, RAD50 и NBN) связывается с gH2AX и с ATM, что способствует накоплению факторов репарации ДНК в хроматине, окружающем повреждение, активируя тем самым процесс репарации двухцепочечных разрывов ДНК [28–30]. Комплекс MRN является единственным сенсором активации ATM при дисфункции теломер [31]. Он играет центральную роль в активации ATM-киназы в сайтах двухцепочечных разрывов ДНК [32].
Протеинкиназа ATM, входящая в семейство киназ P13/P14, играет важную роль в путях передачи сигнала о двухцепочечных разрывах в ДНК высших эукариот. ATM опосредованно контролирует наличие двухцепочечных разрывов ДНК через индуцированные ими изменения в структуре хроматина [33]. Белок ATM может находиться как в ядре, так и в цитоплазме, в том числе в пероксисомах, везикулах и митохондриях. Киназа АТМ имеет сотни мишеней в целом ряде сигнальных путей, участвующих в поддержании клеточного и окислительно-восстановительного гомеостаза, регуляции митохондриальных функций [34–36]. Предполагается, что в ответ на появление двухцепочечных разрывов ДНК ATM активируется с помощью двух независимых путей, вовлекающих TP53BP1, с одной стороны, и NBN, с другой [37]. Кроме того, АТМ может активироваться в митохондриях в ответ на окислительный стресс независимо от клеточного ответа на повреждения ДНК, на одноцепочечные разрывы ДНК, на изменения структуры хроматина [38–41]. Мутации в гене ATM приводят к возникновению атаксии-телеангиэктазии (табл. 1), для которой, помимо основных нейродегенеративных нарушений, характерны сосудистые изменения, а также частые респираторные инфекции. Описаны ассоциации вариантов гена АТМ с долгожительством [42, 43], шизофренией [10], коронарным атеросклерозом, сахарным диабетом [6, 44], инсулинорезистентностью [45] и ответом на терапию метформином больных сахарным диабетом типа 2 [46–48].
Таблица 1.
Моногенные заболевания, вызываемые мутациями в генах белков комплекса BASC
| Тип репарации ДНК | Ген | Менделевское заболевание/синдром | OMIM* |
|---|---|---|---|
| Репарация двухцепочечных разрывов ДНК (гомологичная рекомбинация, негомологичное соединение концов) | ATM | Атаксия телеангиоэктазия | 208 900 |
| NBN (NBS1) | Синдром хромосомных поломок Ниймегена | 251 260 | |
| RAD50 | Заболевание, подобное синдрому Ниймегена | 613 078 | |
| MRE11 | ATLD1 (Ataxia-telangiectasia-like disorder 1, заболевание (тип 1), подобное атаксии-телеангиоэктазии) | 604 391 | |
| Гомологичная рекомбинация | BRCA1 | Наследственный рак молочной железы и яичников (familial breast-ovarian cancer-1, BROVCA1) | 604 370 |
| Рак поджелудочной железы (Pancreatic cancer, susceptibility to 4) | 614 320 | ||
| Анемия Фанкони (Fanconi anemia, complementation group S), группа комплементации S | 617 883 | ||
| BLM | Синдром Блума | 210 900 | |
| Мисматч-репарация | MLH1 | Синдром Линча (наследственный неполипозный колоректальный рак типа 2) | 609 310 |
| Синдром Туркота (синдромальный рак мисматч- репарации, сочетание колоректального полипоза и первичных опухолей ЦНС) | 276 300 | ||
| Синдром Муира–Торре (Muir–Torre syndrome, MRTES) | 158 320 | ||
| PMS2 | Синдром Линча (наследственный неполипозный колоректальный рак типа 4) | 614 337 | |
| Синдром Туркота (синдромальный рак мисматч- репарации, сочетание колоректального полипоза и первичных опухолей ЦНС) | 276 300 | ||
| MSH2 | Синдром Линча (наследственный неполипозный колоректальный рак типа 1) | 120 435 | |
| Синдром Туркота (синдромальный рак мисматч- репарации, сочетание колоректального полипоза и первичных опухолей ЦНС) | 276 300 | ||
| Синдром Муира–Торре (Muir–Torre syndrome, MRTES) | 158 320 | ||
| MSH6 | Синдром Линча (наследственный неполипозный колоректальный рак типа 5) | 614 350 | |
| Синдром Туркота (синдромальный рак мисматч- репарации, сочетание колоректального полипоза и первичных опухолей ЦНС) | 276 300 | ||
| Наследственный рак эндометрия | 608 089 | ||
| Мисматч-репарация, эксцизионная репарация | RFC1 | Церебеллярная атаксия, невропатия и синдром вестибулярной арефлексии | 614 575 |
| RFC2 | Локализован в регионе делеции при синдроме Вильямса (RFC2 – один из 28 генов в этом регионе) | 194 050 | |
| PCNA | ATLD2 (Ataxia-telangiectasia-like disorder 2), подобное атаксии-телеангиэктазии заболевание типа 2 | 615 919 |
Примечание. Описаны также мутации в генах MLH1, MSH2, NBN, RAD50 (см. [55]), приводящие к синдромам дефицита антител IgAD и CVID, но не включенные в OMIM.
Комплекс MRN, состоящий из белков MRE11, NBN и RAD50, участвует не только в репарации двухцепочечных разрывов ДНК, но связан и с формированием теломер и проверкой повреждений ДНК [49]. Этот комплекс вовлечен в сигнальные пути, опосредующие развитие врожденного иммунитета [50]. Мутации в гене NBN (NBS1) приводят к синдрому хромосомных поломок Ниймегена, в гене RAD50 – к заболеванию, подобному синдрому Ниймегена (Nijmegen breakage syndrome-like disorder) (табл. 1). При этом полиморфные варианты гена NBN ассоциированы с аутоиммунными заболеваниями [13], нарушениями метаболизма [12] и процессами старения [51]. Варианты гена RAD50 ассоциированы с бронхиальной астмой [14, 52, 53] и сердечно-сосудистой патологией [54]. Мутации гена MRE11 описаны при ATLD1 (Ataxia-telangiectasia-like disorder 1, заболевание (тип 1), сходное с атаксией-телеангиэктазией) (табл. 1) [56, 57], а ассоциации вариантов этого гена – с инфарктом миокарда [7], иммунным старением Т-клеток [58].
Для эффективной передачи сигнала через MRN и ATM требуется еще целый ряд факторов, в том числе TP53BP1 и BRCA1, присутствие которых в комплексе определяет путь, по которому пойдет репарация двухцепочечных разрывов ДНК – гомологичной рекомбинации (HR), опосредуемой BRCA1, или негомологичного воссоединения концов (NHEJ), опосредуемого TP53BP1 [59, 60]. BRCA1 – ядерный фосфопротеин, играющий важную роль в поддержании геномной стабильности в целом и в качестве опухолевого супрессора. Функции BRCA1 чрезвычайно разнообразны: он взаимодействует с компонентами гистондеацетилазного комплекса, обусловливая участие этого фермента в процессах транскрипции, репарации ДНК и рекомбинации [61]; участвует в сборке митотического веретена деления [62]. Cвязываясь с RAD50, BRCA1 блокирует экзонуклеазную активность MRN-комплекса [63, 64]. Из всех генов белков комплекса BASC, BRCA1 изучается наиболее активно – мутации в этом гене ответственны примерно за 40% случаев наследственного рака молочной железы и более чем за 80% случаев наследственного рака молочной железы и яичников (табл. 1) [4, 5]. Полиморфизм гена BRCA1 ассоциирован с характером воспалительного ответа [5, 65].
Один из компонентов системы гомологичной рекомбинации – продукт гена BLM, взаимодействует с топоизомеразой 3α и двумя геликазами того же подсемейства, что и BLM (DExH-бокс-содержащие ДНК- и РНК-геликазы). BLM обладает ДНК-зависимой АТРазной и ДНК-геликазной активностью, вовлечен в процессы рекомбинации, репарации, репликации, сегрегации сестринских хроматид в митозе, участвует в разрешении структуры Холлидея и расплетании G-квадруплексов, в опосредованном рекомбинацией удлинении теломер, регуляции экспрессии генов [66–69]. Мутации в этом гене вызывают развитие синдрома Блума (табл. 1), который характеризуется слишком высокой частотой рекомбинации гомологичных хромосом и сестринских хроматид [70, 71]. Влияя на жизнеспособность клеток и апоптоз, BLM участвует в прогрессии катаракты [72]. На мышиной модели показано, что BLM обеспечивает правильное развитие и функционирование В-лимфоцитов различных подтипов [73]. Показано участие BLM в патогенезе колоректального рака и рака предстательной железы [74, 75].
Следует отметить существование ассоциации полиморфных вариантов в генах NBN, MRE11, RAD50, АТМ, BRСA1 и BLM с различными онкологическими заболеваниями [5, 67, 76–85].
БЕЛКИ КОМПЛЕКСА BASC И МИСМАТЧ-РЕПАРАЦИЯ
Еще один комплекс, ассоциированный с BASC, представлен белками весьма сложной системы пострепликативной мисматч-репарации ДНК (MMR). Основная роль этого комплекса (включающего как минимум 20 белков) заключается в устранении ошибок, связанных с репликацией ДНК (кроме таких мисматчей, как G/T, G/G и A/C, мишенями этой системы служат также и О6-метилгуанин, спаренный с C или Т, межцепочечные сшивки СpG, вызванные действием цисплатина, индуцированные УФ-светом фотопродукты, пуриновые аддукты бензпирена, производные аминофлуорена, 8-оксигуанин, инсерции и делеции) [86]. Кроме того, MMR ингибирует рекомбинацию между неидентичными последовательностями и влияет на многие процессы, связанные с метаболизмом ДНК, включая передачу сигналов о повреждении ДНК, экспансию тринуклеотидных повторов, переключения синтеза классов иммуноглобулинов, соматическую гипермутабельность [87]. MMR, по-видимому, играет двойную роль в ответе на повреждения ДНК (непосредственно мисматч-репарация и передача сигнала о повреждении ДНК), а также вовлечена в активацию индукции апоптоза [88]. В экспериментах in vivo показано, что MMR опосредует клеточную реакцию на дисфункцию теломер через ослабление индукции белка p21 [89].
Мисматчи опознаются и связываются субъединицами MutS, представляющими собой гетеродуплексы трех типов: комплекс MutSα, образуемый белками MSH2 и MSH6, распознает преимущественно одиночные мисматчи и небольшие инсерции/делеции; комплекс MutSβ (MSH2/MSH3), связывающий протяженные инсерции/делеции; и комплекс MutSγ (MSH4/MSH5), участвующий в процессе мейотической рекомбинации [90–93]. С MutS связывается субъединица MutL, также представленная несколькими гетеродимерными формами: MutLα, MutLβ, MutLγ. Белки MLH1 и PMS2 формируют субъединицу MutLα. Показаны физические взаимодействия MutLα с ДНК-полимеразой III, которая присоединяется к участку репарации. MLH1 может также образовывать гетеродимеры с MLH3, формируя субъединицу MutLγ, участвующую в процессе мейоза. PMS2 и MLH3 обладают слабой эндонуклеазной активностью, критичной для функционирования субъединицы MutL. PMS2 не может разрезать метилированную ДНК, поэтому, вероятно, таким путем могут исправляться только de novo нарушения, однако в клетках человека это пока не доказано [17, 18, 93, 94]. MLH1 в комплексе с PMS1 (MutLβ) или PMS2 подавляют гомологичную рекомбинацию, особенно когда это касается коротких гомологичных отрезков ДНК. MutL-комплекс человека к тому же защищает от формирования геномных перестроек, участвуя в процессах, не имеющих отношения непосредственно к мисматч-репарации [95]. Так PMS2 принимает участие в сперматогенезе [96]. Мутации в генах MLH1, MLH3, PMS2, PMS1, MSH2, MSH6 могут быть причиной некоторых наследственных онкологических заболеваний (табл. 1), таких как синдром Линча (наследственный неполипозный колоректальный рак) и синдром Туркота (синдромальный рак мисматч-репарации, синдром полипозной опухоли мозга) [4, 97–101]. Мутации в MLH1 и MSH2 могут быть причиной синдрома Muir–Torre (MRTES), в MLH1, MLH3, MSH2, MSH3, MSH6 – рака эндометрия [97, 102–105]. Показаны ассоциации полиморфных вариантов этих генов и при других онкозаболеваниях [5, 106–111]. Кроме того, выявлены ассоциации полиморфных вариантов генов MLH1 и PMS2 с продолжительностью жизни [112, 113]. Описаны мутации в гене MSH5, приводящие к преждевременному угасанию функции яичников [114, 115], ассоциации полиморфных вариантов с IGAD1 (дефицит IgA) [116]. Ген MSH4 ассоциирован с мужским бесплодием [117]. На мышиной модели показано, что дефицит MSH2 приводит к общегеномному увеличению степени метилирования гистона H3 [118].
Немаловажную роль в мисматч-репарации играет еще один белковый комплекс – репликативный фактор С (RF-C), обладающий АТРазной активностью в присутствии ДНК и ядерного антигена пролиферирующих клеток (PCNA). RF-C представляет собой гетеропентамер, субъединицы которого кодируются генами RFC1, RFC2, RFC3, RFC4 RFC5 [119]. RF-C взаимодействует с 5'-концом последовательности ДНК, затем связывает PCNA, опосредуя его посадку на ДНК. PCNA необходим для сборки комплекса репликации, он стабилизирует комплекс матричной ДНК и ДНК-полимеразы (дельта или эпсилон), что обеспечивает процессивный синтез ДНК [120]. PCNA может связывать также ряд белков (лигазу 1, метилтрансферазу, флэп-эндонуклеазу 1 и т.д.), привлекая их, при необходимости, к репликативному комплексу, он вовлечен в поддержание жизнеспособности пролиферирующих клеток [121–123]. Эндонуклеазная активность MutLα активируется PCNA [124]. RF-С принимает участие в процессах репликации, эксцизионной репарации нуклеотидов, мисматч-репарации, поддержании стабильности теломер [125–127]. Полиморфизм генов PCNA, RFC1, RFC2, RFC3, RFC4, RFC5 активно изучается при онкопатологиях [5, 119, 128–130]. Экспансия пентануклеотидного повтора в гене RFC1 приводит к аутосомно-рецессивной поздневозрастной мозжечковой атаксии (табл. 1) [131]. Выявлена ассоциация полиморфного варианта в гене RFC1 с иммунологическими реакциями [132]. Ген RFC2 локализован в участке делеции при синдроме Вильямса (табл. 1) [133, 134]. Мутации в гене PCNA приводят к развитию ATLD2 (табл. 1) – заболеванию, сходному с атаксией-телеангиэктазией [135]. Уровень PCNA активно используется в качестве белкового маркера пролиферирующих клеток, поэтому изменение его уровня при различных патологиях и в модельных системах отмечено во многих работах (см. [5] и ссылки там). Показана вовлеченность PCNA в патогенез болезни Паркинсона [136, 137].
ГЕНЫ БЕЛКОВ КОМПЛЕКСА BASC И МНОГОФАКТОРНАЯ ПАТОЛОГИЯ
В настоящее время нет достаточно убедительных доказательств участия большинства генов белков этого функционального класса в развитии многофакторной патологии, хотя в этой области накоплено уже немало данных. A. Ciccia и S.J. Elledge, рассматривая 40 синдромов, вызываемых мутациями более чем в 80 генах, пришли к выводу, что дефекты в репарации ДНК в первую очередь влияют на гомеостаз нервной, иммунной и репродуктивной систем, они также могут привести к преждевременному старению или предрасположенности к раку [34]. Действительно, из приведенных этими авторами данных видно, что патология нервной системы наблюдается в 62% случаев синдромов, вызываемых мутациями в генах белков репарации ДНК, повышенная предрасположенность к онкопатологии в – 37%, нарушения в функционировании иммунной системы в – 35%, те или иные признаки преждевременного старения находят у 25% больных [34]. В доступных источниках наибольший объем информации касается вовлеченности генов белков репарации ДНК в развитие онкопатологий (см., например, DisGenet, а также [4, 5, 67, 76–85, 97–101, 106–111, 119, 128–130]). Именно поэтому нам представляется наиболее интересным рассмотреть и другие аспекты.
Дисфункция митохондрий
Хромосомные перестройки и нестабильность генома фактически являются маркерами нарушения процессов репарации ДНК. Однако ряд признаков, общих для некоторых синдромов, связанных с нарушением репарации ДНК (атаксия-телеангиэктазия, синдром Блума, синдром Ниймегена), таких как преждевременное старение, задержка роста, инсулинорезистентность, эндокринные нарушения и иммунодефицит, – могут вызываться дефектами антиоксидантной защиты и, как следствие, накоплением окислительных повреждений ДНК [138]. Эти процессы связаны не только с непосредственным накоплением мутаций в ядрах соматических клеток (процесс корректной репарации ДНК особенно актуален для нейронов), но и с накоплением повреждений в мтДНК. Поскольку мтДНК кодирует важные субъединицы дыхательной цепи митохондрий, то дефекты в ней зачастую приводят к нарушению процессов окислительного фосфорилирования [34, 139]. Дисфункция митохондрий может быть причиной целого ряда патологических состояний, затрагивающих в первую очередь ткани с активным метаболизмом – ЦНС, скелетные мышцы, сердце, а также целый ряд других тканей, вызывая нейродегенерацию, сердечно-сосудистые и метаболические заболевания, старение, рак и т.д. [139, 140]. Нарушения ультраструктуры и функций митохондрий, повышенная продукция митохондриальных АФК характерны для атаксии-телеангиэктазии и сходного с ней ATLD, синдромов Блума, Ниймегена, Кокейна и пигментной ксеродермы [36, 138].
Большинство АФК генерируются во время нормального клеточного метаболизма в основном в митохондриях, пероксисомах и эндоплазматической сети [141]. Непосредственная физическая близость к источникам АФК и отсутствие гистонов приводят к увеличению скорости мутагенеза в митохондриях, которая там в 10–20 раз выше, чем в ядерном геноме [140, 142]. Наиболее стабильное окислительное повреждение ДНК – 8-оксигуанин – во время последующей репликации может образовывать мисматчи с аденином, причем чаще, чем “правильные” пары с цитозином [143]. Таким образом формируется “мутаторный” фенотип, т.е. повреждение ДНК, вызывающее дальнейшее появление мутаций [144, 145]. Окислительные повреждения ДНК исправляются в первую очередь посредством BER (эксцизионная репарация оснований), в митохондриях ошибки также исправляются преимущественно посредством BER. Однако это далеко не единственный механизм. В настоящее время доказано, что в митохондриях активно функционируют также системы NER (эксцизионная репарация нуклеотидов), MMR, а также системы репарации двухцепочечных разрывов ДНК – как основанные на гомологичной рекомбинации, так и на негомологичном воссоединении концов, вероятно, путем сшивания концов, основанном на микрогомологии – MMEJ. Несмотря на то, что детали функционирования этих систем в митохондриях и в ядре могут отличаться [140, 145–147], вполне ожидаемо участие белков комплекса BASC в репарационных процессах в митохондриях.
Некоторые белки комплекса BASC встречаются в митохондриях и необходимы для их нормального функционирования. В частности, на клеточных и животных моделях показано, что потеря активности киназы АТМ приводит к быстрому изменению гомеостаза митохондрий. Фактически это свидетельствует о важной роли АТМ в функционировании митохондрий, не зависящей от повреждений как в ядерной, так и в мтДНК. Предполагается, что увеличение уровня окислительного стресса у мышей с нокаутом гена АТМ обусловлено повышением уровня АФК в митохондриях, возможно, из-за сниженной активности комплекса I [148]. При дефиците комплекса MMR (MLH1 или MSH2) в клетке также снижается активность комплекса I [124, 145], а потеря MLH1 приводит, кроме того, к существенному снижению количества копий мтДНК. При сборке комплекса MMR на мисматчах ДНК происходит взаимодействие MLH1 и ATM, что необходимо для развития дальнейшего клеточного ответа на повреждение ДНК [149]. Дисфункция митохондрий, вызванная дефицитом белков системы мисматч-репарации, может либо опосредоваться именно нарушением взаимодействия MLH1/ATM [145], либо быть независимым событием с неизвестным пока механизмом реализации. Показано участие комплекса MRN и продукта гена BLM в функционировании MMEJ, однако доказательства присутствия и функциональной значимости в митохондриях к настоящему времени получены только для MRE11 и RAD50 [140, 150, 151]. Роль BRCA1 в поддержании стабильности как ядерного, так и митохондриального геномов считается универсальной [152]. Локализация других белков комплекса BASC в митохондриях еще не установлена.
Таким образом, имеются убедительные данные о роли ряда генов систем репарации ДНК в развитии дисфункции митохондрий и ответа на воздействие окислительного стресса. Наиболее полно изучено участие в этих процессах продукта гена АТМ [153–155]. Но вовлеченность в развитие окислительного стресса может объяснять связь гена АТМ с патогенезом ограниченного спектра заболеваний, в частности сердечно-сосудистых или нейродегенеративных. На наш взгляд, более универсальным механизмом, посредством которого белки генов систем репарации ДНК могут вносить вклад в развитие многофакторной патологии различной этиологии, является модификация иммунного ответа и воспаления.
Иммунный ответ
Наличие сопутствующих иммунологических нарушений у пациентов с моногенными заболеваниями, вызываемыми мутациями в генах систем репарации ДНК (наряду с повышенной радиочувствительностью и склонностью к развитию онкопатологии), хорошо известно. Так, у больных атаксией-телеангиэктазией (мутации в гене АТМ) с младенчества наблюдается повышенная частота бактериальных инфекций дыхательных путей, обусловленная нарушением сборки генов иммуноглобулинов и T-клеточных рецепторов (TCR). Аналогичные нарушения обнаружены и у пациентов с синдромами Ниймегена (мутации в гене NBN) и Блума (мутации в гене BLM) [156–158].
Формирование антигенраспознающих участков иммуноглобулинов и ТCR при созревании Т- и В-лимфоцитов происходит в результате V(D)J-рекомбинации (реаранжировки) – соматической негомологичной рекомбинации. Мутации в генах некоторых белков, осуществляющих реаранжировку, приводят к развитию тяжелых комбинированных иммунодефицитов (SCID), обусловленных нарушениями в созревании и/или функционировании лимфоцитов, таких как, например, синдром Оменна. Нарушение реаранжировки – не единственный патогенетический механизм развития SCID, кроме него описаны и такие пути, как накопление токсичных метаболитов, нарушение цитокиновой сигнализации, аномалии тимуса, снижение выживаемости лимфопоэтических клеток-предшественников. Однако стоит подчеркнуть, что повышенная радиочувствительность отмечена только у пациентов со SCID, у которых заболевание вызвано мутациями в генах белков, вовлеченных в V(D)J-рекомбинацию [159–161]. Из белков комплекса BASC в таких перестройках участвуют ATM [162, 163], MRE11 [164], NBN [164–166], RAD50 [164].
На более поздних этапах созревания лимфоцитов формирование дальнейшего разнообразия антител связано с двумя процессами: соматической гипермутацией (somatic hypermutation, SHM) и переключением синтеза классов иммуноглобулинов (class switch recombination, CSR) [167]. Нарушение этих процессов приводит к развитию редких первичных иммунодефицитов, характеризующихся отсутствием продукции переключенного изотипа (IgG, IgA или IgE) [168, 169]. Участие ферментов системы мисматч-репарации в переключении классов иммуноглобулинов описано на мышиной модели еще в 1999 г. [170]. Немного позже показали участие в этом процессе белков MRN-комплекса уже и у человека [171]. Известно, что CSR осуществляется с привлечением, в том числе, белков MRN-комплекса – MRE11, RAD50 и NBN [171, 172], а также MSH2 и MLH1 [170], PMS2 [169, 170], ATM [173], BRCA1 [174]. Все белки системы MMR также вовлечены в процессы соматической гипермутации [175, 176]. Известны мутации в генах системы MMR (MLH1, MSH2) и комплекса MRN (NBN, RAD50), приводящие к развитию синдромов дефицита антител IgAD и CVID, а также полиморфные варианты в генах различных систем репарации ДНК, ассоциированные с нарушениями процесса переключения классов иммуноглобулинов [55].
В результате анализа более миллиона историй болезней Blaire и соавт. составили список, в который вошли более 3000 высоко скоррелированных пар “менделевское заболевание/сложно наследуемое заболевание” [1]. Запрос по ключевому слову “Immunodeficiency” (иммунодефицит) позволил получить список из двух моногенных (иммунодефицит с повышенным уровнем IgM (Immunodeficiency with Increased IgM) и SCID) и 50 многофакторных заболеваний. При этом примерно в трети этих заболеваний ведущим или сопутствующим патогенетическим механизмом является воспаление (например, вирусная инфекция, ревматоидный артрит, болезнь Крона, псориаз, бронхит, угревая сыпь, острый инфаркт миокарда, катаракта, остеоартрит и т.д.).
“Менделевский код”
Опубликован список из 3276 генов, в котором выделяют три группы – гены “комплексных и менделевских заболеваний (СМ)”, “комплексных, но не менделевских (CNM)” и “менделевских, но не комплексных (MNC)” [2]. Из 16 генов комплекса BASC в этом списке представлены восемь, и только один из них – ген ATM – относится к типу “CM”, как приводящий к атаксии-телеангиэктазии и ассоциированный с ревматоидным артритом. Два гена отнесены к категории “CNM” и ассоциированы с астмой/атопическим дерматитом (RAD50) и болезнью Альцгеймера (RFC3). Еще пять генов отнесены к категории “MNC” – MLH1, PMS2, BRCA1, BLM, MSH2 [2]. Действительно, в каждом из этих пяти генов описаны мутации, приводящие к менделевской патологии (табл. 1). Но можно ли утверждать, что эти гены не связаны со сложно наследуемыми заболеваниями, или же такие исследования просто не проводились?
С 13 из 16 белков комплекса BASC связано развитие моногенных заболеваний (табл. 1). Мутации в генах семи из них приводят либо к развитию онкопатологии (BRCA1, MLH1, PMS2, MSH2, MSH6), либо к повышенной предрасположенности к раку (ATM, NBN). Мутации в восьми генах связаны с неврологическими нарушениями (ATM, NBN, RAD50, MRE11, BRCA1 (при анемии Фанкони), BLM, RFC1, PCNA), а девяти – с иммунологическими нарушениями в составе синдрома (иммунодефицитные состояния – ATM, NBN, MRE11, BLM; T-лимфомы при синдроме Туркота – MLH1, MSH2, MSH6, PMS2) или в результате других мутаций в тех же генах (NBN, RAD50, MLH1, MSH2) [4]. Соответственно, можно предположить участие этих генов в развитии многофакторной патологии, в основе которой могут быть нарушения иммунных процессов, окислительный стресс и/или дисфункция митохондрий.
Ориентируясь на гипотезу о вовлеченности полиморфных вариантов в генах белков систем репарации ДНК в развитие многофакторных заболеваний, мы провели пилотное исследование ишемической болезни сердца (ИБС) и бронхиальной астмы (БА) и изучили изменчивость нескольких генов белков комплекса BASC (ATM, MLH1, PMS2, NBN, MRE11) при этих двух патологиях. Выявлен ряд ассоциаций с патогенетически значимыми для развития ИБС признаками: индексом массы тела и наличием периферического атеросклероза (NBN, MLH1), с уровнями липопротеидов низкой плотности (PMS2), с дислипидемией и индексом массы миокарда у больных ИБС (MRE11); с эхокардиографическими показателями (MLH1) [177–179]. Кроме того, выявлены ассоциации генов ATM и MLH1 с БА, показано, что воздействие средовых факторов (курение, паразитозы) значительно модифицирует проявление ассоциаций [178, 180].
Кроме того, используя приведенные в PheWeb данные, мы проанализировали ассоциированность 16 генов белков комплекса BASC с широким спектром фенотипов [181, 182]. PheWeb представляет собой общедоступный репозиторий результатов ассоциативного анализа примерно 28 млн. SNP с 1403 бинарными фенотипическими признаками (phenome-wide association studies, PheWAS). Этот анализ выполнен Биобанком Великобритании на 408961 образце ДНК европеоидов с использованием специально адаптированного статистического подхода [181, 182]. В табл. 2 представлены ассоциации с наибольшими для каждого гена уровнями значимости (в общей сложности 71 патологический фенотип различной этиологии; группировка фенотипов и их названия в тексте приведены согласно PheWeb). Исследования в формате PheWAS стали проводить лишь в последние годы в связи со значительным снижением стоимости генотипирования, но пока еще не получили широкого распространения. При этом данные интернет-ресурса PheWeb являются, пожалуй, самыми крупными.
Таблица 2.
Клинические фенотипы, ассоциированные с регионами генов белков комплекса BASC*
| Ген | Фенотип | Уровень значимости |
|---|---|---|
| BRCA1 | Нарушение кожи и подкожной клетчатки (без других указаний) | 2.10E–07 |
| Перелом лучевой и локтевой костей | 1.30E–06 | |
| Неуточненные заболевания глаз | 1.60E–06 | |
| Симптомы, касающиеся головы и шеи | 1.70E–06 | |
| MSH2 | Другие уточненные диффузные заболевания соединительной ткани | 2.50E–08 |
| Кровоизлияние или гематома | 3.00E–08 | |
| Мышечные дистрофии и другие миопатии | 3.50E–07 | |
| Послеоперационный шок | 4.20E–07 | |
| Дефекты поля зрения | 7.00E–07 | |
| MSH6 | Кровоизлияние или гематома, осложняющие процедуру | 3.00E–08 |
| Заболевания ногтей (без других указаний) | 6.60E–08 | |
| Спайки брюшины или кишечника | 2.90E–07 | |
| Параноидальные расстройства | 3.70E–07 | |
| Врастающий ноготь | 5.10E–07 | |
| Кровоизлияние (без других указаний) | 9.00E–07 | |
| MLH1 | Заболевания волос и волосяных фолликулов | 2.90E–37 |
| Киста сальной железы | 1.80E–35 | |
| Заболевания сальных желез | 2.30E–35 | |
| Гипотиреоз (без других указаний) | 5.20E–07 | |
| PMS2 | Симптомы со стороны дыхательной системы и другие симптомы со стороны грудной клетки | 1.70E–06 |
| Вторичное злокачественное новообразование органов дыхания | 1.80E–06 | |
| Травматическая артропатия | 2.00E–06 | |
| Кровохарканье | 2.20E–06 | |
| RFC1 | Расстройства, связанные с алкоголем | 1.30E–07 |
| Острая почечная недостаточность | 3.10E–06 | |
| Кровоизлияние или гематома, осложняющие процедуру | 3.10E–06 | |
| Атопический/контактный или другой, или неуточненный дерматит | 3.70E–06 | |
| RFC2 | Паховая грыжа | 6.70E–10 |
| Брюшная грыжа | 5.40E–08 | |
| Тучность | 1.00E–06 | |
| Избыточный вес, ожирение и другое переедание | 1.40E–06 | |
| RFC3 | Судороги | 4.00E–08 |
| Диффузное заболевание соединительной ткани, неуточненное | 4.50E–07 | |
| Эпилепсия | 1.10E–06 | |
| Церебральная дегенерация, неуточненная | 1.30E–06 | |
| RFC4 | Отклонения от нормы при исследовании мочи | 2.00E–07 |
| Поверхностная травма без упоминания инфекции | 8.30E–07 | |
| Заболевания периферических сосудов | 1.30E–06 | |
| Рак дыхательной системы | 1.50E–06 | |
| RFC5 | Рак мочевыводящих органов (в том числе почек и мочевого пузыря) | 9.50E–08 |
| Заболевания селезенки | 1.30E–06 | |
| Расстройство мочеиспускания | 2.30E–06 | |
| Осложнения со стороны мочевыводящих путей | 3.20E–06 | |
| PCNA | Другие заболевания сетчатки | 4.90E–08 |
| Эссенциальная гипертензия | 1.80E–07 | |
| Повышенное кровяное давление | 2.00E–07 | |
| Периапикальный абсцесс | 2.10E–07 | |
| Сосудистые изменения и аномалии сетчатки | 4.90E–07 | |
| Рак щитовидной железы | 5.40E–07 | |
| ATM | Лейомиома матки | 8.50E–11 |
| Доброкачественное новообразование матки | 3.00E–10 | |
| Доброкачественное новообразование кожи | 3.00E–07 | |
| Отравление агентами, в первую очередь влияющими на сердечно-сосудистую систему | 7.10E–07 | |
| Старческая катаракта | 7.60E–07 | |
| BLM | Эссенциальная гипертензия | 3.90E–23 |
| Повышенное кровяное давление | 6.50E–23 | |
| Коронарный атеросклероз | 4.90E–20 | |
| Ишемическая болезнь сердца | 1.10E–17 | |
| Стенокардия | 5.20E–11 | |
| Инфаркт миокарда | 1.40E–10 | |
| Другая форма хронической ишемической болезни сердца | 1.40E–10 | |
| Нарушения липидного обмена | 8.80E–09 | |
| Гиперлипидемия | 8.80E–09 | |
| MRE11 | Побочные эффекты седативных средств или других депрессантов центральной нервной системы и анестетиков | 3.40E–07 |
| Слепота и слабое зрение | 5.30E–07 | |
| Церебральный атеросклероз | 1.30E–06 | |
| Нерегулярные менструальные кровотечения | 2.10E–06 | |
| NBN | Алопеция | 5.30E–07 |
| Простой герпес | 6.30E–07 | |
| Фобия | 9.30E–07 | |
| Другие и неуточненные дефекты коагуляции | 1.00E–06 | |
| RAD50 | Астма | 2.70E–16 |
| Повышенное кровяное давление | 3.90E–09 | |
| Эссенциальная гипертензия | 4.00E–09 | |
| Назальные полипы | 4.60E–09 | |
| Воспалительные заболевания кишечника, другие гастроэнтериты и колиты. | 8.00E–08 | |
| Контрактура ладонной фасции (болезнь Дюпюитрена) | 1.50E–07 |
* Составлено по данным PheWeb version 1.1.17. Примечание. В репозитории PheWeb v. 1.1.17 приведены результаты PheWAS Британского биобанка, полученные для европеоидного населения. Методом “случай/контроль” проведен анализ 1403 фенотипов; количество индивидов с разными фенотипами в группе “случай” составляет от 51 до 77977, в группе “контроль” – от 330 366 до 408 908. [182]. Названия фенотипов приведены согласно PheWeb.
Наибольшее количество фенотипов, ассоциированных с генами белков комплекса BASC (табл. 2), относится к нарушениям сердечно-сосудистой системы, таким как инфаркт миокарда, стенокардия, различные варианты ИБС (показаны ассоциации этих фенотипов с геном BLM), коронарный и церебральный атеросклероз (BLM, MRE11), заболевания периферических сосудов (RFC4), кровоизлияния (MSH6), различные варианты гипертензии (BLM, PCNA, RAD50) (табл. 2). В различных источниках описаны также ассоциации как с сердечно-сосудистой патологией в общем, так и ее отдельными эндофенотипами генов ATM, MRE11 и RAD50 [6–9, 54, 177–179]. Большая группа фенотипов, отнесенная к категории дерматологических нарушений (табл. 2), включает алопецию (ассоциация с геном NBN), заболевания волос и волосяных фолликулов (MLH1), ногтей (MSH6), сальных желез (MLH1), нарушения кожи и подкожной клетчатки (BRCA1), диффузные заболевания соединительной ткани (RFC3, MSH2). Большая часть этих признаков встречается при прогерии и/или процессах физиологического старения. Примечательно, что при мутациях в генах АТМ, BML, NBN в комплекс синдромальных признаков входит преждевременное старение [138]. Известны ассоциации гена NBN с процессами старения [51], АТМ, MLH1 и PMS2 – с продолжительностью жизни [42, 43, 112, 113]. К другим дерматологическим нарушениям (согласно PheWeb, табл. 2) отнесены все варианты дерматитов – как неуточненной этиологии, так и атопического/контактного (RFC1), в то время как астма (RAD50) отнесена к заболеваниям дыхательной системы наряду с кровохарканием (PMS2), назальными полипами (RAD50) и другими симптомами, затрагивающими органы дыхания (PMS2) (табл. 2). С БА ассоциированы гены RAD50 [14, 52, 53], АТМ и MLH1 [178, 180]. И атопический дерматит, и атопическая БА относятся к аллергическим заболеваниям (мы упоминали их в контексте иммунологических патологий). Известен ряд ассоциаций рассматриваемых генов с другими иммунологическими нарушениями – аутоиммунными заболеваниями [13, 15], дефицитом IgA [116], иммунным старением T-клеток [58] и т.д. С некоторыми психическими расстройствами (параноидальные расстройства, алкоголизм, фобии и прочие “симптомы, касающиеся головы”), согласно данным PheWeb, ассоциированы гены BRCA1, MSH6, RFC1, NBN (табл. 2); с шизофренией – ген ATM [10]. Вовлечение АТМ-киназы в регуляцию инсулинового ответа [35, 36] логично объясняет ассоциацию этого гена с инсулиновой резистентностью [6, 44–48], сахарным диабетом и ответом больных на лечение метформином. По данным PheWeb (табл. 2) имеются ассоциации и других генов белков репарации ДНК с метаболическими нарушениями (гипотиреоз, MLH1), нарушение липидного обмена и избыточный вес (BLM, RFC2).
Таким образом, анализ результатов ассоциативных исследований генов белков репарации ДНК (включая информацию PheWeb (табл. 2)) позволяет сделать следующие обобщения. Роли генов белков репарации ДНК в патогенезе неоплазий и канцерогенезе посвящено огромное количество публикаций. Кроме того, выявлены ассоциации рассмотренных в настоящем обзоре генов с нарушениями сердечно-сосудистой, пищеварительной, мочеполовой, скелетно-мышечной, гемопоэтической систем, органов чувств, с эндокринными и метаболическими, психическими и неврологическими нарушениями; а также с инфекционными, респираторными, дерматологическими заболеваниями, травматическими повреждениями и реакциями организма на травмы и отравления. Соответственно, рассмотренные гены обладают более широкой сферой компетенции, чем предполагают классические представления. Все это свидетельствует о целесообразности привлечения генов данного функционального класса к исследованию генетической компоненты многофакторных заболеваний. Достаточно четко выраженные плейотропные эффекты рассмотренных генов дают основания предположить возможность их участия в развитии различных состояний организма как в норме, так и при патологии, а следовательно, объяснить ассоциацию маркеров в этих генах с широко распространенными заболеваниями.
Работа выполнена при частичном финансировании Госзадания Министерства науки и высшего образования (№ 075-00603-19-00).
Этические нормы соблюдены. Обзор написан с использованием открытых публикаций.
Авторы сообщают об отсутствии конфликта интересов.
Список литературы
Blair D.R., Lyttle C.S., Mortensen J.M., Bearden C.F., Jensen A.B., Khiabanian H., Melamed R., Rabadan R., Bernstam E.V., Brunak S., Jensen L.J., Nicolae D., Shah N.H., Grossman R.L., Cox N.J., White K.P., Rzhetsky A. (2013) A nondegenerate code of deleterious variants in Mendelian loci contributes to complex disease risk. Cell. 155, 70–80. https://doi.org/10.1016/j.cell.2013.08.030
Spataro N., Rodrıguez J.A., Navarro A., Bosch E. (2017) Properties of human disease genes and the role of genes linked to Mendelian disorders in complex disease aetiology. Hum. Mol. Genet. 26, 489–500. https://doi.org/10.1093/hmg/ddw405
Freund M.K., Burch K.S., Shi H., Mancuso N., Kichaev G., Garske K.M., Pan D.Z., Miao Z., Mohlke K.L., Laakso M., Pajukanta P., Pasaniuc B., Arboleda V.A. (2018) Phenotype-specific enrichment of Mendelian disorder genes near GWAS regions across 62 complex traits. Am. J. Hum. Genet. 103, 535–552. https://doi.org/10.1016/j.ajhg.2018.08.017
OMIM. https://omim.org/
DisGeNet. www.disgenet.org
Li S., Zhang L., Chen T., Tian B., Deng X., Zhao Z., Yuan P., Dong B., Zhang Y., Mo X. (2011) Functional polymorphism rs189037 in the promoter region of ATM gene is associated with angiographically characterized coronary stenosis. Atherosclerosis. 219, 694–697. https://doi.org/10.1016/j.atherosclerosis.2011.08.040
Verschuren J.J., Trompet S., Deelen J., Stott D.J., Sattar N., Buckley B.M., Ford I., Heijmans B.T., Guchelaar H.J., Houwing-Duistermaat J.J., Slagboom P.E., Jukema J.W. (2013) Non-homologous end-joining pathway associated with occurrence of myocardial infarction: gene set analysis of genome-wide association study data. PLoS One. 8, e56262. https://doi.org/10.1371/journal.pone.0056262
Wang L., Chu A., Buring J.E., Ridker P.M., Chasman D.I., Sesso H.D. (2014) Common genetic variations in the vitamin D pathway in relation to blood pressure. Am. J. Hypertens. 27, 1387–1395. https://doi.org/10.1093/ajh/hpu049
Guo Z., Kozlov S., Lavin M.F., Person M.D., Paull T.T. (2010) ATM activation by oxidative stress. Science. 330, 517–521. https://doi.org/10.1126/science.1192912
Zhang F., Xu Y., Liu P., Fan H., Huang X., Sun G., Song Y., Sham P.C. (2008) Association analyses of the interaction between the ADSS and ATM genes with schizophrenia in a Chinese population. BMC Med. Genet. 9, 119. https://doi.org/10.1186/1471-2350-9-119
Qian Y., Chen W., Wu J., Tao T., Bi L., Xu W., Qi H., Wang Y., Guo L. (2010) Association of polymorphism of DNA repair gene XRCC1 with sporadic late-onset Alzheimer’s disease and age of onset in elderly Han Chinese. J. Neurol. Sci. 295, 62–65. https://doi.org/10.1016/j.jns.2010.05.002
He C., Kraft P., Chasman D.I., Buring J.E., Chen C., Hankinson S.E., Pare G., Chanock S., Ridker P.M., Hunter D.J. (2010) A large-scale candidate-gene association study of age at menarche and age at natural menopause. Hum. Genet. 128, 515–527. https://doi.org/10.1007/s00439-010-0878-4
Lin Y.J., Lan Y.C., Wan L., Huang C.M., Lin C.W., Hsueh K.C., Chen D.Y., Lin T.H., Tsai F.J. (2010) The NBS1 genetic polymorphisms and the risk of the systemic lupus erythematosus in Taiwanese patients. J. Clin. Immunol. 30, 643–648. https://doi.org/10.1007/s10875-010-9427-0
Li X., Howard T.D., Zheng S.L., Haselkorn T., Peters S.P., Meyers D.A., Bleecker E.R. (2010 Genome-wide association study of asthma identifies RAD50-IL13 and HLA-DR/DQ regions. J. Allergy Clin. Immunol. 125, 328–335. https://doi.org/10.1016/j.jaci.2009.11.018
Souliotis V.L., Vougas K., Gorgoulis V.G., Sfikakis P.P. (2016) Defective DNA repair and chromatin organization in patients with quiescent systemic lupus erythematosus. Arthritis Res. Ther. 18, 182. https://doi.org/10.1186/s13075-016-1081-3
GenOntology. http://amigo.geneontology.org/amigo/ term/GO:0006281
Kadyrov F.A., Dzantiev L., Constantin N., Modrich P. (2006) Endonucleolytic function of MutL alpha in human mismatch repair. Cell. 126, 297–308. https://doi.org/10.1016/j.cell.2006.05.039
Sacho E.J., Kadyrov F.A., Modrich P., Kunkel T.A., Erie D.A. (2008) Direct visualization of asymmetric adenine-nucleotide-induced conformational changes in MutL alpha. Mol. Cell. 29, 112–121. https://doi.org/10.1016/j.molcel.2007.10.030
Wang Y., Cortez D., Yazdi P., Neff N., Elledge S.J., Qin J. (2000) BASC, a super complex of BRCA1-associated proteins involved in the recognition and repair of aberrant DNA structures. Genes Dev. 14, 927–939.
Daniel R., Ramcharan J., Rogakou E., Taganov K.D., Greger J.G., Bonner W., Nussenzweig A., Katz R.A., Skalka A.M. (2004) Histone H2AX is phosphorylated at sites of retroviral DNA integration but is dispensable for postintegration repair. J. Biol. Chem. 279, 45810–45814. https://doi.org/10.1074/jbc.M407886200
Paull T.T., Rogakou E.P., Yamazaki V., Kirchgessner C.U., Gellert M., Bonner W.M. (2000) A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr. Biol. 10, 886–895. https://doi.org/10.1016/s0960-9822(00)00610-2
Redon C., Pilch D.R., Rogakou E.P., Orr A.H., Lowndes N.F., Bonner W.M. (2003) Yeast histone 2A serine 129 is essential for the efficient repair of checkpoint-blind DNA damage. EMBO J. 4, 678–684. https://doi.org/10.1038/sj.embor.embor871
Furuta T., Takemura H., Liao Z.Y., Aune G.J., Redon C., Sedelnikova O.A., Pilch D.R., Rogakou E.P., Celeste A., Chen H.T., Nussenzweig A., Aladjem M.I., Bonner W.M., Pommier Y. (2003) Phosphorylation of histone H2AX and activation of Mre11, Rad50, and Nbs1 in response to replication-dependent DNA double-strand breaks induced by mammalian DNA topoisomerase I cleavage complexes. J. Biol. Chem. 278, 20303–20312. https://doi.org/10.1074/jbc.M300198200
Lowndes N.F., Toh G.W.(2005) DNA repair: the importance of phosphorylating histone H2AX. Curr. Biol. 15, R99–R102. https://doi.org/10.1016/j.cub.2005.01.029
Morrison A.J., Shen X. (2005) DNA repair in the context of chromatin. Cell Cycle. 4, 568–571.
Ibuki Y., Toyooka T. (2015) Evaluation of chemical phototoxicity, focusing on phosphorylated histone H2AX. J. Radiat. Res. 56, 220–228. https://doi.org/10.1093/jrr/rru105
Georgoulis A., Vorgias C.E., Chrousos G.P., Rogakou E.P. (2017) Genome instability and γH2AX. Int. J. Mol. Sci. 18, E1979. https://doi.org/10.3390/ijms18091979
Kobayashi J., Tauchi H., Sakamoto S., Nakamura A., Morishima K., Matsuura S., Kobayashi T., Tamai K., Tanimoto K., Komatsu K. (2002) NBS1 localizes to gamma-H2AX foci through interaction with the FHA/BRCT domain. Curr. Biol. 12, 1846–1851. https://doi.org/10.1016/s0960-9822(02)01259-9
Roossink F., Wieringa H.W., Noordhuis M.G., ten Hoor K.A., Kok M., Slagter-Menkema L., Hollema H., de Bock G.H., Pras E., de Vries E.G., de Jong S., van der Zee A.G., Schuuring E., Wisman G.B., van Vugt M.A. (2012) The role of ATM and 53BP1 as predictive markers in cervical cancer. Int. J. Cancer. 131, 2056–2066. https://doi.org/10.1002/ijc.27488
Chen C., Zhang L., Huang N.J., Huang B., Kornbluth S. (2013) Suppression of DNA-damage checkpoint signaling by Rsk-mediated phosphorylation of Mre11. Proc. Natl. Acad. Sci. USA. 110, 20605–20610. https://doi.org/10.1073/pnas.1306328110
Dimitrova N., de Lange T. (2009) Cell cycle-dependent role of MRN at dysfunctional telomeres: ATM signaling-dependent induction of nonhomologous end joining (NHEJ) in G1 and resection-mediated inhibition of NHEJ in G2. Mol. Cell. Biol. 29, 5552–5563. https://doi.org/10.1128/MCB.00476-09
Lavin M.F., Kozlov S., Gatei M., Kijas A.W. (2015) ATM-dependent phosphorylation of all three members of the MRN complex: from sensor to adaptor. Biomolecules. 5, 2877–2902. https://doi.org/10.3390/biom5042877
Zgheib O., Huyen Y., DiTullio R.A. Jr., Snyder A., Venere M., Stavridi E.S., Halazonetis T.D. (2005) ATM signaling and 53BP1. Radiother. Oncol. 76, 119–122. https://doi.org/10.1016/j.radonc.2005.06.026
Ciccia A., Elledge S.J. (2010) The DNA damage response: making it safe to play with knives. Mol. Cell. 40, 179–204. https://doi.org/10.1016/j.molcel.2010.09.019
Shiloh Y., Ziv Y. (2013) The ATM protein kinase: regulating the cellular response to genotoxic stress, and more. Nat. Rev. Mol. Cell. Biol. 14, 197–210. https://doi.org/10.1038/nrm3546
Choy K.R., Watters D.J. (2018) Neurodegeneration in ataxia-telangiectasia: multiple roles of ATM kinase in cellular homeostasis. Dev. Dyn. 247, 33–46. https://doi.org/10.1002/dvdy.24522
Mochan T.A., Venere M., DiTullio R.A.Jr., Halazonetis T.D. (2003) 53BP1 and NFBD1/MDC1-Nbs1 function in parallel interacting pathways activating ataxia-telangiectasia mutated (ATM) in response to DNA damage. Cancer Res. 63, 8586–8591.
Morita A., Tanimoto K., Murakami T., Morinaga T., Hosoi Y. (2014) Mitochondria are required for ATM activation by extranuclear oxidative stress in cultured human hepatoblastoma cell line Hep G2 cells. Biochem. Biophys. Res. Commun. 443, 1286–1290. https://doi.org/10.1016/j.bbrc.2013.12.139
Navrkalova V., Kafkova L.R., Divoky V., Pospisilova S. (2015) Oxidative stress as a therapeutic perspective for ATM-deficient chronic lymphocytic leukemia patients. Haematologica. 100, 994–996. https://doi.org/10.3324/haematol.2015.130260
Хороненкова С.В. (2016) Механизмы неканонической активации АТМ-киназы. Успехи биол. химии. 56, 197–210. https://fbras.ru/wp-content/uploads/ 2017/10/Khoronenkova-2016.pdf
Berger N.D., Stanley F.K.T., Moore S., Goodarzi A.A. (2017) ATM-dependent pathways of chromatin remodelling and oxidative DNA damage responses. Philos. Trans. R Soc. Lond. B Biol. Sci. 372, 20160283. https://doi.org/10.1098/rstb.2016.0283
Chen T., Dong B., Lu Z., Tian B., Zhang J., Zhou J., Wu H., Zhang Y., Wu J., Lin P., Zhang J., Xu H., Mo X. (2010) A functional single nucleotide polymorphism in promoter of ATM is associated with longevity. Mech. Ageing Dev. 131, 636–640. https://doi.org/10.1016/j.mad.2010.08.009
Piaceri I., Bagnoli S., Tedde A., Sorbi S., Nacmias B. (2013) Ataxia-telangiectasia mutated (ATM) genetic variant in Italian centenarians. Neurol. Sci. 34, 573–575. https://doi.org/10.1007/s10072-012-1188-5
Schiekofer S., Bobak I., Kleber M.E., Maerz W., Rudofsky G., Dugi K.A., Schneider J.G. (2014) Association between a gene variant near ataxia telangiectasia mutated and coronary artery disease in men. Diab. Vasc. Dis. Res. 11, 60–63. https://doi.org/10.1177/1479164113514232
Joven J., Menendez J.A., Fernandez-Sender L., Espinel E., Rull A., Beltran-Debon R., Rodriguez-Gallego E., Riera-Borrull M., Pedro-Botet J., Alonso-Villaverde C., Camps J., Aragones G. (2013) Metformin: a cheap and well-tolerated drug that provides benefits for viral infections. HIV Med. 14, 233–240. https://doi.org/10.1111/hiv.12000
Florez J.C. (2011) Does metformin work for everyone? A genome-wide association study for metformin response. Curr. Diab. Rep. 11, P. 467–469. https://doi.org/10.1007/s11892-011-0220-0
GoDARTS and UKPDS Diabetes Pharmacogenetics Study Group; Wellcome Trust Case Control Consortium 2, Zhou K., Bellenguez C., Spencer C.C., Bennett A.J., Coleman R.L., Tavendale R., Hawley S.A., Donnelly L.A., Schofield C., Groves C.J., Burch L., Carr F., Strange A., Freeman C., Blackwell J.M., Bramon E., Brown M.A., Casas J.P., Corvin A., Craddock N., Deloukas P., Dronov S., Duncanson A., Edkins S., Gray E., Hunt S., Jankowski J., Langford C., Markus H.S., Mathew C.G., Plomin R., Rautanen A., Sawcer S.J., Samani N.J., Trembath R., Viswanathan A.C., Wood N.W.; MAGIC investigators, Harries L.W., Hattersley A.T., Doney A.S., Colhoun H., Morris A.D., Sutherland C., Hardie D.G., Peltonen L., McCarthy M.I., Holman R.R., Palmer C.N., Donnelly P., Pearson E.R. (2011) Common variants near ATM are associated with glycemic response to metformin in type 2 diabetes. Nat. Genet. 43, P. 117–120. https://doi.org/10.1038/ng.735
van Leeuwen N., Nijpels G., Becker M.L., Deshmukh H., Zhou K., Stricker B.H., Uitterlinden A.G., Hofman A., van’t Riet E., Palmer C.N., Guigas B., Slagboom P.E., Durrington P., Calle R.A., Neil A., Hitman G., Livingstone S.J., Colhoun H., Holman R.R., McCarthy M.I., Dekker J.M., 't Hart L.M., Pearson E.R. (2012) A gene variant near ATM is significantly associated with metformin treatment response in type 2 diabetes: a replication and meta-analysis of five cohorts. Diabetologia. 55, 1971–1977. https://doi.org/10.1007/s00125-012-2537-x
Haber J.E. (1998) The many interfaces of Mre11. Cell. 95, 583–586.
Bhattacharya S., Srinivasan K., Abdisalaam S., Su F., Raj P., Dozmorov I., Mishra R., Wakeland E.K., Ghose S., Mukherjee S., Asaithamby A. (2017) RAD51 interconnects between DNA replication, DNA repair and immunity. Nucl. Acids Res. 45, 4590–4605. https://doi.org/10.1093/nar/gkx126
Pereira-Lopes S., Tur J., Calatayud-Subias J.A., Lloberas J., Stracker T.H., Celada A. (2015) NBS1 is required for macrophage homeostasis and functional activity in mice. Blood. 126, 2502–2510. https://doi.org/10.1182/blood-2015-04-637371
Murk W., Walsh K., Hsu L.I., Zhao L., Bracken M.B., Dewan A.T. (2011) Attempted replication of 50 reported asthma risk genes identifies a SNP in RAD50 as associated with childhood atopic asthma. Hum. Hered. 71, 97–105. https://doi.org/10.1159/000319536
Chen J., Zhang J., Hu H., Jin Y., Xue M. (2015) Polymorphisms of RAD50, IL33 and IL1RL1 are associated with atopic asthma in Chinese population. Tissue Antigens. 86, 443–447. https://doi.org/10.1111/tan.12688
Mathur P., Kaga S., Zhan L., Das D.K., Maulik N. (2005) Antibody-array technique reveals overexpression of important DNA-repair proteins during cardiac ischemic preconditioning. J. Mol. Cell. Cardiol. 38, 99–102. https://doi.org/10.1016/j.yjmcc.2004.11.032
Offer S.M., Pan-Hammarstrom Q., Hammarstrom L., Harris R.S. (2010) Unique DNA repair gene variations and potential associations with the primary antibody deficiency syndromes IgAD and CVID. PLoS One. 5, e12260. https://doi.org/10.1371/journal.pone.0012260
Stewart G.S., Maser R.S., Stankovic T., Bressan D.A., Kaplan M.I., Jaspers N.G., Raams A., Byrd P.J., Petrini J.H., Taylor A.M. (1999) The DNA double-strand break repair gene hMRE11 is mutated in individuals with an ataxia-telangiectasia-like disorder. Cell. 99, 577–587.
Aquino J., Ribeiro V., Alonso I., Ramos F., Vasconcelos M. (2017) Ataxia-telangiectasia like: una adolescente portadora de una nueva variante del gen MRE11A (ataxia telangiectasia-like disorder -a child with a novel variant in MRE11A gene). Rev. Neurol. 65, 143–144.
Li Y., Shen Y., Hohensinner P., Ju J., Wen Z., Goodman S.B., Zhang H., Goronzy J.J., Weyand C.M. (2016) Deficient activity of the nuclease MRE11A induces T cell aging and promotes arthritogenic effector functions in patients with rheumatoid arthritis. Immunity. 45, 903–916. https://doi.org/10.1016/j.immuni.2016.09.013
Yun M.H., Hiom K. (2009) Understanding the functions of BRCA1 in the DNA-damage response. Biochem. Soc. Trans. 37, 597–604. https://doi.org/10.1042/BST0370597
Lee J.H., Goodarzi A.A., Jeggo P.A., Paull T.T. (2010) 53BP1 promotes ATM activity through direct interactions with the MRN complex. EMBO J. 29, 574–585. https://doi.org/10.1038/emboj.2009.372
Yarden R.I., Brody L.C. (1999) BRCA1 interacts with components of the histone deacetylase complex. Proc. Natl. Acad. Sci. USA. 96, 4983–4988.
Joukov V., Groen A.C., Prokhorova T., Gerson R., White E., Rodriguez A., Walter J.C., Livingston D.M. (2006) The BRCA1/BARD1 heterodimer modulates ran-dependent mitotic spindle assembly. Cell. 127, 539–552.
Zhong Q., Chen C.F., Li S., Chen Y., Wang C.C., Xiao J., Chen P.L., Sharp Z.D., Lee W.H. (1999) Association of BRCA1 with the hRad50-hMre11-p95 complex and the DNA damage response. Science. 285, 747–750. https://doi.org/10.1126/science.285.5428.747
Paull T.T., Cortez D., Bowers B., Elledge S.J., Gellert M. (2001) Direct DNA binding by Brca1. Proc. Natl. Acad. Sci. USA. 98, 6086–6091. https://doi.org/10.1073/pnas.111125998
Teoh H., Quan A., Creighton A.K., Bang A.K.W., Singh K.K., Shukla P.C., Gupta N., Pan Y., Lovren F., Leong-Poi H., Al-Omran M., Verma S. (2013) BRCA1 gene therapy reduces systemic inflammatory response and multiple organ failure and improves survival in experimental sepsis. Gene Ther. 20, 51–61. https://doi.org/10.1038/gt.2011.214
Johnson J.E., Cao K., Ryvkin P., Wang L.S., Johnson F.B. (2010) Altered gene expression in the Werner and Bloom syndromes is associated with sequences having G-quadruplex forming potential. Nucl. Acids Res. 38, 1114–1122. https://doi.org/10.1093/nar/gkp1103
Brosh R.M. Jr. (2013) DNA helicases involved in DNA repair and their roles in cancer. Nat. Rev. Cancer. 13, 542–558. https://doi.org/10.1038/nrc3560
Kitano K. (2014) Structural mechanisms of human RecQ helicases WRN and BLM. Front. Genet. 5, 366. https://doi.org/10.3389/fgene.2014.00366
Nguyen G.H., Tang W., Robles A.I., Beyer R.P., Gray L.T., Welsh J.A., Schetter A.J., Kumamoto K., Wang X.W., Hickson I.D., Maizels N., Monnat R.J. Jr, Harris C.C. (2014) Regulation of gene expression by the BLM helicase correlates with the presence of G‑quadruplex DNA motifs. Proc. Natl. Acad. Sci. USA. 111, 9905–9910. https://doi.org/10.1073/pnas.1404807111
Karow J.K., Constantinou A., Li J.L., West S.C., Hickson I.D. (2000) The Bloom’s syndrome gene product promotes branch migration of Holliday junctions. Proc. Natl. Acad. Sci. USA. 97, 6504–6508. https://doi.org/10.1073/pnas.100448097
Lillard-Wetherell K., Machwe A., Langland G.T., Combs K.A., Behbehani G.K., Schonberg S.A., German J., Turchi J.J., Orren D.K., Groden J. (2004) Association and regulation of the BLM helicase by the telomere proteins TRF1 and TRF2. Hum. Mol. Genet. 13, 1919–1932. https://doi.org/10.1093/hmg/ddh193
Xiang J., Kang L., Gao H., Wu J., Qin B., Zhou T., Zhang G., Guan H. (2019) BLM can regulate cataract progression by influencing cell vitality and apoptosis. Exp. Eye Res. 178, 99–107. https://doi.org/10.1016/j.exer.2018.08.022
Babbe H., McMenamin J., Hobeika E., Wang J., Rodig S.J., Reth M., Leder P. (2009) Genomic instability resulting from Blm deficiency compromises development, maintenance, and function of the B cell lineage. J. Immunol. 182, 347–360. https://doi.org/10.4049/jimmunol.182.1.347
Qian X., Feng S., Xie D., Feng D., Jiang Y., Zhang X. (2017) RecQ helicase BLM regulates prostate cancer cell proliferation and apoptosis. Oncol. Lett. 14, 4206–4212. https://doi.org/10.3892/ol.2017.6704
Votino C., Laudanna C., Parcesepe P., Giordano G., Remo A., Manfrin E., Pancione M. (2017) Aberrant BLM cytoplasmic expression associates with DNA damage stress and hypersensitivity to DNA-damaging agents in colorectal cancer. J. Gastroenterol. 52, 327–340. https://doi.org/10.1007/s00535-016-1222-0
Rapakko K., Heikkinen K., Karppinen S.M., Erkko H., Winqvist R. (2007) Germline alterations in the 53BP1 gene in breast and ovarian cancer families. Cancer Lett. 245, 337–340. https://doi.org/10.1016/j.canlet.2006.01.021
Loizidou M.A., Michael T., Neuhausen S.L., Newbold R.F., Marcou Y., Kakouri E., Daniel M., Papadopoulos P., Malas S., Hadjisavvas A., Kyriacou K. (2009) DNA-repair genetic polymorphisms and risk of breast cancer in Cyprus. Breast Cancer Res. Treat. 115, 623–627. https://doi.org/10.1007/s10549-008-0084-4
Schuetz J.M., MaCarthur A.C., Leach S., Lai A.S., Gallagher R.P., Connors J.M., Gascoyne R.D., Spinelli J.J., Brooks-Wilson A.R. (2009) Genetic variation in the NBS1, MRE11, RAD50 and BLM genes and susceptibility to non-Hodgkin lymphoma. BMC Med. Genet. 10, 117. https://doi.org/10.1186/1471-2350-10-117
Pennington K.P., Walsh T., Harrell M.I., Lee M.K., Pennil C.C., Rendi M.H., Thornton A., Norquist B.M., Casadei S., Nord A.S., Agnew K.J., Pritchard C.C., Scroggins S., Garcia R.L., King M.C., Swisher E.M. (2014) Germline and somatic mutations in homologous recombination genes predict platinum response and survival in ovarian, fallopian tube, and peritoneal carcinomas. Clin. Cancer Res. 20, 764–775. https://doi.org/10.1158/1078-0432.CCR-13-2287
Damiola F., Pertesi M., Oliver J., Le Calvez-Kelm F., Voegele C., Young E.L., Robinot N., Forey N., Durand G., Vallee M.P., Tao K., Roane T.C., Williams G.J., Hopper J.L., Southey M.C., Andrulis I.L., John E.M., Goldgar D.E., Lesueur F., Tavtigian S.V. (2014) Rare key functional domain missense substitutions in MRE11A, RAD50, and NBN contribute to breast cancer susceptibility: results from a Breast Cancer Family Registry case-control mutation-screening study. Breast Cancer Res. 16, R58. https://doi.org/10.1186/bcr3669
Janku F., Kaseb A.O., Tsimberidou A.M., Wolff R.A., Kurzrock R. (2014) Identification of novel therapeutic targets in the PI3K/AKT/mTOR pathway in hepatocellular carcinoma using targeted next generation sequencing. Oncotarget. 5, 3012–3022. https://doi.org/10.18632/oncotarget.1687
Ramus S.J., Song H., Dicks E., Tyrer J.P., Rosenthal A.N., Intermaggio M.P., Fraser L., Gentry-Maharaj A., Hayward J., Philpott S., Anderson C., Edlund C.K., Conti D., Harrington P., Barrowdale D., Bowtell D.D., Alsop K., Mitchell G.; AOCS Study Group, Cicek M.S., Cunningham J.M., Fridley B.L., Alsop J., Jimenez-Linan M., Poblete S., Lele S., Sucheston-Campbell L., Moysich K.B., Sieh W., McGuire V., Lester J., Bogdanova N., Durst M., Hillemanns P.; Ovarian Cancer Association Consortium, Odunsi K., Whittemore A.S., Karlan B.Y., Dork T., Goode E.L., Menon U., Jacobs I.J., Antoniou A.C., Pharoah P.D., Gayther S.A. (2015) Germline mutations in the BRIP1, BARD1, PALB2, and NBN genes in women with ovarian cancer. J. Natl. Cancer Inst. 107, pii: djv214. https://doi.org/10.1093/jnci/djv214
Smolarz B., Michalska M.M., Samulak D., Romanowicz H., Wojcik L. (2019) Polymorphism of DNA repair genes via homologous recombination (HR) in ovarian cancer. Pathol. Oncol. Res. 25, 1607–1614. https://doi.org/10.1007/s12253-019-00604-5
Kang Z., Zhu Y., Zhang Q.A., Dong L., Xu F., Zhang X., Guan M. (2019) Methylation and expression analysis of mismatch repair genes in extramammary Paget’s disease. J. Eur. Acad. Dermatol. Venereol. 33, 874–879. https://doi.org/10.1111/jdv.15404
Slavin T.P., Sun C.L., Chavarri-Guerra Y., Sedrak M.S., Katheria V., Castillo D., Herzog J., Dale W., Hurria A., Weitzel J.N. (2020) Older breast cancer survivors may harbor hereditary cancer predisposition pathogenic variants and are at risk for clonal hematopoiesis. J. Geriatr. Oncol. 11, 316–319. https://doi.org/10.1016/j.jgo.2019.09.004
Marra G., Schar P. (1999) Recognition of DNA alterations by the mismatch repair system. Biochem. J. 338, 1–13.
Jiricny J. (2006) The multifaceted mismatch-repair system. Nat. Rev. Mol. Cell. Biol. 7, 335–346. https://doi.org/10.1038/nrm1907
Luo Y., Lin F.T., Lin W.C. (2004) ATM-mediated stabilization of hMutL DNA mismatch repair proteins augments p53 activation during DNA damage. Mol. Cell. Biol. 24, 6430–6444. https://doi.org/10.1128/MCB.24.14.6430-6444.2004
Siegl-Cachedenier I., Munoz P., Flores J.M., Klatt P., Blasco M.A. (2007) Deficient mismatch repair improves organismal fitness and survival of mice with dysfunctional telomeres. Genes Dev. 21, 2234–2247. https://doi.org/10.1101/gad.430107
Paquis-Flucklinger V., Santucci-Darmanin S., Paul R., Saunieres A., Turc-Carel C., Desnuelle C. (1997) Cloning and expression analysis of a meiosis-specific MutS homolog: the human MSH4 gene. Genomics. 44, 188–194.
Bocker T., Barusevicius A., Snowden T., Rasio D., Guerrette S., Robbins D., Schmidt C., Burczak J., Croce C.M., Copeland T., Kovatich A.J., Fishel R. (1999) hMSH5: a human MutS homologue that forms a novel heterodimer with hMSH4 and is expressed during spermatogenesis. Cancer Res. 59, 816–822.
Snowden T., Shim K.S., Schmutte C., Acharya S., Fishel R. (2008) hMSH4-hMSH5 adenosine nucleotide processing and interactions with homologous recombination machinery. J. Biol. Chem. 283, 145–154. https://doi.org/10.1074/jbc.M704060200
Cannavo E., Gerrits B., Marra G., Schlapbach R., Jiricny J. (2007) Characterization of the interactome of the human MutL homologues MLH1, PMS1, and PMS2. J. Biol. Chem. 282, 2976–2986. https://doi.org/10.1074/jbc.M609989200
Raschle M., Marra G., Nystrom-Lahti M., Schar P., Jiricny J. (1999) Identification of hMutLbeta, a hetero-dimer of hMLH1 and hPMS1. J. Biol. Chem. 274, 32368–32375. https://doi.org/10.1074/jbc.274.45.32368
Siehler S.Y., Schrauder M., Gerischer U., Cantor S., Marra G., Wiesmüller L. (2009) Human MutL-complexes monitor homologous recombination independently of mismatch repair. DNA Repair (Amst.). 8, 242–252. https://doi.org/10.1016/j.dnarep.2008.10.011
van Oers J.M., Roa S., Werling U., Liu Y., Genschel J., Hou H. Jr., Sellers R.S., Modrich P., Scharff M.D., Edelmann W. (2010) PMS2 endonuclease activity has distinct biological functions and is essential for genome maintenance. Proc. Natl. Acad. Sci. USA. 107, 13384–13389. https://doi.org/10.1073/pnas.1008589107
Hamilton S.R., Liu B., Parsons R.E., Papadopoulos N., Jen J., Powell S.M., Krush A.J., Berk T., Cohen Z., Tetu B, Burger P.C., Wood P.A., Taqi F., Booker S.V., Petersen G.M., Offerhaus G.J.A., Tersmette A.C., Giardiello F.M., Vogelstein B., Kinzler K.W. (1995) The molecular basis of Turcot’s syndrome. N. Engl. J. Med. 332, 839–847. https://doi.org/10.1056/NEJM199503303321302
Ou J., Rasmussen M., Westers H., Andersen S.D., Jager P.O., Kooi K.A., Niessen R.C., Eggen B.J., Nielsen F.C., Kleibeuker J.H., Sijmons R.H., Rasmussen L.J., Hofstra R.M. (2009) Biochemical characterization of MLH3 missense mutations does not reveal an apparent role of MLH3 in Lynch syndrome. Genes Chromosomes Cancer. 48, 340–350. https://doi.org/10.1002/gcc.20644
Senter L., Clendenning M., Sotamaa K., Hampel H., Green J., Potter J.D., Lindblom A., Lagerstedt K., Thibodeau S.N., Lindor N.M., Young J., Winship I., Dowty J.G., White D.M., Hopper J.L., Baglietto L., Jenkins M.A., de la Chapelle A. (2008) The clinical phenotype of Lynch syndrome due to germline PMS2 mutations. Gastroenterology. 135, 419–428. https://doi.org/10.1053/j.gastro.2008.04.026
Farrell M.P., Hughes D.J., Drost M., Wallace A.J., Cummins R.J., Fletcher T.A., Meany M.A., Kay E.W., de Wind N., Power D.G., Andrews E.J., Green A.J., Gallagher D.J. (2013) Multivariate analysis of MLH1 c.1664T>C (p.Leu555Pro) mismatch repair gene variant demonstrates its pathogenicity. Fam. Cancer. 12, 741–747. https://doi.org/10.1007/s10689-013-9652-9
Rossi L., Le Frere-Belda M.A., Laurent-Puig P., Buecher B., De Pauw A., Stoppa-Lyonnet D., Canlorbe G., Caron O., Borghese B., Colas C., Delhomelle H., Chabbert-Buffet N., Grandjouan S., Lecuru F., Bats A.S. (2017) Clinicopathologic characteristics of endometrial cancer in Lynch syndrome: a French multicenter study. Int. J. Gynecol. Cancer. 27, 953–960. https://doi.org/10.1097/IGC.0000000000000985
Risinger J.I., Umar A., Boyd J., Berchuck A., Kunkel T.A., Barrett J.C. (1996) Mutation of MSH3 in endometrial cancer and evidence for its functional role in heteroduplex repair. Nat. Genet. 14, 102–105.
Simpkins S.B., Bocker T., Swisher E.M., Mutch D.G., Gersell D.J., Kovatich A.J., Palazzo J.P., Fishel R., Goodfellow P.J. (1999) MLH1 promoter methylation and gene silencing is the primary cause of microsatellite instability in sporadic endometrial cancers. Hum. Mol. Genet. 8, 661–666. https://doi.org/10.1093/hmg/8.4.661
Stefansson I., Akslen L.A., MacDonald N., Ryan A., Das S., Jacobs I.J., Salvesen H.B. (2002) Loss of hMSH2 and hMSH6 expression is frequent in sporadic endometrial carcinomas with microsatellite instability: a population-based study. Clin. Cancer Res. 8, 138–143.
Taylor N.P., Powell M.A., Gibb R.K., Rader J.S., Huettner P.C., Thibodeau S.N., Mutch D.G., Goodfellow P.J. (2006) MLH3 mutation in endometrial cancer. Cancer Res. 66, 7502–7508. https://doi.org/10.1158/0008-5472.CAN-06-0248
Chen C.C., Yang S.Y., Liu C.J., Lin C.L., Liaw Y.F., Lin S.M., Lee S.D., Chen P.J., Chen C.J., Yu M.W. (2005) Association of cytokine and DNA repair gene polymorphisms with hepatitis B-related hepatocellular carcinoma. Int. J. Epidemiol. 34, 1310–1318. https://doi.org/10.1093/ije/dyi191
Rubio-Del-Campo A., Salinas-Sanchez A.S., Sanchez-Sanchez F., Gimenez-Bachs J.M., Donate-Moreno M.J., Pastor-Navarro H., Carrion-Lopez P., Escribano J. (2008) Implications of mismatch repair genes hMLH1 and hMSH2 in patients with sporadic renal cell carcinoma. BJU Int. 102, 504–509. https://doi.org/10.1111/j.1464-410X.2008.07581.x
Zhang Y., Shu Y.M., Wang S.F., Da B.H., Wang Z.H., Li H.B. (2010) Stabilization of mismatch repair gene PMS2 by glycogen synthase kinase 3beta is implicated in the treatment of cervical carcinoma. BMC Cancer. 10, 58. https://doi.org/10.1186/1471-2407-10-58
Vilkin A., Niv Y. (2011) Association between hMLH1 hypermethylation and JC virus (JCV) infection in human colorectal cancer (CRC). Clin. Epigenetics. 2, 1–5. https://doi.org/10.1007/s13148-010-0013-3
Shinsato Y., Furukawa T., Yunoue S., Yonezawa H., Minami K., Nishizawa Y., Ikeda R., Kawahara K., Yamamoto M., Hirano H., Tokimura H., Arita K. (2013) Reduction of MLH1 and PMS2 confers temozolomide resistance and is associated with recurrence of glioblastoma. Oncotarget. 4, 2261–2270. https://doi.org/10.18632/oncotarget.1302
Vageli D.P., Zaravinos A., Daniil Z., Dahabreh J., Doukas S.G., Spandidos D.A., Gourgoulianis K.I., Koukoulis G.K. (2013) hMSH2 and hMLH1 gene expression patterns differ between lung adenocarcinoma and squamous cell carcinoma: correlation with patient survival and response to adjuvant chemotherapy treatment. Int. J. Biol. Markers. 27, e400–4. https://doi.org/10.5301/JBM.2012.9420
Kim D.J., Yi S.M., Lee S.Y., Kang H.S., Choi Y.H., Song Y.W., Park S.C. (2006) Association between the MLH1 gene and longevity. Hum. Genet. 119, 353–354. https://doi.org/10.1007/s00439-006-0148-7
Han J., Ryu S., Moskowitz D.M., Rothenberg D., Leahy D.J., Atzmon G., Barzilai N., Suh Y. (2013) Discovery of novel non-synonymous SNP variants in 988 candidate genes from 6 centenarians by target capture and nextgeneration sequencing. Mech. Ageing Dev. 134, 478–485. https://doi.org/10.1016/j.mad.2013.01.005
Mandon-Pepin B., Touraine P., Kuttenn F., Derbois C., Rouxel A., Matsuda F., Nicolas A., Cotinot C., Fellous M. (2008) Genetic investigation of four meiotic genes in women with premature ovarian failure. Eur. J. Endocrinol. 158, 107–115. https://doi.org/10.1530/EJE-07-0400
Guo T., Zhao S., Zhao S., Chen M., Li G., Jiao X., Wang Z., Zhao Y., Qin Y., Gao F., Chen Z.J. (2017) Mutations in MSH5 in primary ovarian insufficiency. Hum. Mol. Genet. 26, 1452–1457. https://doi.org/10.1093/hmg/ddx044
Sekine H., Ferreira R.C., Pan-Hammarstrom Q., Graham R.R., Ziemba B., de Vries S.S., Liu J., Hippen K., Koeuth T., Ortmann W., Iwahori A., Elliott M.K., Offer S., Skon C., Du L., Novitzke J., Lee A.T., Zhao N., Tompkins J.D., Altshuler D., Gregersen P.K., Cunningham-Rundles C., Harris R.S., Her C., Nelson D.L., Hammarstrom L., Gilkeson G.S., Behrens T.W. (2007) Role for Msh5 in the regulation of Ig class switch recombination. Proc. Natl. Acad. Sci. USA. 104, 7193–7198. https://doi.org/10.1073/pnas.0700815104
Aston K.I., Krausz C., Laface I., Ruiz-Castane E., Carrell D.T. (2010) Evaluation of 172 candidate polymorphisms for association with oligozoospermia or azoospermia in a large cohort of men of European descent. Hum. Reprod. 25, 1383–1397. https://doi.org/10.1093/humrep/deq081
Herberg M., Siebert S., Quaas M., Thalheim T., Rother K., Hussong M., Altmuller J., Kerner C., Galle J., Schweiger M.R., Aust G. (2019) Loss of Msh2 and a single-radiation hit induce common, genome-wide, and persistent epigenetic changes in the intestine. Clin. Epigenetics. 11, 65. https://doi.org/10.1186/s13148-019-0639-8
Li Y., Gan S., Ren L., Yuan L., Liu J., Wang W., Wang X., Zhang Y., Jiang J., Zhang F., Qi X. (2018) Multifaceted regulation and functions of replication factor C family in human cancers. Am. J. Cancer Res. 8, 1343–1355. PMID: 30210909
Okumura K., Nogami M., Taguchi H., Dean F.B., Chen M., Pan Z.Q., Hurwitz J., Shiratori A., Murakami Y., Ozawa K., Eki T. (1995) Assignment of the 36.5-kDa (RFC5), 37-kDa (RFC4), 38-kDa (RFC3), and 40-kDa (RFC2) subunit genes of human replication factor C to chromosome bands 12q24.2-q24.3, 3q27, 13q12.3-q13, and 7q11.23. Genomics. 25, 274–278. https://doi.org/10.1016/0888-7543(95)80135-9
Moldovan G.L., Pfander B., Jentsch S. (2007) PCNA, the maestro of the replication fork. Cell. 129, 665–679. https://doi.org/10.1016/j.cell.2007.05.003
Witko-Sarsat V., Mocek J., Bouayad D., Tamassia N., Ribeil J.A., Candalh C., Davezac N., Reuter N., Mouthon L., Hermine O., Pederzoli-Ribeil M., Cassatella M.A. (2010) Proliferating cell nuclear antigen acts as a cytoplasmic platform controlling human neutrophil survival. J. Exp. Med. 207, 2631–2645. https://doi.org/10.1084/jem.20092241
Cazzalini O., Sommatis S., Tillhon M., Dutto I., Bachi A., Rapp A., Nardo T., Scovassi A.I., Necchi D., Cardoso M.C., Stivala L.A., Prosperi E. (2014) CBP and p300 acetylate PCNA to link its degradation with nucleotide excision repair synthesis. Nucl. Acids Res. 42, 8433–8448. https://doi.org/10.1093/nar/gku533
Rashid S. (2017) Targeting the mitochondria for the treatment of MLH1-deficient disease / Queen Mary University of London. PhD Thesis. 1–248. http:// qmro.qmul.ac.uk/xmlui/handle/123456789/30924
KEGG PATHWAY Database. https://www.genome.jp/ kegg/pathway.html
Uchiumi F., Ohta T., Tanuma S. (1996) Replication factor C recognizes 5'-phosphate ends of telomeres. Biochem. Biophys. Res. Commun. 229, 310–315. https://doi.org/10.1006/bbrc.1996.1798
Ohashi E., Tsurimoto T. (2017) Functions of multiple clamp and clamp-loader complexes in eukaryotic. In: DNA Replication. From Old Principles to New Discoveries. Eds Masai H., Foiani M. Adv. Exp. Med. Biol. 135–162. https://doi.org/10.1007/978-981-10-6955-0_7
Liao Y.H., Ren J.T., Zhang W., Zhang Z.Z., Lin Y., Su F.X., Jia W.H., Tang L.Y., Ren Z.F. (2017) Polymorphisms in homologous recombination repair genes and the risk and survival of breast cancer. J. Gene Med. 19, e2988. https://doi.org/10.1002/jgm.2988
Hua Q., Gu X., Chen X., Song W., Wang A., Chu J. (2019) IL-8 is involved in radiation therapy resistance of esophageal squamous cell carcinoma via regulation of PCNA. Arch. Biochem. Biophys. 676, 108158. https://doi.org/10.1016/j.abb.2019.108158
Ye X., Ling B., Xu H., Li G., Zhao X., Xu J., Liu J., Liu L. (2020) Clinical significance of high expression of proliferating cell nuclear antigen in non-small cell lung cancer. Medicine (Baltimore). 99, e19755. https://doi.org/10.1097/MD.0000000000019755
Cortese A., Simone R., Sullivan R., Vandrovcova J., Tariq H., Yau W.Y., Humphrey J., Jaunmuktane Z., Sivakumar P., Polke J., Ilyas M., Tribollet E., Tomaselli P.J., Devigili G., Callegari I., Versino M., Salpietro V., Efthymiou S., Kaski D., Wood N.W., Andrade N.S., Buglo E., Rebelo A., Rossor A.M., Bronstein A., Fratta P., Marques W.J., Zuchner S., Reilly M.M., Houlden H. (2019) Biallelic expansion of an intronic repeat in RFC1 is a common cause of late-onset ataxia. Nat. Genet. 51, 649–658. https://doi.org/10.1038/s41588-019-0372-4
Arora M., Lindgren B., Basu S., Nagaraj S., Gross M., Weisdorf D., Thyagarajan B. (2010) Polymorphisms in the base excision repair pathway and graft-versus-host disease. Leukemia. 24, 1470–1475. https://doi.org/10.1038/leu.2010.139
Peoples R., Perez-Jurado L., Wang Y.K., Kaplan P., Francke U. (1996) The gene for replication factor C subunit 2 (RFC2) is within the 7q11.23 Williams syndrome deletion. Am. J. Hum. Genet. 58, 1370–1373. PMID: 8651315
Martindale D.W., Wilson M.D., Wang D., Burke R.D., Chen X., Duronio V., Koop B.F. (2000) Comparative genomic sequence analysis of the Williams syndrome region (LIMK1-RFC2) of human chromosome 7q11.23. Mamm. Genome. 11, 890–898. https://doi.org/10.1007/s003350010166
Baple E.L., Chambers H., Cross H.E., Fawcett H., Nakazawa Y., Chioza B.A., Harlalka G.V., Mansour S., Sreekantan-Nair A., Patton M.A., Muggenthaler M., Rich P., Wagner K., Coblentz R., Stein C.K., Last J.I., Taylor A.M., Jackson A.P., Ogi T., Lehmann A.R., Green C.M., Crosby A.H. (2014) Hypomorphic PCNA mutation underlies a human DNA repair disorder. J. Clin. Invest. 124, 3137–3146. https://doi.org/10.1172/JCI74593
Zhang Z., Zhang Z., Wang H., Zhang G., Hu D., Xiong J., Xiong N., Wang T., Cao X., Mao L. (2014) Proliferating cell nuclear antigen binds DNA polymerase-β and mediates 1-methyl-4-phenylpyridinium-induced neuronal death. PLoS One. 9, e106669. https://doi.org/10.1371/journal.pone.0106669
Li D.W., Li G.R., Zhang B.L., Feng J.J., Zhao H. (2016) Damage to dopaminergic neurons is mediated by proliferating cell nuclear antigen through the p53 pathway under conditions of oxidative stress in a cell model of Parkinson’s disease. Int. J. Mol. Med. 37, 429–435. https://doi.org/10.3892/ijmm.2015.2430
Maciejczyk M., Mikoluc B., Pietrucha B., Heropolitanska-Pliszka E., Pac M., Motkowski R., Car H. (2017) Oxidative stress, mitochondrial abnormalities and antioxidant defense in ataxia-telangiectasia, Bloom syndrome and Nijmegen breakage syndrome. Redox Biol. 11, 375–383. https://doi.org/10.1016/j.redox.2016.12.030
Greaves L.C., Reeve A.K., Taylor R.W., Turnbull D.M. (2012) Mitochondrial DNA and disease. J. Pathol. 226, 274–286. https://doi.org/10.1002/path.3028
Vasileiou P.V.S., Mourouzis I., Pantos C. (2017) Principal aspects regarding the maintenance of mammalian mitochondrial genome integrity. Int. J. Mol. Sci. 18, 1821. https://doi.org/10.3390/ijms18081821
Gorrini C., Harris I., Mak T. (2013) Modulation of oxidative stress as an anticancer strategy. Nat. Rev. Drug Discov. 12, 931–947. https://doi.org/10.1038/nrd4002
Song S, Pursell Z.F., Copeland W.C., Longley M.J., Kunkel T.A., Mathews C.K. (2005) DNA precursor asymmetries in mammalian tissue mitochondria and possible contribution to mutagenesis through reduced replication fidelity. Proc. Natl. Acad. Sci. USA. 102, 4990–4995. https://doi.org/10.1073/pnas.0500253102
Kalam M.A., Haraguchi K., Chandani S., Loechler E.L., Moriya M., Greenberg M.M., Basu A.K. (2006) Genetic effects of oxidative DNA damages: comparative mutagenesis of the imidazole ring-opened formamidopyrimidines (Fapy lesions) and 8-oxo-purines in simian kidney cells. Nucl. Acids Res. 34, 2305–2315. https://doi.org/10.1093/nar/gkl099
Martin S.A., Hewish M., Sims D., Lord C.J., Ashworth A. (2011) Parallel high-throughput RNA interference screens identify PINK1 as a potential therapeutic target for the treatment of DNA mismatch repair-deficient cancers. Cancer Res. 71, 1836–1848. https://doi.org/10.1158/0008-5472.CAN-10-2836
Rashid S., Freitas M.O., Cucchi D., Bridge G., Yao Z., Gay L., Williams M., Wang J., Suraweera N., Silver A., McDonald S.A.C., Chelala C., Szabadkai G., Martin S.A. (201) MLH1 deficiency leads to deregulated mitochondrial metabolism. Cell Death Dis. 10, 795. https://doi.org/10.1038/s41419-019-2018-y
Storr S.J., Woolston C.M., Martin S.G. (2012) Base excision repair, the redox environment and therapeutic implications. Curr. Mol. Pharmacol. 5, 88–101. PMID: .22122466
Зиновкина Л.А. (2018) Механизмы репарации митохондриальной ДНК млекопитающих. Биохимия. 83, 349–367.
Valentin-Vega Y.A., Maclean K.H., Tait-Mulder J., Milasta S., Steeves M., Dorsey F.C., Cleveland J.L., Green D.R., Kastan M.B. (2012) Mitochondrial dysfunction in ataxia-telangiectasia. Blood. 119, 1490–1500. https://doi.org/10.1182/blood-2011-08-373639
Brown K.D., Rathi A., Kamath R., Beardsley D.I., Zhan Q., Mannino J.L., Baskaran R. (2003) The mismatch repair system is required for S-phase checkpoint activation. Nat. Genet. 33, 80–84. https://doi.org/10.1038/ng1052
Tadi S.K., Sebastian R., Dahal S., Babu R.K., Choudhary B., Raghavan S.C. (2016) Microhomology-mediated end joining is the principal mediator of double-strand break repair during mitochondrial DNA lesions. Mol. Biol. Cell. 27, 223–235. https://doi.org/10.1091/mbc.E15-05-0260
Seol J.H., Shim E.Y., Lee S.E. (2018) Microhomology-mediated end joining: good, bad and ugly. Mutat. Res. 809, 81–87. https://doi.org/10.1016/j.mrfmmm.2017.07.002
Coene E.D., Hollinshead M.S., Waeytens A.A., Schelfhout V.R., Eechaute W.P., Shaw M.K., Van Oostveldt P.M., Vaux D.J. (2005) Phosphorylated BRCA1 is predominantly located in the nucleus and mitochondria. Mol. Biol. Cell. 16, 997–1010. https://doi.org/10.1091/mbc.e04-10-0895
Rotman G., Shiloh Y. (1997) Ataxia-telangiectasia: is ATM a sensor of oxidative damage and stress? Bioessays. 19, 911–917.
Okuno Y., Nakamura-Ishizu A., Otsu K., Suda T., Kubota Y. (2012) Pathological neoangiogenesis dependson oxidative stress regulation by ATM. Nat. Med. 18, 1208–1216. https://doi.org/10.1038/nm.2846
Ambrose M., Gatti R.A. (2013) Pathogenesis of ataxia-telangiectasia: the next generation of ATM functions. Blood. 121, 4036–4045. .https://doi.org/10.1182/blood-2012-09-456897
Bonilla F.A.1, Geha R.S. (2003) Primary immunodeficiency diseases. J. Allergy Clin. Immunol. 111, S571–581. https://doi.org/10.1067/mai.2003.86
Geha R.S., Notarangelo L.D., Casanova J.L., Chapel H., Conley M.E., Fischer A., Hammarstrom L., Nonoyama S., Ochs H.D., Puck J.M., Roifman C., Seger R., Wedgwood J.; International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. (2007) Primary immunodeficiency diseases: an update from the International Union of Immunological Societies Primary Immunodeficiency Diseases Classification Committee. J. Allergy Clin. Immunol. 120, 776–794. https://doi.org/10.1016/j.jaci.2007.08.053
Bonilla F.A., Barlan I., Chapel H., Costa-Carvalho B.T., Cunningham-Rundles C., de la Morena M.T., Espinosa-Rosales F.J., Hammarstrom L., Nonoyama S., Quinti I., Routes J.M., Tang M.L., Warnatz K. (2016) International Consensus Document (ICON): common variable immunodeficiency disorders. J. Allergy. Clin. Immunol. Pract. 4, 38–59. https://doi.org/10.1016/j.jaip.2015.07.025
Lim M.S., Elenitoba-Johnson K.S. (2004) The molecular pathology of primary immunodeficiencies. J. Mol. Diagn. 6, 59–83. https://doi.org/10.1016/S1525-1578(10)60493-X
Cirillo E., Giardino G., Gallo V., D’Assante R., Grasso F., Romano R., Di Lillo C., Galasso G., Pignata C. (2015) Severe combined immunodeficiency–an update. Ann. N.Y. Acad. Sci. 1356, 90–106. https://doi.org/10.1111/nyas.12849
Kumrah R., Vignesh P., Patra P., Singh A., Anjani G., Saini P., Sharma M., Kaur A., Rawat A. (2019) Genetics of severe combined immunodeficiency. Genes Dis. 24, 52–61. https://doi.org/10.1016/j.gendis.2019.07.004
Bredemeyer A.L., Sharma G.G., Huang C.Y., Helmink B.A., Walker L.M., Khor K.C., Nuskey B., Sullivan K.E., Pandita T.K., Bassing C.H., Sleckman B.P. (2006) ATM stabilizes DNA double-strand-break complexes during V(D)J recombination. Nature. 442, 466–470. https://doi.org/10.1038/nature04866
Zha S., Guo C., Boboila C., Oksenych V., Cheng H.-L., Zhang Y., Wesemann D.R., Yuen G., Patel H., Goff P.H., Dubois R.L., Alt F.W. (2011) ATM damage response and XLF repair factor are functionally redundant in joining DNA breaks. Nature. 469, 250–254. https://doi.org/10.1038/nature09604
Helmink B.A., Bredemeyer A.L., Lee B.-S., Huang C.-Y., Sharma G.G., Walker L.M., Bednarski J.J., Lee W.-L., Pandita T.K., Bassing C.H., Sleckman B.P. (2009) MRN complex function in the repair of chromosomal Rag-mediated DNA double-strand breaks. J. Exp. Med. 206, 669–679. https://doi.org/10.1084/jem.20081326
Chen H.T., Bhandoola A., Difilippantonio M.J., Zhu J., Brown M.J., Tai X., Rogakou E.P., Brotz T.M., Bonner W.M., Ried T., Nussenzweig A. (2000) Response to RAG-mediated VDJ cleavage by NBS1 and gamma-H2AX. Science. 290, 1962–1965. https://doi.org/10.1126/science.290.5498.1962
Saidi A., Li T., Weih F., Concannon P., Wang Z.Q. (2010) Dual functions of Nbs1 in the repair of DNA breaks and proliferation ensure proper V(D)J recombination and T-cell development. Mol. Cell. Biol. 30, 5572–5581. https://doi.org/10.1128/MCB.00917-10
Альтшулер Е.П., Серебряная Д.В., Катруха А.Г. (2010) Получение рекомбинантных антител и способы увеличения их аффинности. Успехи биол. химии. 50, 203–258.
Peron S., Pan-Hammarstrom Q., Imai K., Du L., Taubenheim N., Sanal O., Marodi L., Bergelin-Besançon A., Benkerrou M., de Villartay J.P., Fischer A., Revy P., Durandy A. (2007) A primary immunodeficiency characterized by defective immunoglobulin class switch recombination and impaired DNA repair. J. Exp. Med. 204, 1207–1216. https://doi.org/10.1084/jem.20070087
Peron S., Metin A., Gardes P., Alyanakian M.A., Sheridan E., Kratz C.P., Fischer A., Durandy A. (2008) Human PMS2 deficiency is associated with impaired immunoglobulin class switch recombination. J. Exp. Med. 205, 2465–2472. https://doi.org/10.1084/jem.20080789
Schrader C.E., Edelmann W., Kucherlapati R., Stavnezer J. (1999) Reduced isotype switching in splenic B cells from mice deficient in mismatch repair enzymes. J. Exp. Med. 190, 323–330. https://doi.org/10.1084/jem.190.3.323
Lahdesmaki A., Taylor A.M., Chrzanowska K.H., Pan-Hammarstrom Q. (2004) Delineation of the role of the Mre11 complex in class switch recombination. J. Biol. Chem. 279, 16479–16487. https://doi.org/10.1074/jbc.M312796200
Dinkelmann M., Spehalski E., Stoneham T., Buis J., Wu Y., Sekiguchi J.M., Ferguson D.O. (2009) Multiple functions of MRN in end-joining pathways during isotype class switching. Nat. Struct. Mol. Biol. 16, 808–813. https://doi.org/10.1038/nsmb.1639
Pan-Hammarstrom Q., Dai S., Zhao Y., van Dijk-Hard I.F., Gatti R.A., Borresen-Dale A.L., Hammarstrom L. (2003) ATM is not required in somatic hypermutation of VH, but is involved in the introduction of mutations in the switch mu region. J. Immunol. 170, 3707–3716. https://doi.org/10.4049/jimmunol.170.7.3707
Yun M.H., Hiom K. (2009) Understanding the functions of BRCA1 in the DNA-damage response. Biochem. Soc. Trans. 37, 597–604. https://doi.org/10.1042/BST0370597
Chahwan R., Edelmann W., Scharff M.D., Roa S. (2011) Mismatch-mediated error prone repair at the immunoglobulin genes. Biomed. Pharmacother. 65, 529–536. https://doi.org/10.1016/j.biopha.2011.09.001
Chahwan R., van Oers J.M., Avdievich E., Zhao C., Edelmann W., Scharff M.D., Roa S. (2012) The ATPase activity of MLH1 is required to orchestrate DNA double-strand breaks and end processing during class switch recombination. J. Exp. Med. 209, 671–678. https://doi.org/10.1084/jem.20111531
Бабушкина Н.П., Постригань А.Е., Кучер А.Н. (2018) Вовлеченность генов систем репарации ДНК в развитие сердечно-сосудистой патологии. Сб.: “Молекулярно-биологические технологии в медицинской практике”. Под ред. Масленникова А.Б. Новосибирск: Академиздат. 48–62.
Babushkina N., Postrigan A., Kucher A. (2018) Association rs189037 in the ATM gene with bronchial asthma taking into account environmental factors. BGRS\SB-2018: systems biology and biomedicine (SBioMed-2018) Symp. (2018) Novosibirsk, Russia, Abstracts book, P. 16.
Бабушкина Н.П., Постригань А.Е., Кучер А.Н. (2018) Вовлеченность гена MLH1 в формирование клинических особенностей ИБС. Российский национальный конгресс кардиологов. “Новые технологии – в практику здравоохранения”. Москва, Россия, сборник тезисов. С. 1003.
Бабушкина Н.П., Постригань А.Е., Хитринская Е.Ю., Кучер А.Н. (2019) Средовые эффекты на ассоциации генов белков систем репарации ДНК с бронхиальной астмой. VII Съезд Вавиловского общества генетиков и селекционеров (ВОГиС). (2019) Санкт-Петербург, Россия. Сборник тезисов. С. 788
Zhou W., Nielsen J.B., Fritsche L.G., Dey R., Gabrielsen M.E., Wolford B.N., LeFaive J., VandeHaar P., Gagliano S.A., Gifford A., Bastarache L.A., Wei W.Q., Denny J.C., Lin M., Hveem K., Kang H.M., Abecasis G.R., Willer C.J., Lee S. (2018) Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet. 50, 1335–1341. https://doi.org/10.1038/s41588-018-0184-y
PheWeb v. 1.1.17. http://pheweb.sph.umich.edu/SAIGE-UKB
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология