

Молекулярная биология, 2021, T. 55, № 4, стр. 626-633
Аутофагия не влияет на ответ линии клеток хронического миелоидного лейкоза, устойчивой к иматинибу, на ингибиторы тирзинкиназ
S. Baykal-Köse a, *, H. Efe b, Z. Yüce b
a Izmir Biomedicine and Genome Center (IBG), Dokuz Eylul University Health Campus, Inciralti-Balcova
35340 Izmir, Turkey
b Dokuz Eylul University, Medical School, Medical Biology Department
35330 Izmir, Turkey
* E-mail: seda.baykalkose@gmail.com
Поступила в редакцию 30.06.2020
После доработки 27.08.2020
Принята к публикации 03.09.2020
Аннотация
Предполагается, что аутофагия – эволюционно консервативный процесс, с помощью которого компоненты цитоплазмы поступают в лизосомы для деградации, играет роль в развитии устойчивости клеток хронического миелоидного лейкоза к иматинибу. Хронический миелоидный лейкоз – клональное миелопролиферативное заболевание, возникает в результате неопластической трансформации гемопоэтических стволовых клеток. С использованием Bcr-Abl-независимой и устойчивой к иматинибу субпопуляции клеток K562 (K562-IR), полученной нами ранее, показано, что в присутствии иматиниба аутофагия запускается посредством трансформации LC3-I/II, активации белка p62 и накопления закисленных вакуолей в клетках K562, чувствительных к ингибитору тирозинкиназ. При этом в клеточной линии K562-IR, устойчивой к иматинибу и независимой от Bcr-Abl, аутофагия не индуцируется. Это исследование проведено в связи с сочетанным применением ингибиторов тирозинкиназ и ингибиторов аутофагии. Показано, что такие комбинации могут быть неэффективными в случае хронического миелоидного лейкоза, устойчивого к ингибиторам тирозинкиназ. Это может быть связано с отсутствием активации клеточного стресса и аутофагии при обработке таких клеток ингибиторами тирозинкиназ, поскольку для выживания им не требуется передача сигналов Bcr-Abl.
ВВЕДЕНИЕ
Изучение механизмов гибели и выживания клеток имеет решающее значение для разработки новых стратегий и терапевтических средств, применяемых при злокачественных заболеваниях. Хронический миелоидный лейкоз (ХМЛ) – клональное миелопролиферативное заболевание, обусловленное неопластической трансформацией гемопоэтических стволовых клеток [1]. В качестве первичного генетического дефекта при ХМЛ идентифицирован химерный ген BCR-ABL. Этот ген расположен на филадельфийской хромосоме, а его образование связано с транслокацией t(9; 22) (q34; q11). Онкобелок Bcr-Abl, обладающий аномальной тирозинкиназной активностью, связан с чрезмерным расширением миелоидного клеточного компартмента. Онкобелок Bcr-Abl обеспечивает повышенную активацию сигналов выживания в лейкозных клетках, которые способствуют увеличению клеточной пролиферации, придают клеткам злокачественный фенотип и делают их устойчивыми к запрограммированной гибели [2]. Ингибитор тирозинкиназ – мезилат иматиниба – произвел революцию в терапии ХМЛ, поскольку подавляет активность онкобелка Bcr-Abl. Иматиниб блокирует сигналы выживания, обеспечиваемые Bcr-Abl, что в конечном итоге приводит к гибели лейкозных клеток [3]. Программируемой гибели клеток, которая обеспечивает элиминацию поврежденных или нежелательных клеток, отводится важная роль в поддержании клеточного гомеостаза. Программируемая гибель клеток осуществляется преимущественно каспазозависимым путем, известным как апоптоз. Известны еще два механизма клеточной смерти – некроз и зависимая от аутофагии гибель клеток [4]. Аутофагия – это эволюционно консервативный процесс, при котором компоненты цитоплазмы доставляются в лизосомы для деградации. Известны три типа аутофагии: микроаутофагия, аутофагия, опосредованная шаперонами, и макроаутофагия, причем преобладает последняя форма (далее именуемая аутофагией) [5]. Аутофагия отвечает за деградацию белков и обновление органелл. Части цитоплазмы и органеллы заключаются в аутофагосомы – двухмембранные везикулы, локализованные в цитозоле, которые придают клеткам характерный вакуолизированный вид [6]. Иными словами, комплекс Atg1/ULK1 передает сигналы, обеспечивающие его перенос на изолирующую мембрану, инициируя образование аутофагосом [5, 7]. Формирование аутофагосом, в свою очередь, рекрутирует комплекс PI3K/PtdIns(3)K класса III (содержит белки Vps34, Vps15, Atg6 и Atg14), необходимый для продукции фосфатидилинозитол-3-фосфата. После привлечения других молекул происходит сборка убиквитинподобных систем конъюгации – Atg8/LC3 и Atg12-Atg5, необходимых для удлинения и созревания аутофагосомы [8].
В дальнейшем аутофагосома сливается с лизосомой и образует аутолизосому, в которой цитозольный материал разрушается и используется в последующей рециклизации.
Аутофагия способствует выживанию клеток за счет повторного использования основных клеточных компонентов. Сначала аутофагию описывали как механизм гибели клеток. Сегодня связь между аутофагией и гибелью клеток считается более сложной, причем неясно, можно ли рассматривать аутофагию как основную причину гибели клеток. Детальное установление вклада аутофагии в гибель клеток (без участия альтернативных путей) все еще остается предметом дискуссий [5, 9]. Присутствие аутофагосом в погибающих клетках может отражать активацию компенсаторных механизмов, связанных с выживанием [10]. Вопреки ожиданиям, в определенных условиях повышенная вакуолизация может не коррелировать с усилением аутофагии, но скорее отражать дефект созревания аутофагосом, приводящий к снижению аутофагии [11].
Устойчивость к иматинибу и другим ингибиторам тирозинкиназ (ИТК) при некоторых вариантах ХМЛ представляет существенную проблему для успешной терапии опухолей, что отчасти связывают с индукцией аутофагии [12, 13]. Высказано предположение, что аутофагия, обеспечивающая выживание клеток в ответ на терапию ИТК, связана с развитием устойчивости к иматинибу. В ряде исследований отмечено увеличение гибели лейкозных клеток в ответ на применение иматиниба в комбинации с ингибиторами аутофагии [14]. Нами проведен сравнительный анализ активации индуцированной иматинибом аутофагии в чувствительных и резистентных к этому ингибитору клетках ХМЛ.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Культуры клеток. BCR-ABL-позитивные клетки ХМЛ человека линии К562 приобретены из коллекции клеток АТСС. Резистентный субклон K562-IR получен в нашей лаборатории с помощью клональной селекции при увеличении доз иматиниба. Начальная доза составляла 0.1 мкМ, а конечная – 10 мкМ, что в 2 раза выше концентрации в сыворотке крови пациентов, получавших иматиниб в суточной дозе 400 мг [15]. Клетки культивировали в среде RPMI 1640 с добавлением 10% фетальной сыворотки крупного рогатого скота (FBS), пенициллина G (1 ед./мл) и стрептомицина (1 мг/мл) при 37°C и 5% CO2. Клетки K562-IR непрерывно культивировали с добавлением 10 мкМ иматиниба, если не указано иное. В некоторых опытах клетки K562-IR дважды промывали PBS для элиминации препарата.
Аннексин V/исследование мембранного потенциала митохондрий. Набор Invitrogen Mitochondrial Membrane Potential/Annexin V Apoptosis Kit (#V35116, “Invitrogen”, США) использовали в соответствии с инструкциями производителя. Набор содержит красители MitoTracker® Red и Alexa® Fluor 488 annexin V для проточной цитометрии (“BD FACSCanto II”, “BD Biosciences”, США) для определения апоптотических изменений мембранного потенциала митохондрий и клеточной мембраны.
Определение активности каспазы-3. Анализ проводили с помощью набора Sigma Fluorimetric Caspase-3 Assay Kit (#CASP3F, “Sigma-Aldrich”, США) в соответствии с инструкциями производителя. Набор содержит субстрат каспазы-3, Ac-DEVD-AMC, который лизируется активной каспазой-3. Высвобождение AMC, коррелирующее с активностью каспазы-3, определяли по интенсивности флуоресценции, измеряемой с использованием флуоресцентного спектрофотометра (Biotek Synergy HT fluorescence spectrophotometer, 360/460 nm).
Вестерн-блот-анализ. Белковые лизаты получали из клеток K562, которые обрабатывали 1 мкМ иматиниба в течение 24 ч, клеток K562-IR, не обработанных иматинибом, и клеток K562-IR, обработанных иматинибом в концентрации 10 мкМ. С этой целью к осадкам клеток добавляли буфер RIPA (150 мМ NaCl, 1.5% NP-40, 0.5% дезоксихолат натрия, 0.1% SDS, 50 мМ Трис-HCl, рН 8.0; добавляли 1 мМ PMSF и ингибиторы фосфатазы и протеазы) и хранили их при температуре –80°C для дальнейшего использования. Белки анализировали количественно с помощью бицинхонинового метода (BCA). Белки разделяли с помощью электрофореза в полиакриламидном геле и переносили на PVDF-мембраны “мокрым” способом. Мембраны обрабатывали раствором специфических антител с последующей визуализацией с использованием чувствительных рентгеновских пленок (“Carestкeam Health”, США). Использовали следующие антитела: к B-актину HRP (“Sigma”, A3854), к eIF4E (“Cell Signaling”, США, #2067), к LC3A/B (“Cell signaling”, #4108), к фосфо-mTOR (“Cell signaling”, #2971), к фосфо-c-Abl (Tyr245) (“Cell signaling”, #2868), к фосфо-CrkL (Tyr207) (“Cell signaling”, #3181).
Количественное определение аутофагических вакуолей путем окрашивания акридиновым оранжевым. Клетки осаждали центрифугированием, ресуспендировали и инкубировали в среде, содержащей 0.5 мкг/мл акридинового оранжевого (“Invitrogen” A3568), в течение 15 мин. Акридиновый оранжевый удаляли, клетки дважды промывали и ресуспендировали в PBS. Количество клеток с низким значением pH в везикулах (AVO) определяли с помощью проточной цитометрии. Клетки анализировали с использованием BD FACSCanto II и программного обеспечения BD FACSDiva. Проанализированы не менее 20 000 клеток. Все эксперименты выполнены в трех повторностях.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Клетки K562-IR не зависят от Bcr-Abl и не поддаются воздействию высоких доз иматиниба
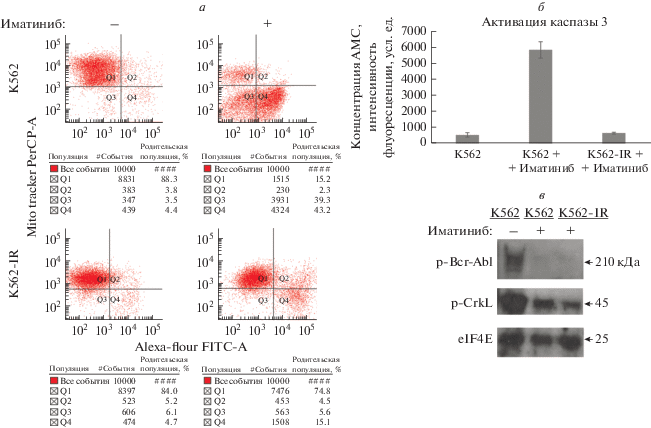
Клетки K562-IR были получены путем клональной селекции клеток, культивируемых в присутствии иматиниба. В предыдущей работе нами показано, что такие клетки устойчивы к первым трем поколениям ИТК [16]. Перед проведением дальнейших экспериментов определили жизнеспособность и ответ на иматиниб резистентных (K562-IR) и чувствительных (K562) клеток ХМЛ после замораживания–оттаивания. Методом проточной цитометрии мы подтвердили, что иматиниб в концентрации 1 мкМ индуцирует гибель клеток К562, тогда как клетки К562-IR, непрерывно культивируемые в присутствии 10 мкМ иматиниба, были устойчивыми к этому препарату; через 72 ч жизнеспособность клеток составила 16 и 86% соответственно (рис. 1а). Активация каспазы в клетках K562, обработанных 1 мкМ иматиниба, оказалась в ~15 раз выше, чем в резистентных клетках K562-IR, обработанных 10 мкМ иматиниба в течение 72 ч (рис. 1б). Была ли внутриклеточная концентрация иматиниба достаточной для ингибирования сигнального пути Bcr-Abl как в клетках K562, так и в клетках K562-IR? Чтобы определить это, нами проведен вестерн-блот-анализ фосфорилированной формы Bcr-Abl (p-Bcr-Abl) и его молекулы-мишени p-CrkL. В обеих клеточных линиях аутофосфорилирование BCR-ABL и фосфорилирование CrkL было подавлено иматинибом, что подтверждает его активность в клетках K562 и K562-IR. Клеточная линия K562-IR в отличие от клеток K562 была устойчивой к иматинибу, несмотря на подавление сигнального пути Bcr-Abl. Это означает, что Bcr-Abl больше не обеспечивает активацию сигнала выживания клеток K562-IR (рис. 1в).
Рис. 1.
Клетки К562-IR, устойчивые к иматинибу и Bcr-Abl-независимые. а – Анализ жизнеспособности клеток К562 и К562-IR с помощью проточной цитометрии (а) и активация каспаз в этих клетках (б). Клетки K562 обрабатывали 1 мкМ иматинибом в течение 72 ч, клетки K562-IR непрерывно культивировали в присутствии 10 мкМ иматиниба. в – Вестерн-блот-анализ пути Bcr-Abl. Клетки K562 обрабатывали 1 мкМ иматинибом в течение 24 ч, а клетки K562-IR непрерывно культивировали с 10 мкМ иматиниба. В качестве контроля загрузки использовали eIF4E.
Иматиниб не вызывает образования закисленных вакуолей в резистентных клетках ХМЛ
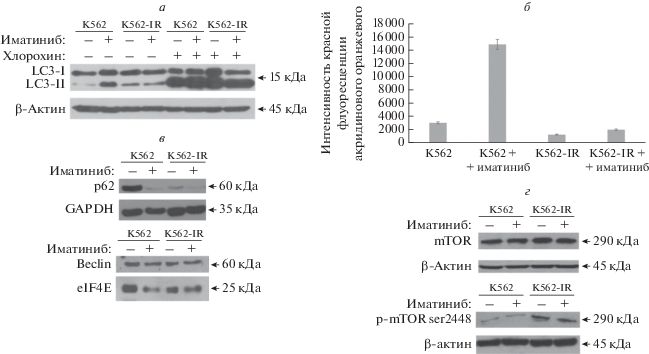
В ходе аутофагии возрастает закисление вакуолей, что можно обнаружить при окрашивании акридиновым оранжевым. Добавление 1 мкМ иматиниба к чувствительным клеткам K562 резко увеличивает образование закисленных вакуолей, в то время как в резистентной линии K562-IR этого не наблюдали даже после обработки 10 мкМ иматиниба в течение 72 ч (рис. 2б).
Рис. 2.
Активация аутофагии в клетках K562 и K562-IR. Клетки K562 обрабатывали 1 мкМ иматиниба в течение 24 ч, а клетки K562-IR непрерывно культивировали с 10 мкМ иматиниба. Образцы без иматиниба: клетки K562-IR дважды промывали PBS и культивировали в течение 24 ч перед экспериментом. а – Вестерн-блот-анализ количества белков LC3-I/II. Положительный контроль ингибирования аутофагии – клетки, обработанные 50 мкМ хлорохином в течение 24 ч перед экспериментом. б – Окрашивание клеток K562 и K562-IR акридиновым оранжевым. в, г – Вестерн-блот-анализ уровней экспрессии p62, Beclin, mTOR и p-mTOR.
Иматиниб не индуцирует аутофагию в резистентных клетках ХМЛ
Количество белков LC3-I/II, p62, Beclin 1 и mTOR определяли с помощью Вестерн-блотинга в чувствительных и резистентных клеточных линиях ХМЛ с иматинибом или без него. При активации аутофагии и образования фагофоры субъединица LC3-I трансформируется в LC3-II. При исследовании ингибирования аутофагии в качестве контроля использовали хлорохин, который ингибирует аутофагию, повышая рН в лизосомах, что приводит к подавлению слияния аутофагосом с лизосомами [17]. При добавлении иматиниба уровень LC3-II в клетках K562 повышается, тогда как в резистентных клетках K562-IR изменения не выявлены (рис. 2а). Хлорохин индуцировал трансформацию LC-I в LC-II как в чувствительных, так и в резистентных клетках независимо от иматиниба. В резистентных клетках количество белка p62 оставалось на низком уровне независимо от наличия или отсутствия иматиниба, в то время как клетки K562 содержали много р62. Изменения количества белка p62 (SQSTM1/sequestosome 1) учитываются при анализе полиубиквитинированных агрегатов белков. p62/SQSTM1 взаимодействует с полиубиквитинированными агрегатами белка через убиквитинсвязывающий домен и с LC3 через его LC3-связывающий домен, тем самым направляя их разрушение в аутолизосоме [18]. С другой стороны, количество белка Beclin1 – маркера индукции аутофагии, было сопоставимым в обеих клеточных линиях независимо от обработки иматинибом. Общий уровень белка mTOR в обеих клеточных линиях не изменялся при обработке иматинибом. Однако в резистентной клеточной линии уровни активной формы mTOR, p-mTOR Ser2448 были выше, чем в чувствительных клетках. Обработка иматинибом не влияла на уровень экспрессии mTOR и p-mTOR в обеих клеточных линиях.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Иматиниб – первый таргетный ингибитор онкобелка Bcr-Abl, введение которого в клиническую практику повлияло на прогноз и лечение ХМЛ. Тем не менее, развитие устойчивости к иматинибу и другим ИТК остается нерешенной терапевтической задачей. Резистентность к ИТК может возникать в результате действия механизмов, связанных или не связанных с Bcr-Abl [15, 19, 20]. Кроме того, пул лейкозных клеток, нечувствительных к ИТК, содержит лейкозные стволовые клетки, которые не зависят от Bcr-Abl и способны выживать при длительной терапии ИТК [21–23]. Ранее показали, что аутофагия активируется при воздействии ИТК на клетки ХМЛ и способствует развитию лекарственной устойчивости [12]. Предположили, что применение ИТК может запускать различные сигнальные пути в лейкозных CD34+CD38− стволовых клетках. Так, ингибирование BCR-ABL с помощью ИТК может вызвать гибель клеток-предшественников, но при этои индуцировать защитную аутофагию [24]. Таким образом, сочетание ИТК с ингибиторами аутофагии рассматривается в качестве новой терапевтической стратегии при ХМЛ.
Аутофагия играет цитопротективную роль, способствуя выживанию раковых клеток. Результат, к которому приводит активация аутофагии, во многом зависит от индивидуального состояния клетки. Осуществляя катаболический процессинг и удаляя поврежденные макромолекулы, аутофагия защищает протеом клетки и обеспечивает поступление строительных блоков в условиях стресса [10, 25]. Аутофагия уменьшает также повреждение ДНК, поскольку удаляет поврежденные органеллы (митохондрии и пероксисомы) с последующим снижением выработки активных форм кислорода [26]. Ингибирование аутофагии в раковых клетках в сочетании с воздействием облучения и обработкой алкилирующими агентами, приводит к усиленной гибели клеток [27]. В ряде исследований показано, что химиотерапевтические агенты индуцируют в раковых клетках как апоптоз, так и аутофагию. [28]. По-видимому, аутофагию, сопутствующую гибели клеток, можно рассматривать как проявление стрессовой реакции, направленной на защиту клеток от повреждения. Аутофагосомы, обнаруженные в таких клетках, могут указывать на попытку предотвращения их гибели [9]. Иматиниб дозозависимым образом активирует аутофагию в клетках млекопитающих, независимо от вида, ткани или статуса иммортализации клеток [29]. При ХМЛ сигналы Bcr-Abl приводят к активации сигнального пути mTOR, который, как известно, ингибирует аутофагию. Кроме того, Bcr-Abl фосфорилирует Beclin-1, что приводит к подавлению аутофагии [30].
Блокирование сигнального пути Bcr-Abl иматинибом и другими ИТК ингибирует путь mTOR [31, 32] и его взаимодействие с белками аутофагии, что приводит к активации аутофагии, которая фактически обеспечивает выживание клеток в ответ на индуцированный иматинибом стресс, способствующий развитию резистентности к терапии. Кроме того, показано, что ингибитор сигнального пути mTOR, отрицательно регулирующего аутофагию, подавляет рост Bcr-Abl-позитивных клеток [33].
Индуцированная ИТК гибель лейкозных клеток может быть усилена путем целенаправленного воздействия на белки аутофагии [34–36]. Новые терапевтические стратегии, направленные на ингибирование аутофагии в клетках ХМЛ, потенциально могут усилить ИТК-индуцированную гибель лейкозных клеток. Показано, что ингибирование аутофагии с помощью гидроксихлорохина (HCQ) способствует повышению чувствительности клеток ХМЛ, включая лейкозные стволовые клетки, к ИТК. Исследование CHOICES (CHlOroquine and Imatinib Combination to Eliminate Stem cells) представляет собой рандомизированное испытание эффективности иматиниба, а также комбинации хлорохина с иматинибом при ХМЛ. Это первое клиническое испытание, направленное на иингибирование аутофагии при ХМЛ. Последние данные свидетельствуют о том, что нетоксичные концентрации ингибиторов аутофагии первого поколения в сыворотке крови некоторых пациентов не способны обеспечить необходимый уровень ингибирования аутофагии, однако ингибиторы аутофагии второго поколения могут улучшить исход заболевания [14].
Однако установлены не все механизмы устойчивости к ИТК, природа устойчивости к ИТК у некоторых пациентов до сих пор неизвестна. Описанный нами терапевтический подход, заключающийся в использовании комбинации ИТК с ингибитором аутофагии, основан на предположении, что иматиниб вызывает клеточный стресс, запускающий аутофагию, которая, в свою очередь, действует в качестве реакции выживания. При активации Bcr-Abl-независимых механизмов клеточного стресса описанные стратегии не будут эффективными.
В этом исследовании мы показали, что ингибирование аутофагии не влияет на жизнеспособность резистентной к ИТК “нестволовой” популяции клеток ХМЛ, полученных из клеточной линии К562. Устойчивые к ИТК клетки линии K562-IR получены методом клональной селекции под воздействием иматиниба. Эти клетки устойчивы к ИТК трех первых поколений. О биологических характеристиках этих клеток сообщалось ранее [16]. Нами показано, что иматиниб блокирует передачу сигналов Bcr-Abl как в клетках K562, так и в клетках K562-IR. Клеточная линия K562-IR устойчива к иматинибу, несмотря на ингибирование Bcr-Abl, что свидетельствует о независимости механизма резистентности от Bcr-Abl. Обработка иматинибом приводила к резкому повышению закисления вакуолей в клетках K562, в то время как в клетках K562-IR такой эффект не наблюдался. Хлорохин индуцировал увеличение количества белка LC3-II как в чувствительных, так и в резистентных клетках независимо от обработки иматинибом. Применение только иматиниба (без хлорохина) приводило к увеличению количества LC3-II в клетках K562, по сравнению с K562-IR. В резистентных клетках (независимо от наличия или отсутствия иматиниба) содержание белка р62 находилось на низком уровне, в то время как в клетках К562 – на высоком. Нами показано также, что синтез активных форм mTOR, р-mTOR Ser2448 был выше в резистентной клеточной линии. Сигнальный путь mTOR – это путь выживания, который регулируется сигналами стресса. В условиях клеточного стресса он ингибируется, что приводит к активации аутофагии, тогда как активный сигнальный путь ингибирует аутофагию, препятствуя формированию и созреванию аутофагосомы [37]. Активный сигнальный путь mTOR коррелирует с отсутствием аутофагии в клетках K562-IR. Полученные нами результаты позволяют предложить модель резистентности, согласно которой обработка клеток ИТК не усиливает аутофагию в клетках ХМЛ, так как не вызывает клеточный стресс. Мы предполагаем, что применение ИТК в комбинации с ингибиторами аутофагии при ХМЛ с Bcr-Abl-независимыми механизмами резистентности к ИТК, не даст желаемого терапевтического эффекта. Выявление независимых от Bcr-Abl механизмов резистентности к ИТК будет способствовать разработке новых терапевтических стратегий при ХМЛ.
Работа получили финансирование Фонда научных исследований Dokuz Eylul University (грант № 2009KBSAG29).
В работе не использовали людей или животных в качестве объектов исследования.
Авторы заявляют об отсутствии конфликта интересов.
Zeynep Yüce и Seda Baykal-Köse участвовали в разработке концепции и дизайна исследования. Подготовка материалов, сбор данных и анализ выполнены Seda Baykal-Köse и Hande Efe. Первый вариант рукописи написан Seda Baykal-Köse и Zeynep Yüce. Все авторы прочитали и одобрили окончательный вариант рукописи.
Список литературы
Quintás-Cardama A., Cortes J. (2009) Molecular biology of bcr-abl1-positive chronic myeloid leukemia. Blood. 113, 1619–1630.
Deininger M.W.N., Goldman J.M., Melo J.V (2000) The molecular biology of chronic myeloid leukemia. Blood. 96, 3343–3356.
Hochhaus A., Larson R.A., Guilhot F., Radich J.P., Branford S., Hughes T.P., Baccarani M., Deininger M.W., Cervantes F., Fujihara S., Ortmann C.-E., Menssen H.D., Kantarjian H., O’Brien S.G., Druker B.J. (2017) Long-term outcomes of imatinib treatment for chronic myeloid leukemia. N. Engl. J. Med. 376, 917–927.
D’Arcy M.S. (2019) Cell death: a review of the major forms of apoptosis, necrosis and autophagy. Cell. Biol. Int. 43, 582–592.
Denton D., Kumar S. (2019) Autophagy-dependent cell death. Cell Death Differ. 26, 605–616.
Levine B., Kroemer G. (2008) Autophagy in the pathogenesis of disease. Cell. 132, 27–42.
Yu L., Chen Y., Tooze S.A. (2018) Autophagy pathway: cellular and molecular mechanisms. Autophagy. 14, 207–215.
Geng J., Klionsky D.J. (2008) The Atg8 and Atg12 ubiquitin-like conjugation systems in macroautophagy. EMBO Rep. 9, 859–864.
Bialik S., Dasari S.K., Kimchi A. (2018) Autophagy-dependent cell death – where, how and why a cell eats itself to death. J. Cell Sci. 131, jcs215152.
Shidoji Y. (2014) Geranylgeranoic acid induces incomplete autophagy but leads to the accumulation of autophagosomes in human hepatoma cells. In: Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging. Elsevier Inc. 3, 173–185.
Kroemer G., Levine B. (2008) Autophagic cell death: the story of a misnomer. Nat. Rev. Mol. Cell. Biol. 9(12), 1–17.
Crowley L.C., O’Donovan T.R., Nyhan M.J., McKenna S.L. (2013) Pharmacological agents with inherent anti-autophagic activity improve the cytotoxicity of imatinib. Oncol. Rep. 29, 2261–2268.
Shao S., Li S., Qin Y., Wang X., Yang Y., Bai H., Zhou L., Zhao C., Wang C. (2014) Spautin-1, a novel autophagy inhibitor, enhances imatinib-induced apoptosis in chronic myeloid leukemia. Int. J. Oncol. 44, 1661–1668.
Mishima Y., Terui Y., Mishima Y., Taniyama A., Kuniyoshi R., Takizawa T., Kimura S., Ozawa K., Hatake K. (2008) Autophagy and autophagic cell death are next targets for elimination of the resistance to tyrosine kinase inhibitors. Cancer Sci. 99, 2200–2208.
Deininger M.W.N., Druker B.J. (2003) Specific targeted therapy of chronic myelogenous leukemia with imatinib. Pharmacol. Rev. 55, 401–423.
Baykal-Köse S., Acikgoz E., Yavuz A.S., Geyik Ö.G., Ateş H., Sezerman O.U., Özsan G.H., Yüce Z. (2020) Adaptive phenotypic modulations lead to therapy resistance in chronic myeloid leukemia cells. PLoS One. 15(2), e0229104. https://doi.org/10.1371/journal.pone.0229104
Mauthe M., Orhon I., Rocchi C., Zhou X., Luhr M., Hijlkema K.J., Coppes R.P., Engedal N., Mari M., Reggiori F. (2018) Chloroquine inhibits autophagic flux by decreasing autophagosome-lysosome fusion. Autophagy. 14, 1435–1455.
Barth S., Glick D., Macleod K.F. (2010) Autophagy: assays and artifacts. J. Pathol. 221, 117–124.
O’Hare T., Zabriskie M.S., Eiring A.M., Deininger M.W. (2012) Pushing the limits of targeted therapy in chronic myeloid leukaemia. Nat. Rev. Cancer. 12, 513–526.
Bubnoff N., Duyster J. (2010) Chronic myelogenous leukemia: treatment and monitoring. Dtsch. Arztebl. Int. 107, 114–121.
Chen Y., Li S. (2013) Molecular signatures of chronic myeloid leukemia stem cells. Biomark. Res. 1, 21.
Zhang H., Li S. (2013) Molecular mechanisms for survival regulation of chronic myeloid leukemia stem cells. Protein Cell. 4, 186–196.
Hu Y., Li S. (2016) Survival regulation of leukemia stem cells. Cell Mol. Life Sci. 73, 1039–1050.
Calabretta B., Salomoni P. (2011) Inhibition of autophagy: A new strategy to enhance sensitivity of chronic myeloid leukemia stem cells to tyrosine kinase inhibitors. Leuk. Lymphoma. 52, 54–59.
Shen S., Codogno P. (2016) The role of autophagy in cell death. In: Autophagy: Cancer, Other Pathologies, Inflammation, Immunity, Infection, and Aging. Elsevier Acad. Press, 139–154.
Auberger P., Puissant A. (2017) Autophagy, a key mechanism of oncogenesis and resistance in leukemia. Blood. 129, 547–552.
Chen N., Karantza V. (2011) Autophagy as a therapeutic target in cancer. Cancer Biol. Ther. 11, 157–168.
Ricci M.S., Zong W. (2006) Chemotherapeutic approaches for targeting cell death pathways. Oncologist. 11, 342–357.
Ertmer A., Huber V., Gilch S., Yoshimori T., Erfle V., Duyster J., Elsässer H.P., Schäzl H.M. (2007) The anticancer drug imatinib induces cellular autophagy. Leukemia. 21, 936–942.
Yu C., Gorantla S.P., Müller-Rudorf A., Müller T.A., Kreutmair S., Albers C., Jakob L., Lippert L.J., Yue Z., Engelhardt M., Follo M., Zeiser R., Huber T.B., Duyster J., Illert AL (2020) Phosphorylation of BECLIN-1 by BCR-ABL suppresses autophagy in chronic myeloid leukemia. Haematologica. 105, 1285–1293.
Kharas M.G., Janes M.R., Scarfone V.M., Lilly M.B., Knight Z.A., Shokat K.M., Fruman D.A. (2008) Ablation of PI3K blocks BCR-ABL leukemogenesis in mice, and a dual PI3K/mTOR inhibitor prevents expansion of human BCR-ABL+ leukemia cells. J. Clin. Invest. 118, 3038–3050.
Klejman A., Rushen L., Morrione A., Slupianek A., Skorski T. (2002) Phosphatidylinositol-3 kinase inhibitors enhance the anti-leukemia effect of STI571. Oncogene. 21, 5868–5876.
Mohi M.G., Boulton C., Gu T.L., Sternberg D.W., Neuberg D., Griffin J.D., Gilliland D.G., Neel B.G. (2004) Combination of rapamycin and protein tyrosine kinase (PTK) inhibitors for the treatment of leukemias caused by oncogenic PTKs. Proc. Natl. Acad. Sci. USA. 101, 3130–3135
Baquero P., Dawson A., Mukhopadhyay A., Kuntz E.M., Mitchell R., Olivares O., Ianniciello A., Scott M.T., Dunn K., Nicastri M.C., Winkler J.D., Michie A.M., Ryan K.M., Halsey C., Gottlieb E., Keaney E.P., Murphy L.O., Amaravadi R.K., Holyoake T.L., Helga-son G.V. (2019) Targeting quiescent leukemic stem cells using second generation autophagy inhibitors. Leukemia. 33, 981–994.
Yu Y., Yang L., Zhao M., Zhu S., Kang R., Vernon P., Tang D., Cao L. (2012) Targeting microRNA-30a-mediated autophagy enhances imatinib activity against human chronic myeloid leukemia cells. Leukemia. 26, 1752–1760.
Bellodi C., Lidonnici M.R., Hamilton A., Helgason G.V., Soliera A.R., Ronchetti M., Galavotti S., Young K.W., Selmi T., Yacobi R., Van Etten R.A., Donato N., Hun-ter A., Dinsdale D., Tirrò E., Vigneri P., Nicotera P., Dyer M.J., Holyoake T., Salomoni P., Calabretta B. (2009) Targeting autophagy potentiates tyrosine kinase inhibitor-induced cell death in Philadelphia chromosome-positive cells, including primary CML stem cells. J. Clin. Invest. 119, 1109–1123.
Jung C.H., Ro S.H., Cao J., Otto N.M., Kim Do-Hyung D.H. (2010) MTOR regulation of autophagy. FEBS Lett. 584, 1287–1295.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология