

Молекулярная биология, 2021, T. 55, № 4, стр. 676-682
Фактор роста эпидермиса участвует в повышении экспрессии онкогена URGCP в клетках гепатомы человека
E. Tokay *
Department of Molecular Biology and Genetics, Project Coordination Office, Balıkesir University
10145 Balıkesir, Turkey
* E-mail: esratokay@balikesir.edu.tr
Поступила в редакцию 01.07.2020
После доработки 16.11.2020
Принята к публикации 18.11.2020
Аннотация
Гепатоцеллюлярная карцинома занимает четвертое место в структуре смертности от злокачественных опухолей в мире. Важную роль в канцерогенезе печени играет сигнальный путь рецептора фактора роста эпидермиса (EGFR). В микроокружении злокачественных опухолей печени наблюдается аберрантная экспрессия фактора роста эпидермиса (EGF). Изучена EGF-зависимая регуляция гена URGCP в клетках гепатомы человека (Hep3B). C помощью метода МТТ показано влияние цитокина EGF на пролиферацию клеток Hep3B. С помощью количественной ПЦР в реальном времени и вестерн-блотинга показано участие EGF в экспрессии URGCP на уровне мРНК и белка. Конструкциями, содержащими промоторы гена URGCP различной длины, транзиентно трансфицировали клетки Hep3B и определяли в них базальную активность промоторов в присутствии EGF. C целью изучения механизма EGF-опосредованной положительной регуляции гена URGCP проанализированы некоторые сигнальные пути. Экспрессия URGCP увеличивается в зависимости от концентрации EGF и времени воздействия. Опосредованное EGF повышение экспрессии URGCP может зависеть от цис-действующего элемента в промоторе этого гена (положение –344 …–482).
ВВЕДЕНИЕ
Гепатоцеллюлярная карцинома (ГЦК), занимающая четвертое место в структуре смертности от злокачественных заболеваний, ежегодно диагностируется более чем у 500 000 человек во всем мире. ГЦК относится в быстрорастущим опухолям [1–3]. Факторы роста и их рецепторы играют важную роль в процессах развития тканей и заживления ран [4, 5]. Нарушение регуляции сигнальных каскадов, инициируемых факторами роста, включая каскад фактора роста эпидермиса (EGF) и его рецептора (EGFR), влияет на прогрессию многих видов рака [6, 7]. Так, сигнальные пути EGFR и Hippo вовлечены в канцерогенез ГЦК [8]. В частности, во внутрипеченочном микроокружении ГЦК наблюдается аберрантная экспрессия EGF [3, 9–11].
В ГЦК выявлена не только экспрессия генов, кодирующих EGF и EGFR, но также и сверхэкспрессия онкогена URGCP [12]. Сверхэкспрессия URGCP стимулирует экспрессию мРНК циклина D1 в клетках HepG2, тогда как сайленсинг URGCP, опосредованный РНК-интерференцией, приводит к снижению экспрессии мРНК циклина D1 [13]. Кроме того, экспрессия URGCP повышена в опухолях различного типа, таких как рак желудка, остеосаркома, лейкоз и немелкоклеточный рак легкого [14]. Тем не менее, механизмы регуляции этого онкогена недостаточно изучены. Ранее мы сообщали, что экспрессию гена URGCP индуцируют цитокины TGF-β и TNF-α [15, 16].
Нами изучено влияние EGF на экспрессию URGCP и ее транскрипционную регуляцию в клетках Hep3B. Уровни экспрессии URGCP определяли на уровне мРНК и белка с помощью количественной ПЦР в реальном времени и вестерн-блотинга соответственно. Четырьмя различными конструкциями, содержащими укороченные с 5′-конца промоторные области гена URGCP, транзиентно трансфицировали клетки Hep3B и определяли EGF-опосредованную транскрипционную активность этих конструкций. Для блокирования некоторых сигнальных каскадов и понимания механизмов регуляции экспрессии, опосредованной EGF, использовали несколько селективных ингибиторов сигнальных путей. Представленная работа может внести вклад в понимание механизма EGF-опосредованной регуляции URGCP при ГЦК. Данные о EGF-опосредованной регуляции URGCP могут быть полезными для поиска новых терапевтических мишеней ГЦК.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Реактивы и материалы. В работе использовали: рекомбинантный EGF человека фирмы “Cell Signaling Technology” (США); антитела к URGCP (“Abcam” (ab103323), Великобритания) и к β‑актину (“Sigma Aldrich” (sc-81178), США); Ready-To-Glow™ Dual Secreted Reporter Assay (Cat no: 631734) приобретен у “Clontech” (США). Праймеры получены в “Macrogen” (Ю. Корея), вортманнин (ингибитор PI3K, 9951) и PD98059 (ингибитор MEK-1, 9900 S) – в “Cell Signaling Technology”. Использовали также ингибитор P38 PD169316 (P-9248) фирмы “Sigma Aldrich”; SP600125 (Sc-200635, ингибитор JNK) и ингибитор Nfkb Bay-117085 (CAS 196309-76-9) фирмы “Santa-Cruz” (США); набор для выделения РНК (Total RNA isolation Kit) компании “Fermentas” (CША).
Культура клеток. Клеточная линия ГЦК человека (Hep3B) предоставлена Dipak Ramji (Cardiff University). Клетки Hep3B культивировали в модифицированной Дульбекко среде Игла (DMEM) с высоким содержанием глюкозы, дополненной 10%-ной эмбриональной сывороткой крупного рогатого скота (“Gibco”, США) и 5 мМ глутамином (“Sigma Aldrich”) при 5% CO2 и 37°C. Клетки подвергали сывороточной депривации с использованием 0.1% БСА (бычий сывороточный альбумин, “Sigma Aldrich”) в течение 1 ч до добавления EGF.
Анализ клеточной пролиферации. Влияние EGF на клеточную пролиферацию Hep3B определяли с использованием МТТ-теста (“Sigma Aldrich”). Клетки Hep3B высевали в 96-луночные планшеты (3 × 104 клеток/лунка), затем обрабатывали EGF (0, 10, 20 и 40 нг/мл) в течение 24 и 48 ч с последующей инкубацией с реагентом MTT (20 мкл/лунка) в течение 4 ч. Оптическую плотность (OD) измеряли при 550 нм с помощью Thermo Microplate Reader [15, 17].
Выделение РНК и количественная ПЦР в реальном времени. Суммарную РНК выделяли с помощью набора GeneJET RNA purification kit (“Thermo ScientificTM”, США, Cat no: K0732) и количественно определяли с помощью системы Qubit. Первую цепь кДНК синтезировали, используя 1000 нг суммарной РНК, в соответствии с протоколом для ПЦР в реальном времени (RevertAid First Strand cDNA Synthesis Kit, “Thermo ScientificTM”, Cat no: K1622). Использовали следующие праймеры для мРНК URGCP (GeneBank NM_017920) (F – CTT CAT CCT GAG TCC CTA CCG; R – GCC GTT CTG CTG CAT TCG) и β2-микроглобулина человека (Hß-2F – TTT CTG GCC TGG AGG CTA TC; Hß-2 R – CAT GTC TCG ATC CCA CTT AAC T). мРНК β-микроглобулина человека добавляли в каждый образец в качестве внутреннего контроля. Количественное определение генов-мишеней и референсных генов (β-микроглобулин человека) проводили в трех повторах с помощью прибора Light Cycler H480. ПЦР в реальном времени проводили в общем объеме 10 мкл (1 мкл кДНК, 0.5 мкл каждой пары праймеров (100 пмоль/мкл), 5 мкл Light Cycler-FastStart DNA Master SYBR Green I mix (“Roche”, Швейцария). Условия реакции в термоциклере включали начальную денатурацию при 95°C (3 мин), за которой следовали 35 циклов при 95°C (30 с), 55°C (30 c) и 72°C (30 с). Уровни экспрессии определяли методом ΔΔCT для целевого гена [18].
Анализ сигнальных путей. Клетки Hep3B высевали по 2 × 106 клеток/см2 в культуральный флакон площадью 25 см2. Клетки подвергали сывороточному голоданию, используя БСА в конечной концентрации 0.1%, перед обработкой цитокинами и ингибиторами. Ингибиторы, включая ингибитор MAPK, Mek-1, ингибитор p38, PD169316, ингибитор JNK, SP600125 и ингибитор NF-кB, Bay-117085, добавляли в культуральные флаконы до конечной концентрации 10 мкМ после инкубации в течение 24 ч. Кроме того, использовали вортманнин – ингибитор PI3K, в концентрации 1 мкМ. После инкубации в течение 30 мин к клеткам добавляли цитокин EGF (40 нг/мл). Осадок клеток собирали после инкубации в течение 6 ч для выделения РНК и белка [15].
Вестерн-блотинг. Клетки лизировали с использованием буфера Laemmli. Вестерн-блот-анализ проводили согласно Tokay и Kockar [15]. Концентрация антител против URG4 составляла 1 : 100 (об./об.); загрузку образцов контролировали с использованием антител к β-актину в концентрации 1 : 1000 (об./об.).
Транзиентная трансфекция промоторных конструкций. Клетки, культивируемые во флаконах площадью 25 см2, трансфицировали четырьмя конструкциями (10 мкг), содержащими промоторные области гена URGCP разной длины (–109…+63/pMET (P1), –261…+63/pMET (P2), –344…+63/pMET (P3) и –482…+63/pMET (P4)), клонированные ранее в репортерный вектор pMetLuc, содержащий ген люциферазы, как описано ранее [19]. Клетки инкубировали в течение 24 ч, а затем высевали в 96-луночные планшеты (по 3 × 104 клеток на лунку). Через 24 ч в лунки добавляли EGF и ингибиторы сигнальных путей в указанных выше концентрациях. Клетки подвергали сывороточному голоданию за 1 ч до обработки EGF и ингибиторами. Для определения активности люциферазы среду удаляли после инкубации клеток в течение 6 ч. С целью контроля эффективности трансфекции клетки трансфицировали также контрольным вектором pMetLuc (2 мкг), экспрессирующим ген люциферазы.
Активность репортерных конструкций. Активность люциферазы определяли с использованием набора для анализа секретируемой люциферазы (“Clontech”, США), измерения проводили на люминометре (Fluoraskan Ascent FL, “Thermo Fisher Scintific”) в соответствии с опубликованным ранее протоколом [20]. Эксперименты повторяли не менее 3 раз.
Статистический анализ. Результаты выражали как среднее ± стандартное отклонение (SD), вычисленные по трем независимым экспериментам. Статистический анализ проводили с помощью программного обеспечения GraphPad Prism версии 7.0 для Windows и однофакторного дисперсионного анализа (ANOVA).
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
EGF влияет на пролиферацию клеток Hep3B в зависимости от сывороточных условий
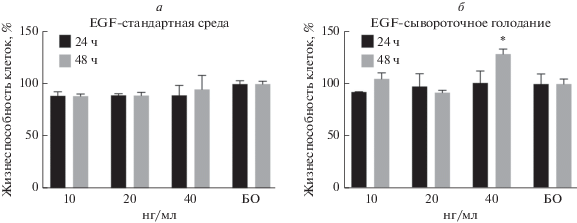
Влияние EGF на пролиферацию клеток Hep3B определяли с помощью МТТ-теста. Клетки высевали на 96-луночные планшеты. Через 24 ч добавляли EGF в конечной концентрации 10, 20 и 40 нг/мл. МТТ-анализ проводили через 24 и 48 ч. Результаты представлены на рис. 1а. Хорошо видно, что в использованных нами условиях (24 и 48 ч, нормальное содержание сыворотки) EGF не влиял на пролиферацию клеток Hep3B. В случае сывороточного голодания клетки подвергали сывороточной депривации, добавляя 0.1% БСА за 1 ч до внесения EGF. Как показано на рис. 1б, EGF в концентрации 40 нг/мл повышал пролиферацию клеток через 48 ч после обработки.
Рис. 1.
Влияние EGF на пролиферацию клеток Hep3B при нормальном содержании сыворотки (а) и в состоянии сывороточного голодания (МТТ-анализ) (б). Клетки рассевали на 96-луночные планшеты (30 000 клеток/лунка). После инкубации в течение 24 ч в лунки добавляли EGF – 10, 20 и 40 нг/мл. Сывороточную депривацию с использованием 0.1% БСА проводили в течение 24 ч до добавления EGF. После инкубации в течение 24 и 48 ч измеряли поглощение при 550 нм. Представленные данные репрезентативны по трем независимым экспериментам. Показано стандартное отклонение, вычисленное по трем повторностям. Значения p получены с помощью t-теста в результате сравнения с контрольными (необработанными) клетками, *p < 0.05 считали статистически значимыми. Здесь и далее БО – клетки без обработки.
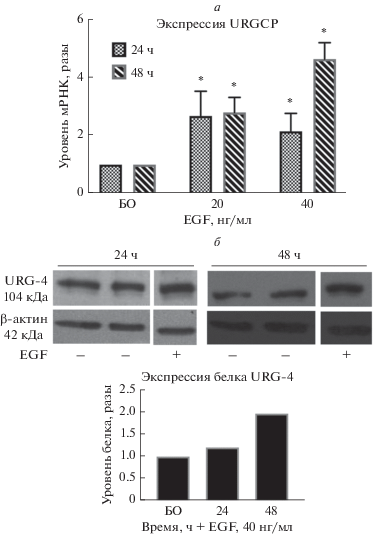
EGF повышает экспрессию URGCP на уровне мРНК и белка в зависимости от концентрации и времени
Влияние EGF на экспрессию URGCP определяли на уровне мРНК и белка с использованием технологии количественной ПЦР в реальном времени и вестерн-блотинга. Клетки высевали во флакон, а затем в условиях сывороточного голодания добавляли EGF в концентрации 20 и 40 нг/мл. Из рис. 2а видно, что обработка клеток EGF (20 нг/мл) в течение 24 и 48 ч приводила к повышению экспрессии URGCP примерно в 3 и 2.5 раза соответственно. Кроме того, через 48 ч стимулирующий эффект EGF в концентрации 40 нг/мл усиливался примерно в 4.5 раза. Стимуляцию экспрессии белка URGCP подтверждали методом вестерн-блотинга. Обнаружено, что воздействие EGF в концентрации 40 нг/мл в течение 48 ч приводило к повышению экспрессии белка URGCP примерно в 2 раза (рис. 2б).
Рис. 2.
Определение EGF-опосредованной экспрессии URGCP в клетках Hep3B на уровне мРНК и белка. а – Уровень мРНК URGCP определяли с помощью ПЦР в реальном времени. Гистограммы строили с помощью программы Graphpad Prism 7.0. Изменение уровня мРНК рассчитывали методом ΔΔCt. Приведены результаты, вычисленные по трем независимым репрезентативным экспериментам. Показано стандартное отклонение для трех повторов. Значения p получены с помощью t-теста. *p < 0.05 считали статистически значимыми. б – Вестерн-блот-анализ экспрессии белка URGCP. В качестве внутреннего контроля использовали β-актин. Денситометрические определения проводили с помощью программы Image J.
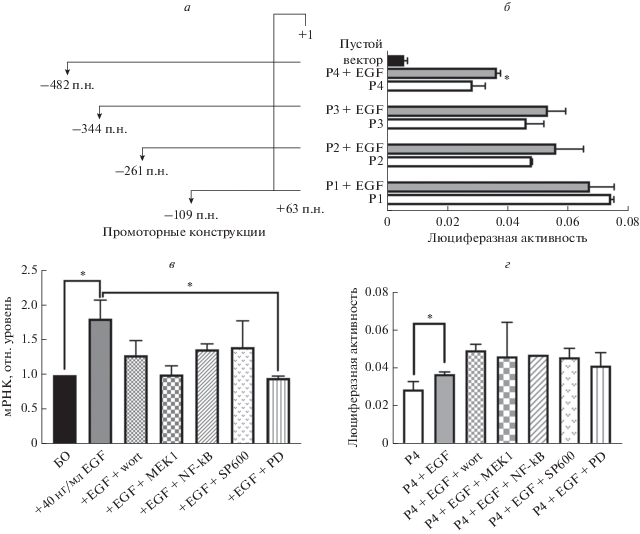
EGF-опосредованная регуляция URGCP контролируется через путь p38/MAPK
Механизм положительной регуляции URGCP изучали с помощью анализа сигнальных путей в присутствии EGF с использованием селективных ингибиторов. Транскрипционный уровень URGCP определяли в клетках Нер3В, транзиентно трансфицированных Са-фосфатным методом четырьмя различными промоторными конструкциями гена URGCP. Добавляли ингибиторы и цитокин EGF и инкубировали в течение 6 ч. После этого собирали среду культивирования и определяли в ней люциферазную активность с использованием системы анализа секретируемой люциферазы. Из рис. 3б видно, что EGF значительно повышает активность промотора Р4 (почти в 1.2 раза) по сравнению с его базальной активностью. Уровень мРНК определяли, проводя количественную ПЦР на кДНК-матрице из клеток, обработанных селективными ингибиторами и цитокином EGF. Из рис. 3в видно, что ингибитор PD169316 значительно снижает уровень мРНК URGCP в клетках, обработанных EGF. Показано также, что упомянутые ингибиторы не снижают существенно активность промотора Р4 (рис. 3г). Таким образом, мы можем сделать вывод, что EGF-опосредованная экспрессия URGCP не регулируется участком ‒482…+63 промотора URGCP.
Рис. 3.
Влияние специфических ингибиторов сигнальных путей и цитокина EGF на уровень мРНК в клетках Hep3B и на активность промоторных конструкций. Люциферазную активность промоторных конструкций URGCP определяли с помощью люминометра. Ингибиторы сигнальных путей (вортманнин (wort), MEK1, NF-кB, SP600125 (SP600) и PD169316 (PD)) были добавлены к клеткам Hep3B для определения регуляторных участков промотора URGCP. Гистограммы строили с помощью программы Graphpad Prism 7.0. Данные репрезентативны для трех независимых экспериментов. Показано стандартное отклонение для трех повторов. *р < 0.05 считали статистически значимыми.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Ранее показали, что экспрессия EGF в ГЦК значительно выше, чем в нормальных тканях [21]. Высокий уровень экспрессии EGF способствует миграции и инвазии ряда опухолевых клеток, включая клетки ГЦК [22]. Мы проверили, может ли стимуляция EGF влиять на экспрессию гена URGCP в клетках гепатомы Нер3В. Huang и Xu показали, что стимуляция клеток HepG2 и HCCLM3 цитокином EGF (50 и 100 нг/мл) приводит к значительному увеличению пролиферации [23]. Однако, как показано в МТТ-тесте, EGF в концентрации 20 и 40 нг/мл не подавлял пролиферацию клеток Hep3B в течение 24 и 48 ч в условиях нормального содержания сыворотки. При сывороточном голодании EGF в концентрации 40 нг/мл индуцировал пролиферацию клеток Hep3B в течение 48 ч. Эти результаты соответствуют данным [23]. Влияние EGF на пролиферацию клеток имеет временную и концентрационную зависимость.
Нас особенно интересовало, влияет ли воздействие EGF на уровни экспрессии URGCP, обычно повышенные в тканях ГЦК, параллельно с уровнями HBxAg (антиген вируса гепатита В) [13]. URGCP способствует пролиферации клеток ГЦК путем воздействия на ген циклина D1 [13]. В клетках Hep3B, подвергнутых 48-часовому воздействию EGF, уровни URGCP были значительно выше, чем в необработанных клетках. Реагирует ли специфический промоторный участок URGCP на EGF? Чтобы ответить на этот вопрос, определили транскрипционный уровень гена URGCP, обеспечиваемый четырьмя различными промоторными конструкциями, укороченными с 5′-конца. Значительное повышение активности промотора Р4 под действием EGF указывает на возможное присутствие в участке, охватывающем область ‒344…–482, сайтов связывания факторов транскрипции, активируемых воздействием EGF.
Установлено, что сигнальные пути EGFR и Hippo играют важную роль в канцерогенезе ГЦК [24]. Механизм повышения экспрессии URGCP под действием EGF изучали с использованием нескольких ингибиторов сигнальных путей, таких как вортманнин (ингибитор PI3K), PD98059 (ингибитор MEK-1), SP600125 (ингибитор JNK) и Bay-117085 (ингибитор NF-кB). Димеризация EGFR активировала три основных нисходящих сигнальных пути – Ras/Raf/MEK/ERK/MAPK, PI3K/AKT (PKB) и JAK/STAT [25]. Каскады MAPK также включали киназу р38. Результаты нашей работы показывают, что EGF усиливает экспрессию URGCP на уровне мРНК через путь p38/MAPK. Подробно изучено, влияет ли ингибитор p38/MAPK на повышение активности промотора P4. Установлено, что люциферазная активность конструкции Р4 не снижалась в присутствии PD169316 (ингибитор р38) в отличие от уровня мРНК. В этих условиях EGF-опосредованная регуляция URGCP не могла контролироваться использованными конструкциями. В нашей работе впервые изучено повышение транскрипции гена URGCP, опосредованное EGF.
Опосредованная EGF регуляция URGCP может быть ответственной за процесс метастазирования ГЦК. Однако для подтверждения этого предположения необходимы дополнительные исследования. Результаты нашей работы позволят лучше понять механизмы EGF-опосредованной регуляции URGCP.
Список литературы
Fuchs B.C., Hoshida Y., Fuji T., Wei L., Yamada S., Lauwers G.Y., McGinn C.M., DePeralta D.K., Chen X., Kuroda T., Lanuti M., Schmitt A.D., Gupta S., Crenshaw A., Onofrio R., Taylor B., Winckler W., Bardeesy N., Caravan P., Golub T.R., Tanabe K.K. (2014) Epidermal growth factor receptor inhibition attenuates liver fibrosis and development of hepatocellular carcino-ma. Hepatology. 59(4), 1577–1590.
Huang P., Xu X., Wang L., Zhu B., Wang X., Xia J. (2014) The role of EGF-EGFR signalling pathway in hepatocellular carcinoma inflammatory microenvironment. J. Cell. Mol. Med., 18(2), 218–230.
Liu Z.C., Ning F., Wang H.F., Chen D.-Y., Cai Y.-N., Sheng H.-Y., Lash G.E., Liu L., Du J. (2017) Epidermal growth factor and tumor necrosis factor α cooperatively promote the motility of hepatocellular carcinoma cell lines via synergistic induction of fibronectin by NF-κB/p65. Biochim. Biophys. Acta Gen. Subj. 1861(11PtA), 2568–2582. https://doi.org/10.1016/j.bbagen.2017.08.010
Nikolova D., Chalovska V., Ivanova M.G., Nikolovska E., Volkanovska A., Orovchanec N., Kunovska S.K., Pet-rushevska G., Janevska V. (2018) Immunohistochemical expression of epidermal growth factor receptor in hepatocellular carcinoma. Pril. (Makedon. Akad. Nauk Umet. Odd Med. Nauki). 39(2–3), 21–28.
Xia H., Dai X., Yu H., Zhou S., Fan Z., Wei G., Tang Q., Gong Q., Bi F. (2018) EGFR-PI3K-PDK1 pathway regulates YAP signaling in hepatocellular carcinoma: the mechanism and its implications in targeted therapy. Cell Death Dis. 9(3), 269. https://doi.org/10.1038/s41419-018-0302-x
Wang W., Ma X.P., Shi Z., Zhang P., Ding D.-L., Huang H.-X., Saiyin H.G., Chen T.Y., Lu P.-X., Wang N.-J., Yu H., Sun J., Zheng S.L., Yu L., Xu J., Jiang D.-K. (2014) Epidermal growth factor receptor pathway polymorphisms and the prognosis of hepatocellular carcinoma. Am. J. Cancer Res. 5(1), 396–410.
Chen B., Wei W., Ma L., Yang B., Gill R.M., Chua M.-S., Butte A.J., So S. (2017) Computational discovery of niclosamide ethanolamine, a repurposed drug candidate that reduces growth of hepatocellular carcinoma cells in vitro and in mice by inhibiting cell division cycle 37 signaling. Gastroenterology. 152(8), 2022–2036.
Lu W.C., Kao S.Y., Yang C.C., Tu H.-F., Wu C.-H., Chang K.-W., Lin S.-C. (2014) Affiliations expandEGF up-regulates miR-31 through the C/EBPβ signal cascade in oral carcinoma. PLoS One. 9(9), e108049.
Hung W., Yang M., Chang C., Tsai J., Chuang L. (1995) Differential regulation of EGF production, EGF receptor-binding, and cellular growth by sodium-butyrate in hep3b and plc/prf/5 human hepatoma-cells. Intern. J. Oncology. 7(5), 1089–1093.
Fan F.T., Shen C.S., Tao L., Tian C., Liu Z.-G., Zhu Z.-J., Liu Y.-P., Pei C.-S., Wu H.-Y., Zhang L., Wang A.-Y., Zheng S.-Z., Huang S.-L., Lu Y. (2014) PKM2 regulates hepatocellular carcinoma cell epithelial-mesenchymal transition and migration upon EGFR activation. Asian Pac. J. Cancer Prev. 15(5), 1961–1970.
Yan F., Bai L.P., Gao H., Zhu C.-M., Lin L., Kang X.-P. (2014) EGF reverses multi-drug resistance via the p‑ERK pathway in HepG2/ADM and SMMC7721/ ADM hepatocellular carcinoma models. Asian Pac. J. Cancer Prev. 15(6), 2619–2623.
Tufan N.L., Lian Z., Liu J., Pan J., Arbuthnot P., Kew M., Clayton M.M., Zhu M., Feitelson M.A. (2002). Hepatitis Bx antigen stimulates expression of a novel cellular gene, URG4, that promotes hepatocellular growth and survival. Neoplasia. 4(4), 355–368.
Satiroglu-Tufan N.L., Dodurga Y., Gok D., Cetinkaya A., Feitelson M.A. (2010) RNA interference-mediated URG4 gene silencing diminishes cyclin D1 mRNA expression in HepG2 cells. Genet. Mol. Res. 9(3), 1557–1567.
Dodurga Y., Seçme M., Şatıroğlu-Tufan N.L. (2018) A novel oncogene URG4/URGCP and its role in cancer. Gene. 668, 12–17.
Tokay E., Kockar F. (2016) Identification of intracellular pathways through which TGF-β1 upregulates URG-4/URGCP gene expression in hepatoma cells. Life Science. 144, 121–128.
Tokay E., Sagkan R.I., Kockar F. (2020) TNF-α induces URG-4/URGCP gene expression in hepatoma cells through starvation dependent manner. Biochem. Genet. https://doi.org/10.1007/s10528-020-09972-z
Alper M., Kockar F. (2014) IL-6 upregulates a disintegrin and metalloproteinase with thrombospondin motifs 2 (ADAMTS-2) in human osteosarcoma cells mediated by JNK pathway, Mol. Cell. Biochemistry. 393(1–2), 165–175.
Livak K.J., Schmittgen T.D. (2001) Analysis of relative gene expression data using real-time quantitative PCR and the 2 (–Delta Delta C(T)) method. Methods. 25(4), 402–408.
Tokay E., Kockar F. (2016) SP1 is a transcriptional regulator of URG-4/URGCP gene in hepatocytes. Mol. Cell. Biochem. 423(1–2), 75–83.
Turkoglu S.A., Kockar F. (2016) SP1 and USF differentially regulate ADAMTS1 gene expression under normoxic and hypoxic conditions in hepatoma cells. Gene. 575, 48–57.
Geng J., Li X., Lang X., Qiao C., Hu M., Yang J., Feng J., Lv M. (2014) Combination of cetuximab and rapamycin enhances the therapeutic efficacy in hepatocellular carcinoma. Technol. Cancer Res. Treat. 13(4), 377–385.
Shehata F., Abdel M.N., Sakr M., Kasem S., Balbaa M. (2013) Epidermal growth factor, its receptor and transforming growth factor-β1 in the diagnosis of HCV-induced hepatocellular carcinoma. Med. Oncol. 30(3), 673. https://doi.org/10.1007/s12032-013-0673-x
Huang P., Xu X.X. (2014) The role of EGF-EGFR signalling pathway in hepatocellular carcinoma inflammatory microenvironment. J. Cell. Mol. Med. 18(2), 218–230.
Xia H., Dai X., Yu H., Zhou S., Fan Z., Wei G., Tang Q., Gong Q., Bi F. (2018) EGFR-PI3K-PDK1 pathway regulates YAP signaling in hepatocellular carcinoma: the mechanism and its implications in targeted therapy. Cell Death Dis. 9(3), 269.
Wee P., Wang Z. (2017) Epidermal growth factor receptor cell proliferation signaling pathway. Cancers (Basel). 9(5), 52.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология