

Молекулярная биология, 2021, T. 55, № 6, стр. 897-926
Противовирусные и противомикробные производные нуклеозидов: структурные особенности и механизмы действия
А. А. Зенченко a, М. С. Дреничев a, *, И. А. Ильичева a, С. Н. Михайлов a
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
* E-mail: mdrenichev@mail.ru
Поступила в редакцию 20.01.2021
После доработки 03.04.2021
Принята к публикации 09.04.2021
Аннотация
Появление новых вирусов и резистентных штаммов патогенных микроорганизмов стало мощным стимулом для поиска новых лекарственных средств. Нуклеозиды – один из перспективных классов природных соединений, на основе которых уже создано более ста лекарственных препаратов, включая противовирусные, антибактериальные и противоопухолевые средства. В обзоре рассмотрены структурно-функциональные особенности и механизмы действия известных аналогов нуклеозидов, обладающих противовирусной, антибактериальной или антипротозойной активностью. Особое внимание уделено механизмам, которые определяют противовирусный эффект аналогов нуклеозидов, содержащих гидрофобные фрагменты. В зависимости от структуры и положения гидрофобного заместителя такие нуклеозиды могут либо блокировать процесс проникновения вирусов в клетки, либо ингибировать стадию репликации их геномов. Проведено сравнение механизмов ингибирования вирусных ферментов соединениями нуклеозидной и ненуклеозидной природы. Рассмотрены стадии создания антипаразитарных препаратов, которые базируются на данных об особенностях метаболических превращений нуклеозидов в организме человека и организме паразита. Описан новый подход к созданию лекарственных препаратов, основанный на использовании пролекарственных форм модифицированных нуклеозидов, которые в результате метаболических процессов конвертируются в эффективный лекарственный препарат непосредственно в целевом органе или ткани. Такая стратегия позволяет уменьшить общую токсичность препарата для человека и увеличить эффективность его действия на клетки, пораженные вирусом.
ВВЕДЕНИЕ
Быстро мутирующие РНК-содержащие вирусы представляют серьезную опасность для человечества. Пандемия COVID-19, вызванная новым коронавирусом тяжелого респираторного синдрома (SARS-CoV-2) в 2020 году, обнажила проб-лему нехватки универсальных противовирусных средств. Вирусы относятся к облигатным паразитам, геном которых кодирует минимальный набор ферментов и белков, необходимых для репликации и сборки вирусных частиц. Для биосинтеза строительных блоков (мономеров) используются ферменты клетки, поэтому воспроизведение вируса возможно только в инфицированной им клетке. Можно выделить два основных типа противовирусных препаратов: первые препятствуют проникновению вируса в клетку, вторые нарушают репликацию вирусного генома в зараженной клетке. Аналоги нуклеозидов могут быть активными противовирусными средствами, причем механизм их действия зависит от типа и положения заместителей. Огромный интерес к созданию противовирусных лекарственных препаратов не ограничивается рассмотрением классических подходов, а по мере появления новых технологий получают развитие и новые направления в этой области [1‒5].
Лекарственные средства на основе нуклеозидов используют и для лечения инфекционных заболеваний, вызываемых бактериями, простейшими и грибами. Возникновение резистентности у патогенных микроорганизмов к применяемым препаратам − это серьезная проблема, с которой столкнулось человечество из-за повсеместного, далеко не всегда оправданного, использования антибиотиков. Именно поэтому разработка новых противомикробных средств была и остается актуальной. В обзоре мы обсуждаем и стратегию поиска антипротозойных соединений для лечения социально значимых инфекционных заболеваний [6‒9].
Создание лекарственных препаратов на основе природных соединений ‒ классический подход, который хорошо себя зарекомендовал. К настоящему времени на основе нуклеозидов создано около ста лекарственных препаратов, причем половина из них относится к противовирусным и четверть ‒ к противоопухолевым препаратам [10]. Природные нуклеозиды имеют разнообразную структуру, они входят в состав нуклеотидов, ДНК, РНК, коферментов. Только из тРНК выделено более 140 минорных нуклеозидов, а из различных природных источников ‒ около 100 дисахаридных нуклеозидов и 200 нуклеозидных антибиотиков, в структуре которых имеются дополнительные функциональные группы и гидрофобные остатки. Библиотека природных нуклеозидов содержит около 600 соединений, которые служат основой для создания новых биологически активных соединений [11].
В предлагаемом обзоре стратегии создания противовирусных и противопаразитарных препаратов проанализированы в контексте особенностей метаболизма нуклеозидов в различных организмах. Детально рассмотрены те особенности структуры аналогов нуклеозидов, которые определяют механизм их лекарственного действия.
МЕТАБОЛИЗМ НУКЛЕОЗИДОВ – КЛЮЧ К ПОНИМАНИЮ МЕХАНИЗМА ДЕЙСТВИЯ ЛЕКАРСТВЕННЫХ СРЕДСТВ
В отличие от большинства лекарственных препаратов нуклеозиды и их аналоги в организме человека сначала подвергаются действию ряда ферментов клеточного метаболизма, которые переводят их в активные формы (нуклеотиды). Схема метаболизма различных модифицированных нуклеозидов на начальном этапе после попадания в организм довольно консервативна. Эти соединения обычно проникают в клетки и в результате ферментативного фосфорилирования последовательно превращаются в нуклеозид-5'-моно-, ди- и трифосфаты (NMP, NDP и NTP соответственно). Эти метаболиты затем ингибируют один или несколько ферментов биосинтеза ДНК или РНК (в том числе вирусные полимеразы). В клетке соотношения нуклеозидфосфатов (NMP : NDP : NTP) для производных разных нуклеозидов отличаются. Оно зависит от скорости фосфорилирования исходного нуклеозида. Первая стадия фосфорилирования (образование NMP) обычно скоростьлимитирующая и, следовательно, наиболее важная для активации. В целом равновесие нуклеозид N ↔ NTP в клетке сдвинуто в сторону образования трифосфатов. Также следует отметить, что концентрации NTP в клетке значительно превышают концентрации NMP, NDP, а также нуклеозидов, участвующих в метаболических процессах, необходимых для роста, поддержания жизнедеятельности организма и для его воспроизведения (табл. 1) [12]. Эти превращения неразрывно связаны с биосинтезом нуклеозидов, нуклеотидов и нуклеиновых кислот в клетке, поэтому знание особенностей биосинтеза нуклеотидов – ключ к пониманию механизма действия биологически активных производных нуклеозидов.
Таблица 1.
Концентрации метаболитов, участвующих в биосинтезе пуриновых нуклеозидовa
| Метаболит | Сb, мкМ |
|---|---|
| ATP | 2102 |
| GTP | 305 |
| ADP | 137 |
| GDP | 36 |
| AMP | 82 |
| GMP | 32 |
| Ado | 0.5 |
| Guo | 0.9 |
| Ade | 0.4 |
| Gua | 97 |
a В таблице обобщены данные, приведенные в обзоре Traut T.W. [12]. b Концентрации нуклеотидов измерены в клетках крови человека, концентрации оснований и нуклеозидов измерены во внеклеточной жидкости.
Пути биосинтеза нуклеозидов
Биосинтез пуриновых и пиримидиновых нуклеозидов и нуклеотидов (рис. 1) реализуется в организме двумя путями. Основной путь (de novo) представляет собой сложный, многоступенчатый и энергоемкий процесс образования гетероциклических оснований в присутствии различных доноров углерода и азота с последующим формированием из них нуклеозидов. Дополнительный путь биосинтеза нуклеозидов, “реутилизационный” (salvage pathways), позволяет клеткам повторно использовать свободные азотистые основания, которые образуются в результате ферментативного расщепления нуклеозидов в обменных процессах. Это обеспечивает экономичность метаболизма в организме.
Рис. 1.
Биосинтез пуриновых и пиримидиновых нуклеозидов/нуклеотидов. Цифрами обозначены ферменты. Биосинтез пуринов: 1) фосфорибозилпирофосфат-синтетаза (EC 2.7.6.1); 2) амидофосфорибозилтрансфераза (EC 2.4.2.14); 3) фосфорибозиламин-глицин-лигаза (EC 6.3.4.13), фосфорибозилглицинамидформилтрансфераза (EC 2.1.2.2), фосфорибозилформилглицинамидинсинтетаза (EC 6.3.5.3), фосфорибозилформилглицинамидин-циклолигаза (EC 6.3.3.1), фосфорибозиламиноимидазолкарбоксилаза (EC 4.1.1.21), 5-(карбоксиамино)имидазол-рибонуклеотидмутаза (EC 5.4.99.18), фосфорибозиламиноимидазолсукцинокарбоксамидсинтетаза (EC 6.3.2.6), аденилосукцинатлиаза (EC 4.3.2.2), фосфорибозиламиноимидазолкарбоксамидформилтрансфераза (EC 2.1.2.3); 4) аденилосукцинатсинтетаза (EC 6.3.4.4); 5) аденилосукцинатлиаза (EC 4.3.2.2); 6) аденилаткиназа (EC 2.7.4.3); 7) нуклеозиддифосфаткиназа (EC 2.7.4.6); 8) аденилатнуклеозидаза (EC 3.2.2.4); 9) аденинфосфорибозилтрансфераза (EC 2.4.2.7); 10) пуриннуклеозидфосфорилаза (EC 2.4.2.1); 11) аденозиндезаминаза (EC 3.5.4.4); 12) гуанозин-инозинкиназа (EC 2.7.1.73); 13) пуриннуклеозидфосфорилаза (EC 2.4.2.1); 14) инозин-5'-монофосфат-дегидрогеназа (EC 1.1.1.205); 15) гуанилатсинтетаза (EC 6.3.5.2); 16) гуанилаткиназа (EC 2.7.4.8); 17) нуклеозиддифосфаткиназа (EC 2.7.4.6); 18) гипоксантин-гуанин-фосфорибозилтрансфераза (EC 2.4.2.8); 19) пуриннуклеозидфосфорилаза (EC 2.4.2.1); 20) гуанозин-инозинкиназа (EC 2.7.1.73); 21) гуаниндезаминаза (EC 3.5.4.3); 22) пуриннуклеозидфосфорилаза (EC 2.4.2.1); 23) ксантингуанинфосфориботрансфераза (EC 2.4.2.22); 24) гуанозин-инозинкиназа (EC 2.7.1.73); 25) гуанозин-5’-монофосфатредуктаза (EC 1.7.1.7); 26) гипоксантин-гуанинфосфорибозилтрансфераза (EC 2.4.2.8); 27) рибонуклеозиддифосфатредуктаза (EC 1.17.4.1); 28) нуклеозиддифосфаткиназа (EC 2.7.4.6); 29) рибонуклеозиддифосфатредуктаза (EC 1.17.4.1); 30) нуклеозиддифосфаткиназа (EC 2.7.4.6). Биосинтез пиримидинов: 1) карбамоилфосфатсинтаза (EC 6.3.5.5); 2) аспартат-карбамоилтрансфераза (EC 2.1.3.2), дигидрооротаза (EC 3.5.2.3), дигидрооротат-дегидрогеназа (EC 1.3.99.11); 3) оротат-фосфорибозилтрансфераза (EC 2.4.2.10); 4) оротидин-5'-фосфатдекарбоксилаза (EC 4.1.1.23); 5) уридилаткиназа (EC 2.7.4.22); 6) нуклеозиддифосфаткиназа (EC 2.7.4.6); 7) цитидин-5'-трифосфат-синтаза (EC 6.3.4.2); 8) рибонуклеозиддифосфатредуктаза (EC 1.17.4.1); 9) нуклеозиддифосфаткиназа (EC 2.7.4.6); 10) дезоксиуридинтрифосфатдифосфатаза (EC 3.6.1.23); 11) тимидилатсинтаза (EC 2.1.1.45); 12) тимидилаткиназа (EC 2.7.4.9); 13) нуклеозиддифосфаткиназа (EC 2.7.4.6); 14) уридин-цитидинкиназа (EC 2.7.1.48); 15) нуклеозиддифосфаткиназа (EC 2.7.4.6); 16) дезоксицитидин-5ʹ-трифосфат-дезаминаза (EC 3.5.4.13); 17) цитидилаткиназа (EC 2.7.4.14); 18) нуклеозиддифосфаткиназа (EC 2.7.4.6); 19) нуклеозидгидролаза (EC 3.2.2); 20) цитозиндезаминаза (EC 3.5.4.1); 21) урацилфосфорибозилтрансфераза (EC 2.4.2.9); 22) цитидиндезаминаза (EC 3.5.4.5); 23) уридин-цитидинкиназа (EC 2.7.1.48); 24) уридинфосфорилаза (EC 2.4.2.3); 25) цитидиндезаминаза (EC 3.5.4.5); 26) тимидинкиназа (EC 2.7.1.21); 27) тимидинфосфорилаза (EC 2.4.2.4); 28) тимидинфосфорилаза (EC 2.4.2.4). Обозначения: ФРПФ ‒ 5-фосфорибозил-1-пирофосфат; IMP ‒ инозин-5'-монофосфат; OMP ‒ оротидин-5'-монофосфат; sAMP ‒ аденилосукцинат; XMP ‒ ксантозин-5'-монофосфата; Xao ‒ ксантозин, Xan ‒ ксантин; Thy ‒ тимин; Thd ‒ тимидин, Hyp ‒ гипоксантин. Упрощенная схема биосинтеза составлена на основании данных по метаболическим превращениям нуклеозидов/нуклеотидов [12‒14].
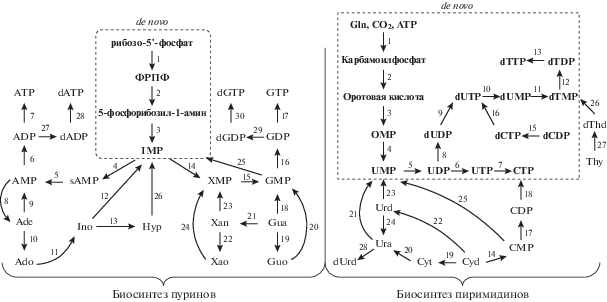
Ключевая реакция биосинтеза de novo пуриновых нуклеотидов ‒ образование 5-фосфорибозил-1-амина в результате переноса амидной группы глутамина на 5-фосфорибозил-1-пирофосфат (ФРПФ) под действием амидофосфорибозилтрансферазы. Далее осуществляется сборка пуринового гетероциклического основания на основе 5-фосфорибозил-1-амина при участии различных доноров углерода и азота с образованием инозин-5'-монофосфата (IMP) ‒ метаболического предшественника пуриновых нуклеотидов. IMP метаболизируется в AMP и GMP через образование промежуточных соединений: аденилосукцината (sAMP) и ксантозин-5'-монофосфата (XMP) ‒ с последующим фосфорилированием специфическими нуклеозидмонофосфат- и нуклеозиддифосфаткиназами до активных трифосфатных форм (ATP и GTP) [13] (см. “Биосинтез пуринов” на рис. 1).
В отличие от синтеза пуринов пиримидиновое гетероциклическое основание изначально синтезируется на основании глутамина, СО2 и ATP с образованием карбамоилфосфата. В ходе ряда превращений образованная из карбамоилфосфата оротовая кислота связывается с рибозо-5'-фосфатом под действием оротатфосфорибозилтрансферазы с образованием оротидин-5'-монофосфата (OMP) – исходного субстрата для биосинтеза пиримидиновых нуклеотидов de novo (см. “Биосинтез пиримидинов” на рис. 1). Дальнейшее декарбоксилирование приводит к образованию промежуточного продукта биосинтеза ‒ уридин-5'-монофосфата (UMP), который в результате последовательного фосфорилирования превращается в активный уридин-5'-трифосфат (UTP). UTP участвует в образовании цитидин-5'-трифосфата (CTP) под действием цитидин-5'-трифосфатсинтазы, катализирующей амидирование UTP путем ATP-зависимого замещения кетогруппы урацила на амидную группу глутамина [13, 14].
Дезоксирибонуклеотиды (dATP, dGTP и dCTP), включаемые в состав ДНК, образуются de novo из рибонуклеозид-5'-дифосфатов под действием рибонуклеозиддифосфатредуктаз и нуклеозиддифосфаткиназ (рис. 1). Тимидин-5'-трифосфат (dTTP) образуется в серии ферментативных реакций, включающих превращение дезоксиуридин-5'-трифосфата (dUTP) в дезоксиуридин-5'-монофосфат (dUMP) под действием дезоксиуридинтрифосфатазы, метилирование dUMP с образованием dTMP, катализируемое тимидилатсинтазой, и последовательное фосфорилирование dTMP нуклеотидкиназами. Бóльшая часть dUTP (около 75%), используемая для синтеза dTTP, образуется в результате дезаминирования dCTP под действием дезоксицитидинтрифосфат-дезаминазы, а не непосредственно из UDP под действием рибонуклеотидредуктазы и нуклеозиддифосфаткиназы [13].
“Реутилизационный” путь биосинтеза основан на присоединении молекулы ФРПФ к свободному азотистому основанию (образующемуся в результате распада нуклеиновых кислот и последующего ферментативного расщепления нуклеотидов), при этом ФРПФ-зависимое фосфорибозилирование пуринов катализируют аденинфосфорибозилтрансфераза, ответственная за образование AMP из аденина, и гипоксантин-гуа-нинфосфорибозилтрансфераза, катализирующая образование IMP и GMP из гипоксантина и гуанина соответственно. Для пути “реутилизации” одна из основных стадий – сопровождающееся фосфоролизом расщепление N-гликозидной связи пуриновых нуклеозидов пуриннуклеозидфосфорилазой, а пиримидиновых нуклеозидов ‒ пиримидиннуклеозидфосфорилазой и уридинфосфорилазой. Все эти ферменты, катализируют обратимое расщепление гликозидной связи соответствующих рибо- и дезоксирибонуклеозидов в присутствии неорганического фосфата с образованием гетероциклического основания и рибозо/дезоксирибозо-1-фосфата. Термодинамическое равновесие реакций смещено в сторону синтеза нуклеозидов [15], причем в случае пуриновых нуклеозидов более значительно [16]. Ключевой фермент фосфоролиза пиримидиновых нуклеозидов ‒ уридинфосфорилаза, катализирующая обратимое расщепление уридина, 2'-дезоксиуридина и тимидина до соответствующих гетероциклических оснований и рибозо-1-фосфата. Дальнейшее превращение нуклеозидов в нуклеотиды катализирует уридин-цитидинкиназа, фосфорилирующая уридин и цитидин до 5'-монофосфатных форм с одинаковой эффективностью [17].
Дополнительный путь биосинтеза пиримидиновых дезоксирибонуклеотидов заключается в фосфорилировании тимидина до dTMP под действием тимидинкиназы. Ключевую роль играет и фосфоролитическое расщепление дезоксиуридина и тимидина под действием тимидинфосфорилазы; при этом термодинамическое равновесие смещено в сторону образования нуклеозидов [13].
Таким образом, метаболизм нуклеозидов/нуклеотидов представляет собой сложный многоступенчатый процесс, включающий большой набор ферментов и реакций, преобразующих нуклеотиды, нуклеозиды и азотистые основания. Обладая структурным сходством с природными субстратами, лекарственные соединения нуклеозидной природы могут влиять на разные стадии биосинтеза нуклеозидов.
Основные различия биосинтеза нуклеозидов de novo у разных организмов. Принципы создания антипаразитарных препаратов
Пуриновый и пиримидиновый биосинтез de novo есть у всех живых организмов всех трех доменов: бактерий, архей и эукариот [18]. Пути биосинтеза у бактерий и эукариот практически одинаковы, а у архей отличаются [19]. В геномах бактерий и архей кластеры генов, кодирующих ферменты биосинтеза de novo, организованы по-разному [20, 21]. Регуляция транскрипции этих генов также различается [19, 20]. Но ферменты, участвующие в биосинтезе, у бактерий и архей одинаковы, в то время как у некоторых эукариот ряд ферментов биосинтеза de novo отличается [18]. Так, у дрожжей Saccharomyces cerevisiae активные центры карбамоилфосфатсинтетазы (CPSase; EC 6.3.5.5) и аспартаткарбамоилтрансферазы (ATCase; EC 2.1.3.2) встроены в общую полипептидную цепь, которая содержит также неактивный вариант дигидрооротазы (DHOase; EC 3.5.2.3). Для пиримидинового биосинтеза клетки млекопитающих используют трифункциональный белок дигидрооротатсинтетазу, который содержит в одной полипептидной цепи три активных центра, выполняющих поочередно функции CPSase, ATCase, и DHOase [18, 20], и бифункциональный белок, в полипептидную цепь которого встроены активные центры оротатфосфорибозилтрансферазы (OPRTase; EC 2.4.2.10) и оротодин-5'-монофосфат-декарбоксилазы (OMPdecase; EC 4.1.1.23) [20]. Ферменты пиримидинового биосинтеза у низших эукариот находятся в митохондриях, а у млекопитающих − в цитозоле клеток [18, 20], что обеспечивает максимальную компактность реакционного пространства при синтезе de novo в клетках млекопитающих. Биосинтез de novo на 95% обеспечивает клетки нуклеотидами IMP и UMP [18]. Вирусы и фаги, как инфекционные агенты, размножающиеся только в живых клетках, используют в своем инфекционном цикле синтезированные хозяином нуклеозиды/нуклеотиды [22].
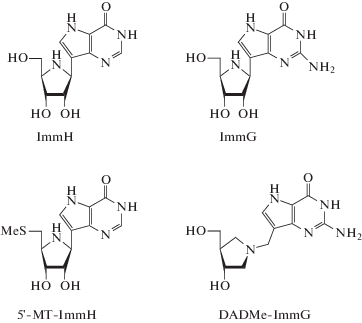
В процессе эволюции некоторые организмы утратили часть генов, необходимых для пуринового либо пиримидинового синтеза de novo. Это ауксотрофы – их существование возможно только при наличии в питательной среде производных пуринов/пиримидинов. Они могут паразитировать в других организмах, в частности в организме человека, вызывая различные заболевания. Ключевое значение для них имеет “реутилизационный” путь биосинтеза нуклеозидов. Следовательно, ферменты, катализирующие реакции дополнительного пути нуклеотидного обмена (среди них нуклеозидфосфорилазы), можно рассматривать в качестве мишеней для создания антибактериальных и антипротозойных лекарственных средств [23‒26]. К пуриновым ауксотрофам относятся такие организмы, как гельминт Schistosoma mansoni, вызывающий у человека эндемическое заболевание – шистосомоз [25, 26], и малярийный плазмодий, Plasmodium falciparum, ‒ возбудитель тяжелой формы малярии [23, 24]. К пиримидиновым ауксотрофам относится жгутиковый протист Giardia lamblia [27‒29], который может паразитировать в тонком кишечнике млекопитающих и птиц, вызывая заболевание лямблиоз. Поиск эффективных и высокоспецифичных ингибиторов нуклеозидфосфорилаз ‒ одна из стратегий создания препаратов для лечения заболеваний, вызываемых перечисленными паразитами. На этом пути уже достигнуты определенные результаты. Детально были исследованы производные иммуциллина (рис. 2), конформация которых близка предполагаемой переходной конформации субстрата пуриннуклеозидфосфорилаз [30, 31]. Ранее сообщалось, что 5'-метилтиоиммуцилин-H (5'‑methylthio-immucillin-H; 5'-MT-ImmH) связывается с пуриннуклеозидфосфолазой Plasmodium falciparum (PfPNP) в 112 раз эффективнее, чем с пуриннуклеозидфосфорилазой эритроцитов человека (Kd 2.7 и 303 нМ соответственно). Скорее всего, это обусловлено различным строением активных центров этих двух пуриннуклеозидфосфорилаз [31]. Предложено использовать 5'-MT-ImmH в качестве ингибитора PfPNP при лечении малярии у человека [9, 31, 32]. Другое производное иммуциллина, DADMe-Immucillin-G (BCX4945), в настоящее время проходит доклинические испытания. Уже продемонстрирована его высокая эффективность при лечении малярии на модели обезьян [33]. Структура его комплекса с пуриннуклеозидфосфорилазой эритроцитов человека исследована методом ЯМР [34].
МЕХАНИЗМ ПРОТИВОВИРУСНОГО ДЕЙСТВИЯ НУКЛЕОЗИДОВ
Ингибиторы полимераз
Механизм биологической активности большинства противовирусных лекарственных препаратов на основе нуклеозидов и их аналогов связан с их внутриклеточным превращением в 5′-моно-, ди- и трифосфаты под действием вирусных и клеточных киназ с последующим включением в нуклеиновые кислоты, что приводит к ингибированию репликации вирусного генома [3]. Геном вируса кодирует ряд белков и ферментов, необходимых для размножения вируса в инфицированной клетке, при этом поддержание жизненного цикла вируса осуществляется за счет метаболических путей клетки-хозяина с вовлечением соответствующих ферментов.
Вирусные ферменты, участвующие в репликации вирусных нуклеиновых кислот, представлены РНК-зависимой РНК-полимеразой (РНК-содержащие вирусы), РНК-зависимой ДНК-полимеразой (ретровирусы), ДНК-зависимой ДНК-полимеразой и ДНК-зависимой РНК-полимеразой для ДНК-содержащих вирусов. Вирусы простого герпеса (HSV), помимо ДНК-полимеразы как основного фермента репликации, кодируют тимидинкиназу, которая в клетке осуществляет 5'-монофосфорилирование нуклеозидов и их аналогов [35]. Вирусные ферменты обладают, как правило, меньшей специфичностью, чем ферменты клетки-хозяина, что обусловливает высокую скорость размножения вирусов и быстрое развитие устойчивости к действию лекарственных препаратов.
Действие нуклеозидов и их аналогов может быть нацелено на ингибирование синтеза нуклеиновых кислот вирусов, увеличение частоты летальных мутаций вирусных геномов (летальный мутагенез), возможны и другие механизмы их действия, направленные на ингибирование биосинтеза нуклеозидов [4]. У вирусов в синтезе нуклеиновых кислот участвуют ферменты, отличные от клеточных: ДНК-зависимая ДНК-полимераза, РНК-зависимая ДНК-полимераза (обратная транскриптаза, ОТ), РНК-зависимая РНК-полимераза. Нуклеозидные аналоги в форме NTP нацелены на подавление синтеза вирусных РНК/ДНК различными путями, которые представлены на рис. 3.
Рис. 3.
Основные пути действия лекарственных препаратов нуклеозидной природы на синтез вирусных РНК/ДНК. Трифосфатная форма аналога нуклеозида может выступать в качестве ингибитора РНК/ДНК-полимеразы (а), терминатора роста цепи РНК/ДНК (б), кинетического терминатора (в), а также действовать по механизму летального мутагенеза (г). Обозначения: N-киназа ‒ нуклеозидкиназа; НК ‒ нуклеиновая кислота; М ‒ мутация; pppM – аналог нуклеозид-5'-трифосфата; pppN – природный нуклеозид-5'-трифосфат; B – гетероциклическое основание; R = H, OH; R' = H, OH.
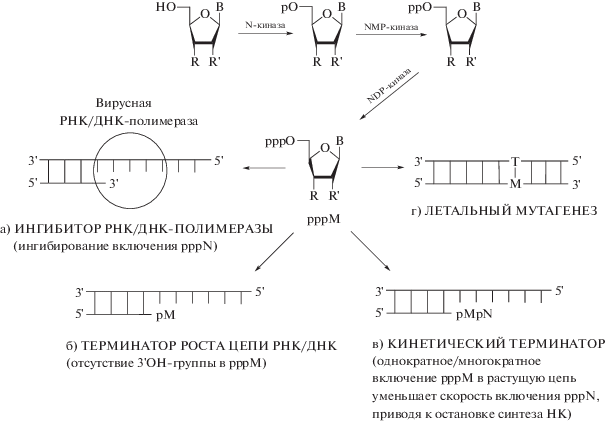
Любой модифицированный нуклеозид-5'-трифосфат может ингибировать синтез нуклеиновых кислот либо как конкурентный ингибитор включения природного NTP в растущую цепь нуклеиновой кислоты (рис. 3а), либо как терминатор роста цепи за счет встраивания нуклеозид-5'-трифосфата, не содержащего гидроксильной группы в 3'-положении, в растущую цепь РНК/ДНК (рис. 3б). В качестве терминаторов роста цепи могут выступать также нуклеозид-5'-трифосфаты, содержащие стерически малодоступную для протекания ферментативной реакции гидроксильную группу в 3'-положении. Например 3'-метил-UTP, который представляет собой терминирующий субстрат РНК-полимеразы Escherichia coli, может быть использован для секвенирования нуклеиновых кислот. В этом соединении 3'-метильная группа, которая замещает протон, существенно уменьшает реакционную способность 3'‑гидроксильной группы [36, 37].
Возможно также кинетическое терминирование роста цепи нуклеиновой кислоты ‒ когда модифицированный нуклеозид-5'-трифосфат может быть встроен в растущую цепь, но скорость его включения и последующая элонгация цепи сильно замедлены в сравнении с природным NTP. Кинетическое терминирование может быть результатом внутримолекулярных или межмолекулярных стерически неоптимальных взаимодействий фермента с модифицированным нуклеозид-5'-трифосфатом, а также нарушением водородных связей между матрицей и праймером (рис. 3в). В результате синтез цепи нуклеиновой кислоты останавливается. Это явление было впервые описано на рубеже 90-х годов XX века [37‒39]. В условиях односубстратной реакции, когда к предсинтезированной праймерной РНК вместе с природным UTP добавляли D-алло-диастереомер или L-тало-диастереомер 5'-метил-UTP (5'-aMe-UTP и 5'-tMe-UTP), 5'-aMe-UTP включался примерно в 100 раз, а 5'-tMe-UTP – в 1000 раз менее эффективно, чем природный субстрат UTP [38, 39]. В отсутствие UTP, природного субстрата РНК-полимеразы, к предсинтезированной РНК в этом случае присоединялось не более двух 5'-С-метил-уридиновых остатков, то есть эффективность последующего включения аналогов снижалась еще в большей степени по сравнению с условиями, в которых к ферменту добавляли смесь UTP и 5'‑aMe-UTP или 5'-tMe-UTP [38, 39].
Еще один механизм антивирусной активности соединений на основе нуклеозидов – это “летальный мутагенез”. Мутации при включении модифицированного нуклеозида в цепь РНК/ДНК могут приводить к образованию нежизнеспособных штаммов вируса (рис. 3г) [40 ] . Кроме того, наличие в клетке модифицированных нуклеозидов, а также соответствующих им NMP, NDP и NTP, может вызывать ингибирование биосинтеза нуклеиновых кислот за счет изменения концентраций природных NMP, NDP и NTP и их соотношений. Ниже рассмотрены основные механизмы действия ряда известных лекарственных препаратов.
Большинство нуклеозидных/нуклеотидных аналогов лекарственных препаратов, используемых в качестве противовирусных агентов, действуют как ингибиторы и терминаторы синтеза ДНК или РНК [2, 5].
Клинически одобренные лекарственные препараты ацикловир, ганцикловир, пенцикловир (табл. 2), представляющие собой ациклические аналоги нуклеозидов, а также их пролекарства, увеличивающие пероральную биодоступность (валацикловир, валганцикловир и фамцикловир), активны в отношении ДНК-содержащих вирусов: HSV и цитомегаловируса (CMV) [1]. Эти лекарственные препараты ингибируют полимеразы HSV и CMV, а также терминируют репликацию, действуя в форме трифосфатов. Геномы вирусов семейства Herpesviridae кодируют ряд ферментов метаболизма нуклеиновых кислот: тимидинкиназу, тимидилатсинтазу, дезоксиуридинтрифосфатазу и рибонуклеотидредуктазу [35]. Наибольший интерес с точки зрения внутриклеточной активации нуклеозидов и их аналогов представляет тимидинкиназа, катализирующая первую, скоростьлимитирующую, стадию ‒ монофосфорилирование. Тимидинкиназа HSV не относится к высокоспецифичным ферментам, поэтому может фосфорилировать не только тимидин, но и аналоги пуриновых нуклеозидов. Лекарственные препараты, одобренные Управлением по санитарному надзору за качеством пищевых продуктов и медикаментов США (FDA) для лечения HSV, ацикловир и пенцикловир после проникновения в клетку фосфорилируются вирусными тимидинкиназами, до соответствующих монофосфатов [41‒45]. Последующее двухступенчатое фосфорилирование ферментами клетки-хозяина: гуанилаткиназой и нуклеозиддифосфаткиназой – приводят к образованию их трифосфорилированных форм, которые конкурируют с природным субстратом dGTP за встраивание в ДНК и блокируют ее синтез [41]. Ганцикловир-5'-трифосфат и валганцикловир-5'-трифосфат включаются в вирусную ДНК и блокируют репликацию вируса, “работая” как кинетические терминаторы [43].
Таблица 2.
Противовирусные лекарственные препараты нуклеозидной природы [1, 2, 41‒45, 47, 50, 51, 54‒59, 61, 62, 65‒101]
| № | Вирус | Лекарственный препарат | Молекулярная мишень IC50 (Ki или Кd)a |
EC50b | Токсичность и побочные эффекты | Статус препарата | |
|---|---|---|---|---|---|---|---|
| I. Конкурентные ингибиторы вирусной ДНК-полимеразы | |||||||
| 1 | HSV, VZV | Видарабин (Vira-A®)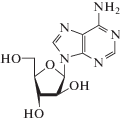 |
ДНК-полимераза | 62.7 ± 8.6 мкMс [90] |
34.7 ± 2.4 мкM (HSV-1/Vero) [90] 42.1 ± 1.8 мкM (HSV-2/Vero) [90] 1.6 ± 0.4 мкM (VZV/HEL) [91] |
LD50d > 5 020 мг/кг (крысы) [86] | Одобрен 1976 г. |
| Тимидин-киназа | ‒ | ||||||
| 2 | HBV | Телбивудин (Tyzeka®)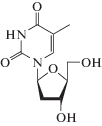 |
Полимераза HBV | 243 ± 9 нМe [61] | 8.950 ± 4.803 нМ [61] | Не описано [86] | Одобрен 2006 г. |
| II. Конкурентные ингибиторы вирусной ДНК-полимеразы и терминаторы роста цепи | |||||||
| 3 | HSV, VZV | Ацикловир (Acyclovir®)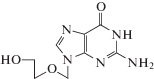 |
ДНК-полимераза | Kd 6 ± 1 мкMe (HSV) [92] | <0.56‒3.3 мкM (HSV/Vero) [85] 3.78 ± 0.58 мкM (HSV-1/Vero) [90] 8 ± 0.44 нМ (VZV/HEL) [91] |
Возбуждение, кома, судороги, вялость, осадок в почечных канальцах [86] | Одобрен 1982 г. |
| Тимидинкиназа | ‒ | ||||||
| 4 | HSV | Пенцикловир (Denavir®)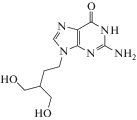 |
ДНК-полимераза, тимидинкиназа |
‒ | 0.16‒1.97 мкМ (HSV-1/WISH), 0.79‒2.37 мкМ (HSV-1/MRC-5), 0.39‒3.16 мкМ (HSV-2/WISH), 3.55‒8.29 мкМ (HSV-2/MRC-5) [93] |
Головная и эпигастральная боль, повышение уровня сывороточной липазы, тошнота, диспепсия, головокружение, гипербилирубинемия [86] | Одобрен 1993 г. |
| 5 | HSV, VZV, CMV | Валацикловир (Valtrex®)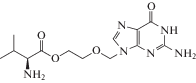 |
ДНК-полимераза, тимидинкиназа |
‒ | 0.09‒60 мкM(HSV-1) 0.04‒44 мкM (HSV-2) 0.53‒48 мкM (VZV) [85] | LD50 = 903.5 мг/кг (крысы) [86] | Одобрен 1995 г. |
| 6 | HSV, VZV | Фамцикловир (Famvir®)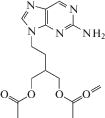 |
ДНК-полимераза, тимидинкиназа |
‒ | 1.2‒2.6 мкM (HSV-1/2) 6.7‒71 мкM (VZV) [93] | ‒ | Одобрен 1994 г. |
| 7 | HSV, CMV (у пациентов со СПИД) | Цидофовир (Vistide®)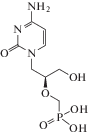 |
ДНК-полимераза | 6.6 ± 0.8 мкMf (CMV) [94] | 0.82‒1.29 мкМ (HSV/Vero) [95] |
Канцерогенез, эритропения, лимфопения, нефротоксичность, тератогенность [88] | Одобрен 1996 г. |
| Тимидинкиназа | ‒ | ||||||
| 8 | HBV | Адефовир дипивоксил (Hepsera®)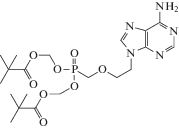 |
Полимераза HBV | 31 ± 2.7 нМf [61] | 2.636 ± 1.549 нМ (HBV/HepG2) [61] |
Нефротоксичность [86] | Одобрен 2002 г. |
| III. Конкурентные ингибиторы вирусной РНК/ДНК-полимеразы и кинетические терминаторы роста цепи | |||||||
| 9 | CMV | Ганцикловир (Zirgan®,
Cytovene®, Vitrasert®)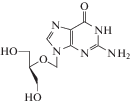 |
ДНК-полимераза, тимидинкиназа | ‒ | 0.08‒13.6 мкM [93] | Панцитопения, ухудшение работы ЖКТ, острая почечная недостаточность [86] | Одобрен 1989 г. |
| 10 | CMV | Валганцикловир (Valcyte®)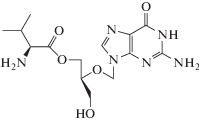 |
ДНК-полимераза, тимидинкиназа |
‒ | 0.08‒22.94 мкM [93] | Нефротоксичность [86] | Одобрен 2001 г. |
| 11 | VZV | Бривудинh (Zostex®, Brivirac®)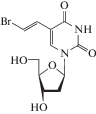 |
ДНК-полимераза, тимидинкиназа |
‒ | 3.00‒9.00 мМ (VZV) [50] |
‒ | Одобрен 2000 г. |
| 12 | HBV | Энтекавир (Baraclude®)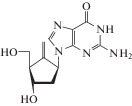 |
Полимераза HBV | 0.5 ± 0.1 нМe [61] | 5.3 ± 2.5 нМ (HBV/HepG2) [61] |
Не описано [86] | Одобрен 2005 г. |
| 13 | HCV | Софосбувир (Sovaldi®)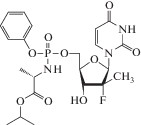 |
РНК-зависимая РНК-полимераза (NS5B) | 0.7‒2.6 мкMe [85] | 0.014‒0.11 мкM [85] | Головная боль, усталость, брадикардия (редко) [86] | Одобрен 2013 г. |
| 14 | EBOV, MERS-CoV, SARS-CoV-1, SARS-CoV-2 |
Ремдесивирi (Veklury®)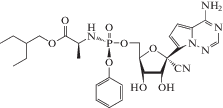 |
РНК-зависимая РНК-полимераза | 0.032 мкMf (MERS-CoV) [85] |
9.9 нM (SARS-CoV-2/HAE), 280 нM (SARS-CoV-2/CALU) [85] |
Не описано [86] | Одобрен для экстрен-ного ис-пользования в США (SARS-CoV-2) 2020 г. |
| IV. Конкурентные ингибиторы ОТ-ВИЧ и терминаторы роста цепи | |||||||
| 15 | ВИЧ | Зидовудин, Азидотимидин
(Azidothymidin, Retrovir®)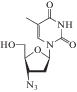 |
ОТ-ВИЧ, ОТ-теломеразы (TERT) |
270 ± 3.7 нMe [96] | 0.01‒0.49 мкM (моноциты, лимфоциты) [93] | LD50 = 3 084 мг/кг (перорально, мыши); усталость, головная боль, тошнота, рвота [86] | Одобрен 1987 г. |
| 16 | ВИЧ | Диданозин (Videx®) |
ОТ-ВИЧ | ‒ | 2.5‒10 мкМ (лимфобласты) 0.01‒0.1 мкМ (моноциты, макрофаги) [93] | Панкреатит, периферическая нейропатия, диарея, гиперурикемия, печеночная недостаточность [86] | Одобрен 1991 г. |
| 17 | ВИЧ | Зальцитабин (Hivid®) |
ОТ-ВИЧ | ‒ | 30‒500 нМ (лимфобласты, моноциты, лимофоциты) [85] | Передозировка >1.5 мг/кг; сыпь, лихорадка [86] |
Одобрен 1992 г. |
| 18 | ВИЧ | Ставудин (Zerit®)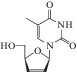 |
ОТ-ВИЧ | 0.0083‒0.032 мкMe [85] | 0.009‒4 мкM (моноциты лимфобласты, одноядерные клетки) [85] |
Периферическая нейропатия, жжение, онемение, боль в руках или ногах [86] | Одобрен 1994 г. |
| 19 | ВИЧ, HBV | Ламивудин (Epivir®)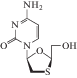 |
ОТ-ВИЧ, полимераза HBV | 6.2 ± 4.4 нMe (HBV) [61] | 1.491 ± 1.033 нM (HBV) [61] |
Головная боль, тошнота, недомогание, усталость, диарея, кашель [86] | Одобрен 1995 г. |
| 20 | ВИЧ | Абакавир (Ziagen®)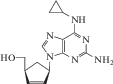 |
ОТ-ВИЧ | ‒ | 0.26 ± 0.18 мкM (лимфобласты, моноциты) [93] |
Головная боль, недомогание, усталость, тошнота, рвота, нарушение сна [86] | Одобрен 1995 г. |
| 21 | ВИЧ, HBV | Эмтрицитабин (Emtriva®)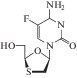 |
ОТ-ВИЧ | ‒ | 0.0013–0.64 мкM (лимфобласты, MAGI-CCR5, одноядерные клетки периферической крови) [93] |
Гепатотоксичность, ацидоз [86] | Одобрен 2003 г. |
| 22 | ВИЧ, HBV | Тенофовир дизопроксил фумарат (Viread®)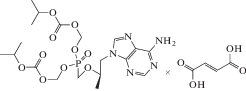 |
ОТ-ВИЧ, полимераза HBV |
‒ | 0.04‒8.5 мкM (ВИЧ/лимфобласты, моноциты, макрофаги, лимфоциты) [93] 2.482 ± 1.938 нM (HBV) [61] |
Карциногенез [86] | Одобрен 2001 г. (ВИЧ), 2008 г. (HBV) |
| V. Конкурентные ингибиторы вирусной РНК-полимеразы + ингибиторы IMPDHj + летальный мутагенез | |||||||
| 23 | HCV, RSVk, хантавирус, вирус гриппа |
Рибавирин (Copegus®, Rebetol®,
Virazole®)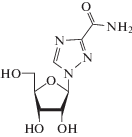 |
IMPDH | Ki 200 мкMg [98] | 8.4 мкM (HCV/Huh-7.5) [99] |
LD50 = 2 700 мг/кг (крысы) LD50 = 1 300 мг/кг (мыши) Эритропения, нарушение сна, раздражительность, ане-мия, нарушение функции печени (больные ВИЧ) [86] |
Одобрен 1985 г. |
| РНК-зависимая РНК-полимераза | ‒ | ||||||
| 24 | Вирус гриппа, SARS-CoV-2 | Фавипиравирl (Avigan®)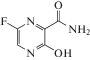 |
РНК-зависимая РНК-полимераза | ‒ | 0.43‒0.61 мкМ (вирус гриппа А (H1N1)/MDCK) 1.34‒1.97 мкМ (вирус гриппа А (H3N2)/MDCK) [100] |
LD50 > 2 000 мг/кг (крысы) LD50 > 1 000 мг/кг (собаки, обезьяны) У человека не описано; у животных: угнетение гематопоэза, эритропения, влияние на функцию гепатоцитов, тератогенность [86] |
Одобрен в Японии
в 2014 г. (вирус гриппа A, B, C); в России и КНР в 2020 г. (SARS-CoV-2) |
| VI. Ингибиторы синтеза вирусной ДНК, механизм полностью не изучен | |||||||
| 25 | HSV, VZV | Идоксуридин (Herplex®)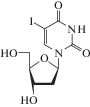 |
ДНК-полимераза | ‒ | 2.8 мкM (HSV-1) [101] 8.5-11.2 мкМ (BV) [101] |
LD50=3080 мг/кг (мыши); гиперчувствитель-ность глаз к свету [86] | Одобрен 1963 г. |
| Тимидинкиназа | 1.5 ± 0.2 мкMm [101] | ||||||
| 26 | HSV | Трифлуридин (Viroptic®)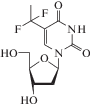 |
Тимидилатсинтаза | ‒ | < 1.7 мкM (HSV-1) [101] 1.7‒3.4 мкМ (BV) [101] |
LD50 = 2 946 мг/кг (крысы, внутривенно), LD50 > 4 379 мг/кг (перорально); по-давление функций костного мозга [86] |
Одобрен 1980 г. |
| Тимидинкиназа | 2.0 ± 0.1 мкMm [101] | ||||||
а IC50 ‒ концентрация 50%-ного ингибирования активности ферментата в присутствии NTP; Ki ‒ константа ингибирования, Kd ‒ константа диссоциации. В скобках указан вирус/клеточная культура, на которых проведено указанное исследование. b EC50 ‒ концентрация 50%-ного ингибирования репродукции вируса на культурах клеток в присутствии нуклеозидного аналога. c Дефицитный по тимидинкиназе штамм HSV-1 (B2006, мутантный ген тимидинкиназы). d LD50 ‒ доза, вызывающая гибель 50% популяции лабораторных животных. e 5'-трифосфатная форма. f 5'-дифосфатная форма. g Метиловый эфир рибавирин-5'-фосфата. h Препарат получил одобрение для лечения HSV/VZV в Европейском Союзе, но не одобрен FDA [59, 86]. i Препарат получил разрешение FDA на “экстренное использование” в США для лечения COVID-19 в ноябре 2020 года. j IMPDH (inosine-5'-monophosphate dehydrogenase) ‒ инозин-5'-монофосфатдегидрогеназа. k RSV (respiratory-syncitial virus) ‒ респираторно-синцитиальный вирус. l Препарат получил одобрение для лечения вирусов гриппа А, B, C в Японии в 2014 году, одобрен в Российской Федерации и Китае, но не одобрен FDA. m Тимидинкиназа вируса герпеса В обезьян (BV).
Аналог пуринового нуклеозида видарабин (табл. 2), нацеленный на HSV и вирус варицелла-зостер (VZV), в активной 5'-трифосфатной форме останавливает репликацию вирусной ДНК по механизму конкурентного ингибирования вирусной ДНК-полимеразы, замещая dATP [46].
Следует отметить, что наряду с основным механизмом действия аналогов нуклеозидов для них известны другие пути и мишени (рибонуклеозидредуктаза, полиаденилирование РНК и S-аденозил-гомоцистеингидролаза) [43]. Механизм противовирусного действия аналогов 2'-дезоксиуридина (идоксуридин, трифлуридин; табл. 2) подробно не изучен. Предполагается, что эти соединения ингибируют синтез ДНК вирусов герпеса. После монофосфорилирования вирусными специфичными тимидинкиназами, а затем клеточными киназами до 5'-трифосфатных форм эти лекарственные препараты конкурируют с dTTP за встраивание в вирусную ДНК [47]. После монофосфорилирования вирусными тимидинкиназами 5-(трифторметил)-2'-дезоксиуридин-5'-монофосфат необратимо ингибирует тимидилатсинтазу, а его трифосфат конкурирует с dTTP за включение в цепь ДНК, синтезируемую ДНК-полимеразой [43]. Механизм действия другого аналога 2'-дезоксиуридина – бривудина (лекарственный препарат одобрен для лечения вируса герпеса в ряде европейских стран) ‒ заключается в кинетическом терминировании синтеза ДНК вирусов HSV-1 и VZV [47‒50].
В качестве терминаторов репликации вирусов выступают также ациклические нуклеозидфосфонатные аналоги (цидофовир, адефовир дипивоксил, тенофовир дизопроксил фумарат; табл. 2), механизм действия которых основан на наличии фосфонатной связи (–PCO–) вместо обычной фосфатной (–POC–). Это характерная особенность нуклеотидных аналогов, активных в отношении полимераз CMV, ВИЧ и вируса гепатита B (HBV) [51]. Эти препараты изначально фосфорилируются клеточными киназами до фосфонодифосфатных форм, поскольку их фосфонатная группа имитирует 5'-монофосфат в нуклеотидах. Как и в случае других модифицированных нуклеозид-5'-трифосфатов, они конкурентно ингибируют включение природного субстрата в растущую цепь ДНК и, как следствие, активность ДНК-полимеразы. Фосфонатная связь, в отличие от фосфатной, не может быть расщеплена фосфодиэстеразой, что обеспечивает высокую устойчивость этих соединений в клетке [52, 53].
Лекарственные препараты адефовир и тенофовир способны обходить первую лимитирующую стадию фосфорилирования во внутриклеточном метаболизме нуклеозидных аналогов. В дифосфорилированных формах адефовир и тенофовир действуют в качестве альтернативных субстратов и терминаторов цепи в реакции синтеза ДНК, катализируемой обратной транскриптазой ВИЧ (ОТ-ВИЧ) [54‒56]. Ингибирующее действие адефовира и тенофовира на репликацию HBV может быть связано с терминированием роста цепи вирусной ДНК в присутствии ОТ. Адефовир и тенофовир в виде дифосфатов, очевидно, обладают большей аффинностью к ОТ-ВИЧ (и ОТ-HBV), чем к клеточным ДНК-полимеразам α, β, γ, δ и ε [57, 58], и это может объяснить их селективность в отношении ретровирусов и гепаднавирусов.
Все препараты в группе нуклеозидных ингибиторов ОТ-ВИЧ (зидовудин, диданозин, зальцитабин, ставудин, ламивудин, абакавир, эмтрицитабин; табл. 2), известные как 2',3'-дидезоксинуклеозидные аналоги, в виде 5'-трифосфатов терминируют синтез вирусной ДНК. Механизм их действия заключается в конкуренции с природными субстратами за встраивание в вирусную ДНК, синтезируемую ОТ-ВИЧ. Встроившись, они терминируют рост цепи вследствие отсутствия 3'-ОН группы [59].
Недавно сообщалось, что ОТ ретровирусов и гепаднавирусов имеют сходную структуру нуклеотидсвязывающего кармана [60]. Вот почему аналоги нуклеозидов, ингибирующие репродукцию ВИЧ: ламивудин и эмтрицитабин, ‒ оказываются эффективными и для лечения пациентов с гепатитом B.
Механизм действия других нуклеозидных препаратов (телбивудин и энтекавир; табл. 2) против HBV также заключается в каскаде трифосфорилирования исходных нуклеозидов с последующим ингибированием полимеразы HBV. Хотя телбивудин содержит 3'-OH группу, обеспечивающую удлинение цепи вирусной ДНК, альтернативный механизм действия может заключаться в терминации роста цепи ДНК [2]. Энтекавир-5'-трифосфат ингибирует обратную транскрипцию, осуществляемую HBV-полимеразой, тремя путями: (1) препятствует образованию ковалентного комплекса HBV-полимеразы с растущей (–)цепью вирусной ДНК (ингибирование прайминговой активности HBV-полимеразы); (2) кинетически терминирует синтез (–)цепи вирусной ДНК, встраиваясь в нее вместо dGTP; (3) кинетически терминирует синтеза (+)цепи вирусной ДНК на матрице (–)цепи, встраиваясь вместо dGTP. Данные молекулярного моделирования и изучения ферментативной активности позволяют предположить, что уникальное свойство энтекавира ингибировать сразу три вида активности HBV-полимеразы связано с оптимальным соответствием пространственному расположению боковых групп аминокислотных остатков в хвостовой части сайта связывания NTP при сохранении всех ключевых взаимодействий, характерных для природного субстрата dGTP [61, 62]. Энтекавир-5'-трифосфат ингибирует ДНК-полимеразу HBV (Ki 0.0012 мМ), будучи слабым ингибитором клеточных ДНК-полимераз α, β, δ (Ki 18–40 мМ) и митохондриальной ДНК-полимеразы (Ki > 160 мМ) [63], а телбивудин-5'-трифосфат практически не ингибирует полимеразы клеток человека [64]. В отличие от других ингибиторов HBV-полимеразы (ламивудин, адефовир дипивоксил), энтекавир содержит 3'-ОН группу, что не предотвращает включения еще нескольких нуклеотидов в синтезируемую полимеразой цепь вирусной ДНК (кинетическая терминация).
Софосбувир представляет собой ингибитор РНК-зависимой РНК-полимеразы (NS5B) вируса гепатита С (HCV). Софосбувир подвергается быстрому внутриклеточному метаболизму в печени с образованием активного аналога UTP, который связывается с активным сайтом NS5B и далее действует по механизму обрыва цепи, синтезируемой вирусной РНК [65‒69]. Аминокислотная последовательность активного сайта РНК-полимеразы NS5B высококонсервативна, что обеспечивает высокую селективность и низкую токсичность действия cофосбувира по результатам доклинических и клинических исследований [67].
Помимо ингибитора HBV-полимеразы энтекавира, к кинетическим терминаторам синтеза вирусных нуклеиновых кислот в ряду аналогов нуклеозидов относится ингибитор HSV-полимеразы бривудин, а также активный в отношении ряда РНК-вирусов: Эбола, коронавируса ближневосточного респираторного синдрома (MERS-CoV), коронавирусов тяжелого острого респираторного синдрома (SARS-CoV-1 и SARS-CoV-2) ‒ лекарственный препарат ремдесивир (табл. 2) [70‒74].
Еще один лекарственный препарат − рибавирин (табл. 2) ‒ ингибирует отдельные стадии биосинтеза нуклеотидов de novo, что снижает скорость репликации вирусов в клетке. Исторически считалось, что основной механизм действия рибавирина против ряда РНК-вирусов (HCV, респираторно-синцитиального вируса) заключается в ингибировании инозин-5'-монофосфатдегидрогеназы и связанном с ним снижении уровня GTP [75], необходимого для трансляции, транскрипции и репликации генома РНК-вируса. Однако важными альтернативными механизмами противовирусной активности рибавирин-5'-трифосфата может быть конкурентное ингибирование РНК-полимеразы, а также летальный мутагенез [76], при котором не происходит мгновенного ингибирования, а генерируются вирусные геномы, содержащие ошибочные нуклеотиды. Рибавирин-5'-трифосфат включается в РНК вместо ATP и GTP и образует комплементарные пары с уридином и цитидином, что резко увеличивает частоту мутаций, приводящих к появлению нежизнеспособных популяций РНК-вирусов [77‒79].
Лекарственный препарат фавипиравир (табл. 2), первоначально одобренный в Японии в 2014 году для лечения гриппа, оказался эффективным средством против SARS-CoV-2 [80]. Существует несколько мнений о механизме действия этого соединения. Некоторые исследователи полагают, что человеческая гипоксантин-гуанин-фосфорибозилтрансфераза играет ключевую роль в процессе активации фавипиравира как аналога азотистого основания, превращая фавипиравир в 5'‑фосфорибозилированный метаболит, который фосфорилируется киназами до активного 5'-трифосфата и затем ингибирует РНК-зависимую РНК-полимеразу вируса, конкурируя с ее нативными субстратами GTP и ATP [81, 82]. В других исследованиях показано, что фавипиравир может действовать и по механизму летального мутагенеза [83, 84]. Фавипиравир, как и рибавирин, несет карбоксамидную группу, присоединенную к остатку триазола и свободно вращающуюся вокруг С‒С связи, поэтому он может имитировать как гуанин, так и аденин. Соответственно, концепция летального мутагенеза вируса, впервые описанная для рибавирина [79], может быть справедлива и для фавипиравира.
Таким образом, аналоги нуклеозидов представляют собой класс противовирусных агентов с широким спектром биологической активности. Уже доказана их эффективность в отношении ряда РНК-вирусов, включая ретровирус ВИЧ, пикорно-, тога-, рабдо-, покс- и герпесвирусы.
Пролекарственные формы 5'-нуклеозидфосфатов
Специфичность киназ, катализирующих последовательные стадии 5'-фосфорилирования нуклеозидов в клетке, ограничивает возможности применения нуклеозидов для лечения инфекционных заболеваний. Применение в качестве медицинских препаратов 5'-фосфорилированных форм модифицированных нуклеозидов позволяет избежать первой скоростьлимитирующей стадии фосфорилирования нуклеозидов и повысить эффективность их действия на зараженные вирусом клетки [102, 103]. Подобная стратегия имеет ряд недостатков, так как введение полярной фосфатной группы в состав модифицированных нуклеозидов существенно затрудняет проникновение таких соединений в клетку, а внутри они подвергаются действию хозяйских фосфатаз. В связи с этим применение вместо 5'-фосфатных форм нуклеозидов 5'‑фосфонатных производных, не подверженных дефосфорилированию, повышает устойчивость целевого соединения и улучшает его проникновение через клеточную мембрану. На основе фосфонатных производных нуклеозидов и их ациклических аналогов разработан ряд лекарственных средств, способных превращаться в трифосфорилированные формы и подавлять репликацию вирусов в клетке (рис. 4, табл. 2) [102, 104].
Рис. 4.
Фосфонатные пролекарственные формы аналогов нуклеотидов. (S)-HPMPA ‒ (S)-9-(3-гидрокси-2-фосфонилметоксипропил)аденин.
Фосфазид – нуклеозидный ингибитор обратной транскриптазы (НИОТ), применяемый в антиретровирусной терапии ВИЧ [104]. Отечественный препарат на его основе Никавир® (Nikavir) зарекомендовал себя как эффективный нетоксичный препарат для проведения антиретровирусной терапии. Ациклические аналоги фосфазида, препараты адефовир и тенофовир, применяемые в антиретровирусной терапии, подавляют репродукцию ВИЧ и HBV [102]. Для тенофовира дизопроксил фумарата, выпущенного на рынок в 2001 году, продемонстрирована повышенная эффективность по сравнению с тенофовиром, что связано с наличием двух биодеградируемых гидрофобных остатков, улучшающих проникновение этого соединения в клетку. Цидофовир (S-HPMPC) представляет собой производное цитидина, модифицированное по рибофуранозному фрагменту с сохранением 2'- и 5'-гидроксильных групп, активное против большинства ДНК-вирусов, в том числе полиомавирусов, папилломавирусов, ортопоксвирусов, HCV, VZV, вируса Эпштейна–Барр, аденовирусов и CMV [102].
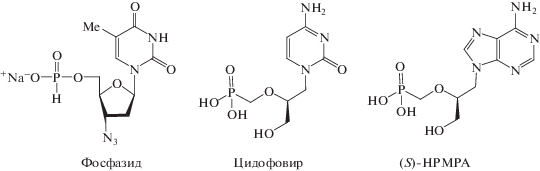
Альтернативой фосфонатному методу считается ПРОТИД-технология (ПРОлекарство + нуклеоТИД), которая была предложена в начале 90-х годов XX века [105, 106]. В основе этого подхода лежит химическая модификация нуклеозид-5'‑фосфатов путем введения двух липофильных биодеградируемых остатков в состав фосфатной группы (рис. 5). Полученные производные легко проникают через клеточную мембрану, эффективно накапливаются в клетке, постепенно метаболизируются до биологически активных нуклеозид-5'-фосфатов. К преимуществам ПРОТИД-технологии по сравнению с фосфонатным методом относится снижение токсичности модифицированных нуклеозидов. Первые эксперименты по применению ПРОТИД-технологии в антивирусной терапии связаны с применением арилфосфатных и фосфорамидатных производных зидовудина [106]. На T-лимфоцитах (линия клеток JM) было показано, что 5'-диарилфосфат зидовудина в концентрации порядка 10 мкM подавлял репликацию ВИЧ-1, в то время как исходный зидовудин был практически не активен на этой линии клеток. Замена одной арильной группы на остаток аланина приводила к существенному увеличению ингибиторной активности 5'-аланил(арил)фосфорамидата зидовудина (EC50 0.8 мкM) по сравнению 5'-диарилфосфатом (EC50 10 мкM). Введение аминокислотных остатков, содержащих гидрофобную боковую цепь, представляется перспективным ввиду повышенной способности проникать в клетку и возможности гидролитического расщепления ферментами внутри клетки.
Рис. 5.
Пролекарственные формы аналогов нуклеозидфосфатов, полученные с применением ПРОТИД-технологии. AZT ‒ азидотимидин.
Модифицированное фосфонатное нуклеозидное пролекарство тенофовира алафенамид обладает более высокой активностью в отношении ВИЧ и HCV и лучше распределяется в тканях по сравнению с тенофовиром. В ряду пуриновых и пиримидиновых 2'C-метилнуклеозидов синтезированы такие производные, как 7-деаза-2'C-метил-аденозин, 2'-метил-2'-фторцитидин, 2'-метил-2'-фторуридин, которые подавляют репликацию ряда РНК- и ДНК-вирусов [103]. Модификации 5'-фосфатного звена приводят к повышению противовирусной активности 2'С-метилнуклеозидов в клетке. Соединение IDX-184, содержащее S-пивалоил-2-тиоэтильный фрагмент и N-бензилфосфамидную группу, оказалось мощным ингибитором репликации HCV (EC50 0.03‒0.2 мкM). Аналог IDX184 ‒ INX189 ‒ проявлял высокую противовирусную активность в отношении HCV (EC50 0.01 мкM). Наличие модифицированного фосфатного остатка способствует большей эффективности INX189 за счет создания более высокой концентрации NTP в клетках по сравнению с исходным 2'С-метилгуанозином (внутриклеточная концентрация NTP из INX189 в 4 раза больше концентрации NTP из 2'С-метилгуанозина через 24 ч после обработки культуры клеток). Применение другого производного 2'С-метилгуанозина, BMS-986094 (INX-189 в комбинации с рибавирином), давало значительный синергетический эффект в ингибировании репликации вируса Денге in vitro (EC90 0.11 мкM для INX-189, EC90 100 мкM для рибавирина, при комбинации двух препаратов: EC90 0.022 мкM для INX-189 и EC90 10 мкM для рибавирина) [107].
В ходе исследования зависимости “структура‒активность” на модифицированных производных пиримидиновых нуклеозидов было показано, что ПРОТИД-производное 2'-метил-2'-фторуридина (рис. 5) эффективно ингибировало репликацию HSV, в то время как исходный нуклеозид, 2'-метил-2'-фторуридин, проявлял очень слабую противовирусную активность. Sp-изомер (расположение заместителей при атоме фосфора соответствует S-конфигурации) этого ПРОТИД-производного (лекарственный препарат софосбувир) намного активнее (EC90 0.42 мкM) Rp-изомера (расположение заместителей при атоме фосфора соответствует R-конфигурации, EC90 7.5 мкM) и используется для лечения гепатита С в комбинации с другими лекарственными препаратами (рис. 5).
Ремдесивир, аналог аденозин-5ʹ-монофосфата, считается перспективным пролекарством, способным ингибировать РНК-зависимую РНК-полимеразу коронавирусов. Этот препарат изначально разрабатывали для терапии лихорадки Эбола и лихорадки Марбург и обнаружили его противовирусную активность в отношении SARS-CoV (EC50 0.069 мкМ) и MERS-CoV (EC50 0.074 мкМ). Ремдесивир метаболизируется в трифосфатную форму намного быстрее, чем его аналог 1'-циано-4-аза-7,9-дидеазааденозин, обладающий значительно более низким показателем противовирусной активности в отношении вирусов SARS-CoV и MERS-CoV. Согласно недавним исследованиям, ремдесивир эффективно ингибирует SARS-CoV-2 в культуре клеток Vero E6 (EC50 0.77 мкМ), при этом обладает низкой цитотоксичностью (концентрация 50%-ной гибели клеток CC50 > > 100 мкМ) и высоким индексом селективности (SI > 129.87) [80].
При модификации структуры ремдесивира путем введения 2'С-метильной группы и 3'O-изобутироильной группы получен новый пролекарственный нуклеозидный аналог (GS-6620, Sp-изомер), который эффективно накапливается в гепатоцитах и подавляет репродукцию репликонов HCV в низкой концентрации (EC50 в диапазоне 68‒427 нM) [108, 109].
В клетке фосфамидный остаток ПРОТИД-лекарства подвергается ферментативному расщеплению в присутствии эстеразы (CES1) и катепсина А (CTSA) c образованием соответствующих нуклеозид-5'-монофосфатов. Именно поэтому ПРОТИД-лекарства эффективны для борьбы с вирусами, инфицирующими клетки с экспрессией CES1 и CTSA (например, HCV в гепатоцитах) [110].
В некоторых случаях стадия монофосфорилирования не лимитирует общую скорость реакции трифосфорилирования, что может быть связано со структурными особенностями производного нуклеозида. Например, для азидотимидина скоростьлимитирующая стадия ‒ образование 5'-дифосфатной формы под действием тимидилаткиназы. Нуклеозиддифосфаткиназа работает с низкой эффективностью на неоптимальных для нее субстратах: уридин-5'-дифосфате и 2',3'-дидегидро-2',3'-дидезоксиуридин-5'-дифосфате [111].
В связи с вышеперечисленными факторами разработка пролекарственных три- и дифосфатных форм нуклеозидов считается более универсальным подходом, позволяющим обойти потенциальные скорость-лимитирующие стадии и побочные метаболические процессы, протекающие в основном на уровне нуклеозидов и их 5'-монофосфатов (например, дезаминирование), и в конечном счете эффективно доставлять биологически активные NDP и NTP в клетку. Ввиду низкой устойчивости и высокой полярности NDP и NTP основные сложности при разработке таких лекарственных форм связаны с низкой устойчивостью пирофосфатной связи и доставкой таких соединений в клетку.
Новый синтетический подход, разработанный в лаборатории проф. Криса Мейера (Chris Meier), предполагает: 1) использование частично заряженных форм NDP и NTP, обладающих устойчивостью к гидролизу; 2) введение биодеградируемых липофильных остатков по концевой фосфатной группе. Таким способом уже получен ряд биологически активных транспортных форм AZT и ставудина (рис. 6) и изучена их противовирусная активность [111]. Химический гидролиз пролекарственной формы AZT в виде δ-циклосалигенилдифосфата (рис. 6а) приводит к образованию смеси 5'-моно- и 5'-дифосфатной форм AZT с преобладанием в смеси монофосфатной формы. С учетом этого факта в качестве более перспективных пролекарственных форм предложено использовать бис(ацилоксибензил)нуклеозидди- и -трифосфаты ‒ DiP- и TriP-пролекарства (рис. 6б). Обработка DiP- и TriP-производных ставудина (II) клеточными экстрактами, сывороткой крови или карбоксиэстеразой из печени свиньи приводила к образованию преимущественно соответствующих ди- и трифосфатов; при этом несимметричные пролекарственные формы (R1 ≠ R2, R1-Me или Bu, R2 > С4H9) были более устойчивы к ферментативному гидролизу пирофосфатной группы по сравнению с симметричными (R1 = R2).
Рис. 6.
Гидролиз дифосфатных и трифосфатных пролекарственных форм AZT (а) и cтавудина d4T (б) (составлено по данным работы [111]).
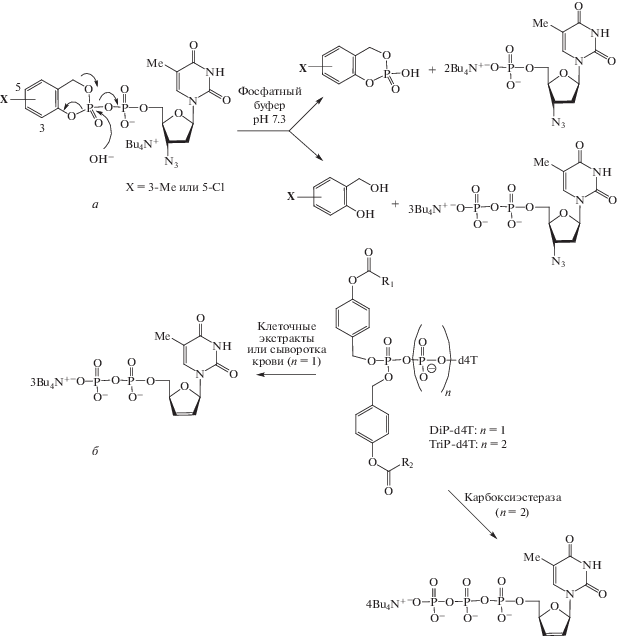
В опытах на T-лимфоцитах DiP- и TriP-производные ставудина (рис. 6б) более эффективно ингибировали ОТ-ВИЧ, чем нефосфорилированный препарат (d4T). Пролекарственные формы нуклеозидов, содержащие ацильные остатки с R1,2 = C8H17, проявляли высокую анти-ВИЧ-активность в опытах на культуре клеток CEM, что доказывает их способность проникать через клеточные мембраны с последующим высвобождением фосфорилированных метаболитов d4T.
Таким образом, фосфатные пролекарственные формы на основе модифицированных нуклеозидов ‒ уже хорошо зарекомендовавший себя подход к созданию новых противовирусных средств. Введение гидрофобных остатков в 5'-фосфатную группу модифицированного нуклеозида (ПРОТИД-технология) позволяет повысить эффективность его действия как ингибитора репликации вируса. ПРОТИД-технология с применением фосфамидных производных нуклеозидов ‒ еще один современный подход к созданию эффективных лекарственных средств для терапии вирусных и онкологических заболеваний. Именно на основе ПРОТИД-технологии получено 3 эффективных противовирусных лекарственных препарата: софосбувир для лечения гепатита С, алафенамид тенофовира для антиретровирусной терапии ВИЧ и гепатита С и ремдесивир для лечения COVID-19.
В качестве альтернативы рассмотренным пролекарственным формам исследуют нуклеозидные аналоги, содержащие остатки никотиновой кислоты [112]. Введение остатков никотиновой кислоты в углеводное звено нуклеозида позволяет снизить токсичность препарата, повысить его устойчивость в клетке, продлить время жизни в кровотоке, а также улучшить проницаемость через гематоэнцефалический барьер [113, 114].
ГИДРОФОБНЫЕ СОЕДИНЕНИЯ С ПРОТИВОВИРУСНЫМИ СВОЙСТВАМИ
Гидрофобные соединения ненуклеозидной природы с противовирусными свойствами
В основе механизма биологической активности ряда противовирусных лекарственных препаратов с выраженными гидрофобными свойствами лежит способность блокировать работу вирусных ферментов, связываясь с их определенным участком. В отличие от лекарств, созданных на нуклеозидной основе, мишенями которых служит нуклеозидсвязывающий активный центр полимераз, эти соединения не подвергаются химическим превращениям внутри клетки. Их мишени ‒ гидрофобные участки вирусных ферментов.
В структуре ОТ-ВИЧ-1 есть гидрофобная область, так называемый “гидрофобный карман”, которая находится на расстоянии около 15 Å от нуклеотидсвязывающего центра [115]. При попадании гидрофобного соединения в этот “карман” и формировании с ним прочной связи фермент теряет конформационную подвижность, что ведет к аллостерическому ингибированию его активности [116]. Ингибиторы гидрофобной природы, которые способны связаться в гидрофобном кармане ОТ-ВИЧ-1, ‒ это ненуклеозидные ингибиторы (ННИОТ ВИЧ-1). Они специфичны по отношению к структуре гидрофобного кармана ОТ-ВИЧ-1 и не обладают активностью в отношении ОТ-ВИЧ-2 и других ретровирусов. В клинической практике эти ингибиторы используют в комплексной терапии ВИЧ-1-инфекции [45]. К одобренным FDA препаратам ННИОТ ВИЧ-1 первого поколения относятся: невирапин, делавиридин, эфавиренц, ‒ к препаратам второго поколения: этравирин и рилпивирин. Класс диарилпиримидинов, препаратов второго поколения, оказался перспективным для создания ингибиторов такого рода в силу конформационной гибкости, в результате которой они могут “перестраиваться” и “адаптироваться” к различным аминокислотным заменам внутри гидрофобного кармана. В 2018 году FDA был одобрен еще один препарат этого класса – доравирин (рис. 7).
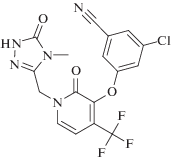
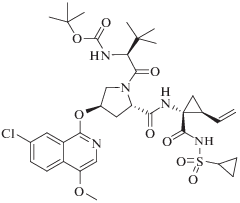
Новые варианты ингибиторов класса диарилпиримидинов активно исследуют в настоящее время в качестве потенциальных противовирусных средств [117‒121]. Так, HCV – это РНК-содержащий оболочечный вирус, который относится к семейству Flaviviridae. В структуре РНК-зависимой РНК-полимеразы этого вируса, NS5B, есть гидрофобный карман, который находится на расстоянии 30 Å от нуклеотидсвязывающего активного центра [122]. Исследовано большое число гидрофобных ароматических соединений, способных связаться с этим сайтом NS5B [123‒129]. В настоящее время при лечении HCV используют препарат дасабувир ‒ аллостерический ингибитор ферментативной активности NS5B из HCV генотипа 1а и 1b (IC50 2.8 и 10.7 нМ соответственно). Этот препарат получил одобрение FDA и включен в список жизненно важных препаратов. Другой препарат класса пиперазинкарбоксамидов – беклабувир (рис. 8); он тоже “работает” как аллостерический ингибитор РНК-полимеразы HCV. Эффективность и безопасность его применения исследована в составе комплексов: беклабувир + пегилированный интерферон + рибавирин [130] и беклабувир + асунапревир (ингибитор сериновой протеазы NS3) + даклатасвир (ингибитор белка NS5A) [131]. На основании клинической оценки эффективности, безопасности и фармакокинетических свойств этих комплексов Бюро фармацевтики и безопасности пищевых продуктов Японии (Pharmaceutical and Food Safety Bureau, PFSB) одобрило применение препарата Ximency. Этот препарат представляет собой второй из приведенных выше комплексов: беклабувир + асунапревир + даклатасвир ‒ с фиксироваными дозами всех компонентов, для лечения пациентов, инфицированных HCV генотипа 1 [132].
Влияние гидрофобных заместителей на противовирусную активность производных нуклеозидов
Производные пиримидинов, такие как 2',5 '-бис-O-(трет-бутилдиметилсилил)-пентафуранозил-3'-спиро-5"-(4"-амино-1",2"-оксатиол-2",2"-диоксид)пиримидин (TSAO), способны связываться с ОТ-ВИЧ-1 in vitro и ингибировать активность фермента по аллостерическому механизму [133‒136]. Однако при сравнении эффективности ингибирования ОТ-ВИЧ этими производными нуклеозидов с эффективностью ННИОТ-ВИЧ эти производные пиримидинов существенно проигрывают. Их ингибиторный эффект теряется даже при минорных аминокислотных заменах в структуре ОТ-ВИЧ-1, не влияющих на ингибиторные свойства известных ННИОТ-ВИЧ [137]. Кроме того, эти аналоги нуклеозидов высокотоксичны, а попытки снизить токсичность, варьируя структуру заместителей, приводят к снижению их ингибиторной активности [138].
Исследования противовирусной активности различных производных нуклеозидов были проведены в клеточных системах в отношении большой группы флавивирусов, которые переносятся членистоногими (арбовирусов) и относятся к возбудителям социально значимых эндемических заболеваний [139‒144]. Среди них вирус клещевого энцефалита, вирус Денге, вирус желтой лихорадки, вирус японского энцефалита, вирус Западного Нила. Оказалось, что наиболее высокую противовирусную активность in vitro проявляют нуклеозиды, содержащие трифенилметильные заместители в разных положениях пентафуранозного цикла. Механизм их действия пока нельзя считать окончательно установленным, хотя Eyer L. и соавт. [145], основываясь на данных по ингибированию вируса Денге в клеточных системах, высказали предположение, что такие производные нуклеозидов ингибируют процесс репликации вирусной РНК, взаимодействуя с активным центром РНК-зависимой РНК-полимеразы.
В тех случаях, когда объемные гидрофобные заместители находятся в аналоге нуклеозида при агликоне, противовирусное действие определяется амфипатическим характером этих соединений. Они ингибируют начальную стадию слияния наружной мембраны оболочечных вирусов с оболочками инфицируемых клеток. Противовирусная эффективность таких производных нуклеозидов продемонстрирована на примере HSV-1 и HSV-2 (IC50 48 нМ) [146], а также вируса клещевого энцефалита (IC50 18‒24 нМ) [147]. В концентрациях, достаточных для создания противовирусного эффекта, такие нуклеозиды не оказывают ни цитотоксического, ни цитостатического эффекта на клетки.
ЗАКЛЮЧЕНИЕ
В обзоре рассмотрены структурно-функциональные характеристики известных аналогов нуклеозидов, которые обусловливают их противовирусные или противопаразитарные свойства. Знание особенностей клеточного метаболизма природных нуклеозидов необходимо, чтобы идентифицировать те стадии обменных процессов, которые могут быть чувствительны к лекарственным препаратам, созданным на основе нуклеозидов. Мишенями для противопаразитарных средств могут быть нуклеозидфосфорилазы ‒ в тех случаях, когда эти ферменты у человека и паразита имеют заметные отличия. Противовирусное действие аналогов нуклеозидов в основном связано с их способностью подвергаться ферментативному фосфорилированию внутри клетки с образованием модифицированных нуклеозид-5'-моно-, ди- и трифосфатов. Эти метаболиты затем конкурируют с природными NTP за связывание с активным центром вирусных полимераз и либо остаются только конкурентными ингибиторами, либо становятся терминаторами роста цепи вирусной ДНК/РНК или триггерами мутагенеза вирусного генома. Аналоги нуклеозидов, несущие гидрофобные заместители, могут быть как ингибиторами вирусных ферментов, участвующих в репликации нуклеиновых кислот вируса, так и ингибиторами процесса проникновения вируса в клетку, а также могут действовать на клеточные ферменты, косвенно задействованные в репликации вирусов (рибавирин). Конкретный механизм противовирусного действия зависит от структуры и положения заместителя в нуклеозиде.
Специфическое ингибирование вирусных ферментов соединениями нуклеозидной и ненуклеозидной природы, содержащими гидрофобное звено, может происходить и по аллостерическому механизму, если в структуре фермента присутствуют гидрофобные участки на относительно небольшом расстоянии от активного центра. К ферментам, обладающим такой структурой, принадлежат ОТ-ВИЧ-1 и РНК-зависимая РНК-полимераза ряда вирусов, что позволяет считать их перспективными мишенями при поиске новых противовирусных средств.
Практически все лекарственные препараты помимо желаемого действия вызывают и побочные эффекты, связанные с токсическим действием препарата на здоровые клетки, на системы инактивации и выведения лекарственного препарата печенью и почками. Лекарства на основе нуклеозидов могут оказывать токсический эффект из-за ингибирования ферментов, метаболизирующих природные нуклеозиды. Еще одна серьезная проблема, ограничивающая применение нуклеозидных препаратов, ‒ развитие у инфекционного агента лекарственной резистентности. Это, как правило, следствие длительного применения препарата при лечении латентных и хронических инфекций. Резистентность возникает при накоплении в геноме патогена мутаций, приводящих к образованию лекарственноустойчивых штаммов [148]. Для повышения эффективности и снижения дозировки препарата важна избирательность его действия на целевые мишени в организме.
В настоящее время при создании препаратов на основе нуклеозидов придерживаются стратегии, которая позволяет снизить их токсичность для человека и повысить эффективность действия на инфицированные клетки. И эта стратегия базируется на использовании пролекарственных форм модифицированных нуклеозидов. Комбинирование гидрофобных остатков при 5'-фосфатной группе модифицированного нуклеозида (ПРОТИД-технология) позволяет усилить его действие на зараженные вирусом клетки. ПРОТИД-технология с применением фосфорамидатных производных нуклеозидов – это высокоэффективный подход к созданию новых лекарств для терапии вирусных заболеваний. С помощью этой технологии получено три эффективных противовирусных препарата: софосбувир для лечения гепатита С, алафенамид тенофовира для антиретровирусной терапии ВИЧ-инфекции и гепатита С и ремдесивир для лечения COVID-19. В настоящее время разрабатываются подходы, связанные с применением ди- и трифосфатных форм нуклеозидных пролекарств, способных оказывать селективное действие за счет их накопления в клетке.
Авторы выражают благодарность к.х.н. Зенченко В.О. за помощь в подготовке графических материалов, к.х.н. Ословскому В.Е. и к.х.н. Осолодкину Д.И. за ценные замечания и рекомендации при подготовке обзора.
Исследование выполнено при финансовой поддержке РФФИ в рамках научных проектов № 18-29-09037 и № 20-34-70116.
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
De Clercq E., Field H.J. (2006) Antiviral prodrugs – the development of successful prodrug strategies for antiviral chemotherapy. Br. J. Pharmacol. 147(1), 1‒11.
De Clercq E., Neyts J. (2009) Antiviral agents acting as DNA or RNA chain terminators. In: Antiviral Strategies. Eds Krausslich H.G., Bartenschlager R. Berlin, Heidelberg: Springer, 53‒84.
Jordheim L.P., Durantel D., Zoulim F., Dumontet C. (2013) Advances in the development of nucleoside and nucleotide analogues for cancer and viral diseases. Nat. Rev. Drug Discov. 12(6), 447‒464.
Jordan P.C., Stevens S.K., Deval J. (2018) Nucleosides for the treatment of respiratory RNA virus infections. Antiviral. Chem. Chemother. 26, 2040206618764483.
De Clercq E. (2019) New nucleoside analogues for the treatment of hemorrhagic fever virus infections. Chem. Asian J. 14(22), 3962–3968.
Postigo M.P., Guido R.V., Oliva G., Castilho M.S., da R. Pitta I., de Albuquerque J.F., Andricopulo A.D. (2010) Discovery of new inhibitors of Schistosoma mansoni PNP by pharmacophore-based virtual screening. J. Chem. Inf. Model. 50(9), 1693‒1705.
Tavares N.C., de Aguiar P.H.N., Gava S.G., Oliveira G., Mourão M.M. (2016) Schistosomiasis: setting routes for drug discovery. In: Special Topics in Drug Discovery. Eds Chen T., Chai S.C. London: InTechOpen, 105‒132.
El Kouni M.H. (2017). Pyrimidine metabolism in schistosomes: a comparison with other parasites and the search for potential chemotherapeutic targets. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 213, 55‒80.
Evans G.B., Tyler P.C., Schramm V.L. (2018) Immucillins in infectious diseases. ACS Inf. Dis. 4(2), 107‒117.
Drenichev M.S., Oslovsky V.E., Mikhailov S.N. (2016) Cytokinin nucleosides − natural compounds with a unique spectrum of biological activities. Curr. Top. Med. Chem. 16(23), 2562‒2576.
Oslovsky V.E., Drenichev M.S., Sun L., Kurochkin N.N., Kunetsky V.E., Mirabelli C., Neyts J., Leyssen P., Mikhailov S.N. (2017) Fluorination of naturally occurring N6-benzyladenosine remarkably increased its antiviral activity and selectivity. Molecules. 22(7), 1219.
Traut T.W. (1994) Physiological concentrations of purines and pyrimidines. Moll. Cell. Biochem. 140(1), 1‒22.
Jensen K.F., Dandanell G., Hove-Jensen B., WillemoËs M. (2008) Nucleotides, nucleosides, and nucleobases. EcoSal Plus. 3(1), 1‒39.
Mathews C.K., van Holde K.E. (1996) Biochemistry, 2nd ed. California: Benjamin Cummings Publishing Co., 784‒817.
Lewkowicz E.S., Iribarren A.M. (2006) Nucleoside phosphorylases. Curr. Org. Chem. 10(11), 1197‒1215.
Alexeev C.S., Kulikova I.V., Gavryushov S., Tara-rov V.I., Mikhailov S.N. (2018) Quantitative prediction of yield in transglycosylation reaction catalyzed by nucleoside phosphorylases. Adv. Synth. Catal. 360(16), 3090‒3096.
Valentin-Hansen P. (1978) Uridine-cytidine kinase from Escherichia coli. Methods Enzymol. 51, 308‒314.
Moran L.A., Horton H.R., Scrimgeour K.G., Per-ry M.D. (2012) Principles of Biochemistry, 5th ed. Ed. Jaworski A. London: Pearson Education.
Brown A.M., Hoopes S.L., White R.H., Sarisky C.A. (2011) Purine biosynthesis in archaea: variations on a theme. Biol. Direct. 6, 63.
Thia-Toong T.L., Roovers M., Durbecq V., Gigot D., Glansdorff N., Charlier D. (2002) Genes of de novo pyrimidine biosynthesis from the hyperthermoacidophilic crenarchaeote Sulfolobus acidocaldarius: novel organization in a bipolar operon. J. Bacteriol. 184(16), 4430‒4441.
He B., Zalkin H. (1992) Repression of Escherichia coli purB is by a transcriptional roadblock mechanism. J. Bacteriol. 174(22), 7121–7127.
Hayes W. (1964) The Genetics of Bacteria and Their Viruses. Studies in Basic Genetics and Molecular Biology, 2nd ed. New York: John Wiley and Sons.
Hyde J.E. (2007) Targeting purine and pyrimidine metabolism in human apicomplexan parasites. Curr. Drug Targets. 8(1), 31‒47.
Hazleton K.Z., Ho M.C., Cassera M.B., Clinch K., Crump D.R., Rosario I., Jr., Schramm V.L. (2012) Acyclic immucillin phosphonates: second-generation inhibitors of Plasmodium falciparum hypoxanthine-guanine-xanthine phosphoribosyltransferase. Chem. Biol. 19(6), 721‒730.
Senft A.W., Crabtree G.W. (1983) Purine metabolism in the schistosomes: potential targets for chemotherapy. Pharmacol. Ther. 20(3), 341‒356.
Lago E.M., Xavier R.P., Teixeira T.R., Silva L.M., da Silva Filho A.A., de Moraes J. (2018) Antischistosomal agents: state of art and perspectives. Future Med. Chem. 10(1), 89‒120.
Lindmark D.G., Jarroll E.L. (1982) Pyrimidine metabolism in Giardia lamblia trophozoites. Mol. Biochem. Parasitol. 5(5), 291‒296.
Aldritt S.M., Tien P., Wang C.C. (1985) Pyrimidine salvage in Giardia lamblia. J. Exp. Med. 161(3), 437‒445.
Vitti G.F., O’Sullivan W.J., Gero A.M. (1987) The biosynthesis of uridine 5′-monophosphate in Giardia lamblia. Int. J. Parasitol. 17(3), 805‒812.
Kicska G.A., Tyler P.C., Evans G.B., Furneaux R.H., Kim K., Schramm V.L. (2002) Transition state analogue inhibitors of purine nucleoside phosphorylase from Plasmodium falciparum. J. Biol. Chem. 277(5), 3219‒3225.
Shi W., Ting L.M., Kicska G.A., Lewandowicz A., Tyler P.C., Evans G.B., Schramm V.L. (2004) Plasmodium falciparum purine nucleoside phosphorylase: crystal structures, immucillin inhibitors, and dual catalytic function. J. Biol. Chem. 279(18), 18103‒18106.
Schramm V.L. (2018) Enzymatic transition states and drug design. Chem. Rev. 118(22), 11194‒11258.
Cassera M.B., Hazleton K.Z., Merino E.F., Obal-dia III N., Ho M.C., Murkin A.S., DePinto R., Gutierrez J.A., Almo S.C., Evans G.B., Babu Y.S., Schramm V.L. (2011) Plasmodium falciparum parasites are killed by a transition state analogue of purine nucleoside phosphorylase in a primate animal model. PLoS One. 6(11), e26916.
Suarez J., Haapalainen A.M., Cahill S.M., Ho M.C., Yan F., Almo S.C., Schramm V.L. (2013) Catalytic site conformations in human PNP by 19F-NMR and crystallography. Chem. Biol. 20(2), 212‒222.
Куханова М.К., Коровина А.Н., Кочетков С.Н. (2014) Вирус простого герпеса человека: жизненный цикл и поиск ингибиторов. Успехи биол. химии. 54, 457‒494.
Айвазашвили В.А., Михайлов С.Н., Падюкова Н.Ш., Карпейский М.Я., Бибилашвили Р.Ш. (1986) 1‑(3'-С-метил-β-D-рибофуранозил)урацил-5'-трифосфат – терминатор синтеза РНК, катализируемого РНК-полимеразой E. coli. Биоорган. химия. 12(5), 708‒710.
Tunitskaya V.L., Rusakova E.E., Padyukova N.S., Ermolinsky B.S., Chernyi A.A., Kochetkov S.N., Mikhailov S.N. (1997) Substrate properties of C′‑methyl UTP derivatives in T7 RNA polymerase reactions. Evidence for N-type NTP conformation. FEBS Lett. 400(3), 263‒266.
Айвазашвили В.А., Михайлов С.Н., Падюкова Н.Ш., Бибилашвили Р.Ш., Карпейский М.Я. (1987) 5'‑C-метилнуклеозидтрифосфаты в реакции синтеза РНК, катализируемой РНК-полимеразой Escherichia coli. Молекуляр. биология. 21(4), 1080‒1091.
Савочкина Л.П., Свиряева Т.В., Бейгельман Л.Н., Падюкова Н.Ш., Кузнецов Д.А., Лысов Ю.П., Михайлов С.Н., Бибилашвили Р.Ш. (1989) Субстратные свойства С-метилнуклеозидтрифосфатов в реакции синтеза РНК, катализируемой РНК-полимеразой Escherichia coli. Молекуляр. биология. 23(6), 1700‒1710).
Chen P., Shakhnovich E.I. (2009) Lethal mutagenesis in viruses and bacteria. Genetics. 183(2), 639‒650.
Elion G.B., Furman P.A., Fyfe J.A., De Miranda P., Beauchamp L., Schaeffer H.J. (1977) Selectivity of action of an antiherpetic agent, 9-(2-hydroxyethoxymethyl)guanine. Proc. Natl. Acad. Sci. USA. 74(12), 5716–5720.
Hodge R.V. (1993) Famciclovir and penciclovir. The mode of action of famciclovir including its conversion to penciclovir. Antiviral Chem. Chemother. 4(2), 67–84.
Aoki F.Y. (2015) Antivirals against herpes viruses. In: Mandell, Douglas, and Bennett’s Principles and Practice of Infectious Diseases, 8th ed. Eds Bennett J.E., Dolin R., Blaser M.J. Philadelphia: Elsevier, 546‒562.
Matthews T., Boehme R. (1988) Antiviral activity and mechanism of action of ganciclovir. Rev. Infect. Dis. 10(Suppl. 3), S490–S494.
De Clercq E., Li G. (2016) Approved antiviral drugs over the past 50 years. Clin. Microbiol. Rev. 29(3), 695‒747.
North T.W., Cohen S.S. (1979) Aranucleosides and aranucleotides in viral chemotherapy. Pharmacol. Ther. 4(1), 81‒108.
De Clercq E. (1997) In search of a selective antiviral chemotherapy. Clin. Microbiol. Rev. 10(4), 674–693.
Descamps J., De Clercq E. (1981) Specific phospho-rylation of E-5-(2-iodovinyl)-2'-deoxyuridine by herpes simplex virus-infected cells. J. Biol. Chem. 256(12), 5973‒5976.
Allaudeen H.S., Kozarich J.W., Bertino J.R., De Clercq E. (1981) On the mechanism of selective inhibition of herpesvirus replication by (E)-5-(2-bromovinyl)-2'-deoxyuridine. Proc. Natl. Acad. Sci. USA. 78(5), 2698‒2702.
De Clercq E. (2004) Discovery and development of BVDU (brivudin) as a therapeutic for the treatment of herpes zoster. Biochem. Pharmacol. 68(12), 2301‒2315.
De Clercq E. (2011) The clinical potential of the acyclic (and cyclic) nucleoside phosphonates. The magic of the phosphonate bond. Biochem. Pharmacol. 82(2), 99‒109.
De Clercq E. (2007) Acyclic nucleoside phosphonates: past, present and future. Bridging chemistry to HIV, HBV, HCV, HPV, adeno-, herpes-, and poxvirus infections: the phosphonate bridge. Biochem. Pharmacol. 73(7), 911‒922.
De Clercq E., Holý A. (2005) Acyclic nucleoside phosphonates: a key class of antiviral drugs. Nat. Rev. Drug Discov. 4(11), 928‒940.
Balzarini J., Hao Z., Herdewijn P., Johns D.G., De Clercq E. (1991) Intracellular metabolism and mechanism of anti-retrovirus action of 9-(2-phosphonylmethoxyethyl)adenine, a potent anti-human immunodeficiency virus compound. Proc. Natl. Acad. Sci. USA. 88(4), 1499‒1503.
Balzarini J., Holy A., Jindrich J., Dvorakova H., Hao Z., Snoeck R., De Clercq E. (1991) 9-[(2RS)-3-fluoro-2-phosphonylmethoxypropyl] derivatives of purines: a class of highly selective antiretroviral agents in vitro and in vivo. Proc. Natl. Acad. Sci. USA. 88(11), 4961‒4965.
De Clercq E. (1991) Broad-spectrum anti-DNA virus and anti-retrovirus activity of phosphonylmethoxyalkylpurines and -pyrimidines. Biochem. Pharmacol. 42(5), 963–972.
Cherrington J.M., Allen S.J.W., Bischofberger N., Chen M.S. (1995) Kinetic interaction of the diphosphates of 9-(2-phosphonylmethoxyethyl)adenine and other anti-HIV active purine congeners with HIV reverse transcriptase and human DNA polymerases α, β and γ. Antiviral Chem. Chemother. 6(4), 217‒221.
Kramata P., Votruba I., Otová B., Holý A. (1996) Different inhibitory potencies of acyclic phosphonomethoxyalkyl nucleotide analogs toward DNA polymerases alpha, delta and epsilon. Mol. Pharmacol. 49(6), 1005‒1011.
Beigel J.H. (2012) Antiviral therapy (Non-HIV). In: Goldman’s Cecil Medicine, 24th ed. Eds Goldman L., Schafer A.I. Philadelphia: Elsevier.
Beck J., Vogel M., Nassal M. (2002) dNTP versus NTP discrimination by phenylalanine 451 in duck hepatitis B virus P protein indicates a common structure of the dNTP-binding pocket with other reverse transcriptases. Nucleic Acids Res. 30(7), 1679‒1687.
Langley D.R., Walsh A.W., Baldick C.J., Eggers B.J., Rose R.E., Levine S.M., Kapur A.J., Colonno R.J., Tenney D.J. (2007) Inhibition of hepatitis B virus polymerase by entecavir. J. Virol. 81(8), 3992‒4001.
Seifer M., Hamatake R.K., Colonno R.J., Standring D.N. (1998) In vitro inhibition of hepadnavirus polymerases by the triphosphates of BMS-200475 and lobucavir. Antimicrob. Agents Chemother. 42(12), 3200–3208.
Scott L.J., Keating G.M. (2009) Entecavir. Drugs. 69(8), 1003‒1033.
Ruiz-Sancho A., Sheldon J., Soriano V. (2007) Telbivudine: a new option for the treatment of chronic hepatitis B. Expert Opin. Biol. Ther. 7(5), 751‒761.
Denning J., Cornpropst M., Flach S.D., Berrey M.M., Symonds W.T. (2013) Pharmacokinetics, safety, and tolerability of GS-9851, a nucleotide analog polymerase inhibitor for hepatitis C virus, following single ascending doses in healthy subjects. Antimicrob. Agents Chemother. 57(3), 1201‒1208.
Murakami E., Tolstykh T., Bao H., Niu C., Steu-er H.M.M., Bao D., Chang W., Espiritu C., Bansal S., Lam A.M., Otto M.J., Sofia M.J., Furman P.A. (2010) Mechanism of activation of PSI-7851 and its diastereoisomer PSI-7977. J. Biol. Chem. 285(45), 34337‒34347.
Asselah T. (2014) Sofosbuvir for the treatment of hepatitis C virus. Expert Opin. Pharmacother. 15(1), 121‒130.
Fung A., Jin Z., Dyatkina N., Wang G., Beigelman L., Deval J. (2014) Efficiency of incorporation and chain termination determines the inhibition potency of 2′‑modified nucleotide analogs against hepatitis C virus polymerase. Antimicrob. Agents Chemother. 58(7), 3636‒3645.
Koch U., Narjes F. (2007) Recent progress in the development of inhibitors of the hepatitis C virus RNA-dependent RNA polymerase. Curr. Top. Med. Chem. 7(13), 1302–1329.
Sheahan T.P., Sims A.C., Graham R.L., Mena-chery V.D., Gralinski L.E., Case J.B., Leist S.R., Pyrc K., Feng J.Y., Trantcheva I., Bannister R., Park Y., Babusis D., Clarke M.O., Mackman R.L., Spahn J.E., Palmiotti C.A., Siegel D., Ray A.S., Cihlar T., Jordan R., Denison M.R., Baric R.S. (2017) Broad-spectrum antiviral GS-5734 inhibits both epidemic and zoonotic coronaviruses. Sci. Transl. Med. 9(396), eaal3653.
Gordon C.J., Tchesnokov E.P., Woolner E., Perry J.K., Feng J.Y., Porter D.P., Götte M. (2020) Remdesivir is a direct-acting antiviral that inhibits RNA-dependent RNA polymerase from severe acute respiratory syndrome coronavirus 2 with high potency. J. Biol. Chem. 295(20), 6785‒6797.
Eastman R.T., Roth J.S., Brimacombe K.R., Simeo-nov A., Shen M., Patnaik S., Hall M.D. (2020) Remdesivir: a review of its discovery and development leading to emergency use authorization for treatment of COVID-19. ACS Cent. Sci. 6(5), 672–683.
Ferner R.E., Aronson J.K. (2020) Remdesivir in covid-19. Br. Med. J. 369, m1610.
Gordon C.J., Tchesnokov E.P., Feng J.Y., Porter D.P., Götte M. (2020) The antiviral compound remdesivir potently inhibits RNA-dependent RNA polymerase from Middle East respiratory syndrome coronavirus. J. Biol. Chem. 295(15), 4773‒4779.
Muller W.E., Maidhof A., Taschner H., Zahn R.K. (1977) Virazole (1-β-D-ribofuranosyl-1, 2, 4-triazole-3-carboxamide); a cytostatic agent. Biochem. Pharmacol. 26(11), 1071‒1075.
Parker W.B. (2005) Metabolism and antiviral activity of ribavirin. Virus Res. 107(2), 165‒171.
Graci J.D., Too K., Smidansky E.D., Edathil J.P., Barr E.W., Harki D.A., Galarraga J.E., Bollinger J.M., Jr., Peterson B.R., Loakes D., Brown D.M., Cameron C.E. (2008) Lethal mutagenesis of picornaviruses with N‑6-modified purine nucleoside analogues. Antimicrob. Agents Chemother. 52(3), 971‒979.
Crotty S., Cameron C., Andino R. (2002) Ribavirin’s antiviral mechanism of action: lethal mutagenesis? J. Mol. Med. 80(2), 86‒95.
Crotty S., Maag D., Arnold J.J., Zhong W., Lau J.Y.N, Hong Z., Andino R., Cameron C.E. (2000) The broad-spectrum antiviral ribonucleoside ribavirin is an RNA virus mutagen. Nat. Med. 6(12), 1375‒1379.
Wang M., Cao R., Zhang L., Yang X., Liu J., Xu M., Shi Z., Hu Z., Zhong W., Xiao G. (2020) Remdesivir and chloroquine effectively inhibit the recently emerged novel coronavirus (2019-nCoV) in vitro. Cell Res. 30(3), 269‒271.
Naesens L., Guddat L.W., Keough D.T., van Kuilenburg A.B.P., Meijer J., Voorde J.V., Balzarini J. (2013) Role of human hypoxanthine guanine phosphoribo-syltransferase in activation of the antiviral agent T-705 (favipiravir). Mol. Pharmacol. 84(4), 615‒629.
Jin Z., Smith L.K., Rajwanshi V.K., Kim B., Deval J. (2013) The ambiguous base-pairing and high substrate efficiency of T-705 (favipiravir) ribofuranosyl 5′-triphosphate towards influenza A virus polymerase. PLoS One. 8(7), e68347.
Baranovich T., Wong S.S., Armstrong J., Marjuki H., Webby R.J., Webster R.G., Govorkova E.A. (2013) T-705 (favipiravir) induces lethal mutagenesis in influenza A H1N1 viruses in vitro. J. Virol. 87(7), 3741‒3751.
Smee D.F., Hurst B.L., Egawa H., Takahashi K., Kadota T., Furuta Y. (2009) Intracellular metabolism of favipiravir (T-705) in uninfected and influenza A (H5N1) virus-infected cells. J. Antimicrob. Chemother. 64(4), 741‒746.
U.S. Food and Drug Administration (fda.gov): FDA-Approved Drugs, clinical trials [Электронный ресурс]. https://www. accessdata.fda.gov/ (дата обращения: 1.03.2021).
DrugBank database. Clinical Drug Data [Электронный ресурс]. https://www.drugbank.com (дата обращения: 1.03.2021).
Approved antiviral compounds and mechanisms of drug actions of approved antiviral drugs [Электронный ресурс]. http://virusface.com (дата обращения: 1.03.2021).
ChEMBL database of bioactive molecules with drug-like properties [Электронный ресурс]. https:// www.ebi.ac.uk/chembl/ (дата обращения: 25.03.2021).
Database of privately and publicly funded clinical studies conducted around the world [Электронный ресурс]. https://clinicaltrials.gov/ (дата обращения: 1.03.2021).
Suzuki M., Okuda T., Shiraki K. (2006) Synergistic antiviral activity of acyclovir and vidarabine against herpes simplex virus types 1 and 2 and varicella-zoster virus. Antiviral Res. 72(2), 157‒161.
Miwa N., Kurosaki K., Yoshida Y., Kurokawa M., Saito S., Shiraki K. (2005) Comparative efficacy of acyclovir and vidarabine on the replication of varicella-zoster virus. Antiviral Res. 65(1), 49‒55.
Hanes J.W., Zhu Y., Parris D.S., Johnson K.A. (2007) Enzymatic therapeutic index of acyclovir. Viral versus human polymerase gamma specificity. J. Biol. Chem. 282(34), 25159‒25167.
The DailyMed database containing labeling, submitted to the Food and Drug Administration (FDA) by companies. [Электронный ресурс]. https://dailymed.nlm.nih.gov/dailymed/ (дата обращения: 25.03.2021).
Xiong X., Smith J.L., Kim C., Huang E.S., Chen M.S. (1996) Kinetic analysis of the interaction of cidofovir diphosphate with human cytomegalovirus DNA polymerase. Biochem. Pharmacol. 51(11), 1563‒1567.
Sergerie Y., Boivin G. (2008) Hydroxyurea enhances the activity of acyclovir and cidofovir against herpes simplex virus type 1 resistant strains harboring mutations in the thymidine kinase and/or the DNA polymerase genes. Antiviral Res. 77(1), 77‒80.
Yoshida Y., Honma M., Kimura Y., Abe H. (2021) Structure, synthesis and inhibition mechanism of nucleoside analogues as HIV-1 reverse transcriptase inhibitors (NRTIs). Chem. Med. Chem. 16(5), 743‒766.
Куханова М.К. (2012) Анти-ВИЧ нуклеозидные препараты: история создания и взгляд в будущее. Молекуляр. биология. 46(6), 860‒860.
Drabikowska A.K., Dudycz L., Shugar D. (1979) Studies on the mechanism of antiviral action of 1-(β-D-ribofuranosyl)-1,2,4-triazole-3-carboxamide (ri-bavirin). J. Med. Chem. 22(6), 653‒657.
Ortega-Prieto A.M., Sheldon J., Grande-Pérez A., Tejero H., Gregori J., Quer J., Esteban J.I., Domingo E., Perales C. (2013) Extinction of hepatitis C virus by ri-bavirin in hepatoma cells involves lethal mutagenesis. PLoS One. 8(8), e71039.
Sheu T. G., Deyde V. M., Okomo-Adhiambo M., Garten R. J., Xu X., Bright R. A., Butler E.N., Wal-lis T.R., Klimov A.I., Gubareva L. V. (2008) Surveillance for neuraminidase inhibitor resistance among human influenza A and B viruses circulating worldwide from 2004 to 2008. Antimicrob. Agents Chemother. 52(9), 3284‒3292.
Focher F., Lossani A., Verri A., Spadari S., Maioli A., Gambino J.J., Wright G.E., Eberle R., Black D.H., Medveczky P., Medveczky M., Shugar D. (2007) Sensitivity of monkey B virus (Cercopithecine herpesvirus 1) to antiviral drugs: role of thymidine kinase in antiviral acti-vities of substrate analogs and acyclonucleosides. Antimicrob. Agents Chemother. 51(6), 2028‒2034.
Seley-Radtke K.L., Yates M.K. (2018) The evolution of nucleoside analogue antivirals: a review for chemists and non-chemists. Part 1: Early structural modifications to the nucleoside scaffold. Antiviral Res. 154, 66‒86.
Yates M.K., Seley-Radtke K.L. (2019) The evolution of antiviral nucleoside analogues: a review for chemists and non-chemists. Part II: Complex modifications to the nucleoside scaffold. Antiviral Res. 162, 5‒21.
Галегов Г.А. (2017) Фосфазид (никавир) – высокоэффективный лекарственный препарат для лечения ВИЧ/СПИД инфекции. Вопросы вирусологии. 62(1), 5‒11.
McGuigan C., Pathirana R.N., Mahmood N., Devine K.G., Hay A.J. (1992) Aryl phosphate derivatives of AZT retain activity against HIV1 in cell lines which are resistant to the action of AZT. Antiviral Res. 17(4), 311–321.
Cahard D., McGuigan C., Balzarini J. (2004) Aryloxy phosphoramidate triesters as pro-tides. Mini Rev. Med. Chem. 4(4), 371‒381.
Yeo K.L., Chen Y.L., Xu H.Y., Dong H., Wang Q.Y., Yokokawa F., Shi P.Y. (2015) Synergistic suppression of dengue virus replication using a combination of nucleoside analogs and nucleoside synthesis inhibitors. Antimicrob. Agents Chemother. 59(4), 2086–2093.
Cho A., Zhang L., Xu J., Lee R., Butler T., Metobo S., Aktoudianakis V., Lew W., Ye H., Clarke M., Doerffler E., Byun D., Wang T., Babusis D., Carey A.C., German P., Sauer D., Zhong W., Rossi S., Fenaux M., McHutchison J.G., Perry J., Feng J., Ray A.S., Kim C.U. (2014) Discovery of the first C-nucleoside HCV polymerase inhibitor (GS-6620) with demonstrated antiviral response in HCV infected patients. J. Med. Chem. 57(5), 1812–1825.
Siegel D., Hui H.C., Doerffler E., Clarke M.O., Chun K., Zhang L., Neville S., Carra E., Lew W., Ross B., Wang Q., Wolfe L., Jordan R., Soloveva V., Knox J., Perry J., Perron M., Stray K.M., Barauskas O., Feng J.Y., Xu Y., Lee G., Rheingold A L., Ray A.S., Bannister R., Strickley R., Swaminathan S., Lee W.A., Bavari S., Cihlar T., Lo M.K., Warren T.K., Mackman R.L. (2017) Discovery and synthesis of a phosphoramidate prodrug of a pyrrolo [2,1-f][triazin-4-amino] adenine C-nucleoside (GS-5734) for the treatment of Ebola and emerging viruses. J. Med. Chem. 60(5), 1648−1661.
Murakami E., Wang T., Babusis D., Lepist E.I., Sauer D., Park Y., Vela J.E., Shih R., Birkus G., Stefanidis D., Kim C.U., Cho A., Ray A.S. (2014) Metabolism and pharmacokinetics of the anti-hepatitis C virus nucleotide prodrug GS-6620. Antimicrob. Agents Chemother. 58(4), 1943–1951.
Meier C. (2017) Nucleoside diphosphate and triphosphate prodrugs – an unsolvable task? Antiviral Chem. Chemother. 25(3) 69–82.
Aggarwal S.K., Gogu S.R., Rangan S.R., Agrawal K.C. (1990) Synthesis and biological evaluation of prodrugs of zidovudine. J. Med. Chem. 33(5), 1505‒1510.
Morin K.W., Wiebe L.I., Knaus E.E. (1993) Synthesis of brain-targeted 1-(2-deoxy-2-fluoro-β-D-ribofuranosyl)-(E)-5-(2-iodovinyl)uracil coupled to a dihydropyridine ⇋ pyridinium salt redox chemical-deli-very system. Carbohydr. Res. 249(1), 109‒116.
Kraevskii A.A. (1992) Search for new drugs for the treatment of HIV infections among nucleosides and nucleotides, II. Pharm. Chem. J. 26(1), 33–47.
Esnouf R., Ren J., Ross C., Jones Y., Stammers D., Stuart D. (1995) Mechanism of inhibition of HIV-1 reverse transcriptase by non-nucleoside inhibitors. Nat. Struct. Biol. 2(4), 303‒308.
(2005) Nucleoside Triphosphates and Their Analogs: Chemistry, Biotechnology, and Biological Applications, 1st ed. Ed. Vaghefi M. Boca Raton, FL: CRC press.
Zhou Z., Liu T., Wu G., Kang D., Fu Z., Wang Z., De Clercq E., Pannecouque C., Zhan P., Liu X. (2019) Targeting the hydrophobic channel of NNIBP: discovery of novel 1,2,3-triazole-derived diarylpyrimidines as novel HIV-1 NNRTIs with high potency against wild-type and K103N mutant virus. Org. Biomol. Chem. 17(12), 3202‒3217.
Kang D., Feng D., Sun Y., Fang Z., Wei F., De Clercq E., Pannecouque C., Liu X., Zhan P. (2020) Structure-based bioisosterism yields HIV-1 NNRTIs with improved drug-resistance profiles and favorable pharmacokinetic properties. J. Med. Chem. 63(9), 4837‒4848.
Kang D., Feng D., Ginex T., Zou J., Wei F., Zhao T., Huang B., Sun Y., Desta S., De Clercq E., Pannecouque C., Zhan P., Liu X. (2020) Exploring the hydrophobic channel of NNIBP leads to the discovery of novel piperidine-substituted thiophene[3,2-d]pyrimidine derivatives as potent HIV-1 NNRTIs. Acta Pharm. Sin. B. 10(5), 878‒894.
Jin K., Liu M., Zhuang C., De Clercq E., Pannecouque C., Meng G., Chen F. (2020) Improving the positional adaptability: structure-based design of biphenyl-substituted diaryltriazines as novel non-nucleoside HIV-1 reverse transcriptase inhibitors. Acta Pharm. Sin. B. 10(2), 344‒357.
Wang Z., Kang D., Feng D., Cherukupalli S., Jiang X., Fu Z., De Clercq E., Pannecouque C., Liu X., Zhan P. (2020) Targeting dual tolerant regions of binding pocket: discovery of novel morpholine-substituted diarylpyrimidines as potent HIV-1 NNRTIs with significantly improved water solubility. Eur. J. Med. Chem. 206, 112811.
Bressanelli S., Tomei L., Rey F.A., De Francesco R. (2002) Structural analysis of the hepatitis C virus RNA polymerase in complex with ribonucleotides. J. Virol. 76(7), 3482‒3492.
Tomei L., Altamura S., Bartholomew L., Biroccio A., Ceccacci A., Pacini L., Narjes F., Gennari N., Bisbocci M., Incitti I., Orsatti L., Harper S., Stansfield I., Rowley M., De Francesco R., Migliaccio G. (2003) Mechanism of action and antiviral activity of benzi-midazole-based allosteric inhibitors of the hepatitis C virus RNA dependent RNA polymerase. J. Virol. 77(24), 13225–13231.
Chan L., Reddy T.J., Proulx M., Das S.K., Pereira O., Wang W., Siddiqui A., Yannopoulos C.G., Poisson C., Turcotte N., Drouin A., Alaoui-Ismaili M.H., Bethell R., Hamel M., L’Heureux L., Bilimoria D., Nguyen-Ba N. (2003) Identification of N,N-disubstituted phenylalanines as a novel class of inhibitors of hepatitis C NS5B polymerase. J. Med. Chem. 46(8), 1283–1285.
Tomei L., Altamura S., Bartholomew L., Bisbocci M., Bailey, C., Bosserman M., Cellucci A., Forte E., Incitti I., Orsatti L., Koch U., De Francesco R., Olsen D.B., Carroll S.S., Migliaccio G. (2004) Characterization of the inhibition of hepatitis C virus RNA replication by nonnucleosides. J. Virol. 78(2), 938–946.
Chan L., Das S.K., Reddy T.J., Poisson C., Proulx M., Pereira O., Courchesne M., Roy C., Wang W., Siddiqui A., Yannopoulos C.G., Nguyen-Ba N., Labrecque D., Bethell R., Hamel M., Courtemanche-Asselin P., L’Heureux L., David M., Nicolas O., Brunette S., Bilimoria D., Bedard J. (2004) Discovery of thiophene-2-carboxylic acids as potent inhibitors of HCV NS5B polymerase and HCV subgenomic RNA replication. Part 1: Sulfonamides. Bioorg. Med. Chem. Lett. 14(3), 793–796.
Chan L., Pereira O., Reddy T.J., Das S.K., Poisson C., Courchesne M., Proulx M., Siddiqui A., Yannopoulos C.G., Nguyen-Ba N., Roy C., Nasturica D., Moinet C., Bethell R., Hamel M., L’Heureux L., David M., Nicolas O., Courtemanche-Asselin P., Brunette S., Bilimoria D., Bedard J. (2004) Discovery of thiophene-2-carboxylic acids as potent inhibitors of HCV NS5B polymerase and HCV subgenomic RNA replication. Part 2: Tertiary amides. Bioorg. Med. Chem. Lett. 14(3), 797–800.
Harper S., Avolio S., Pacini B., Di Filippo M., Altamura S., Tomei L., Paonessa G., Di Marco S., Carfi A., Giuliano C., Padron J., Bonelli F., Migliaccio G., De Francesco R., Laufer R., Rowley M., Narjes F. (2005) Potent inhibitors of subgenomic hepatitis C virus RNA replication through optimization of indole-N-acetamide allosteric inhibitors of the viral NS5B polymerase. J. Med. Chem. 48(14), 4547–4557.
Biswal B.K., Wang M., Cherney M.M., Chan L., Yannopoulos C.G., Bilimoria D., James M.N.G. (2006) Non-nucleoside inhibitors binding to hepatitis C virus NS5B polymerase reveal a novel mechanism of inhibition. J. Mol. Biol. 361(1), 33‒45.
Tatum H., Thuluvath P.J., Lawitz E., Martorell C., DeMicco M., Cohen S., Rustgi V., Ravendhran N., Ghalib R., Hanson J., Zamparo J., Zhao J., Cooney E., Treitel M., Hughes E. (2015) A randomized, placebo-controlled study of the NS5B inhibitor beclabuvir with peginterferon/ribavirin for HCV genotype 1. J. Viral Hepat. 22(8), 658–664.
Toyota J., Karino Y., Suzuki F., Ikeda F., Ido A., Tanaka K., Takaguchi K., Naganuma A., Tomita E., Chayama K., Fujiyama, S., Inada Y., Yoshiji H., Watanabe H., Ishikawa H., Hu W., McPhee F., Linaberry M., Yin P.D., Swenson E.S., Kumada H. (2017) Daclatasvir/asunaprevir/beclabuvir fixed-dose combination in Japanese patients with HCV genotype 1 infection. J. Gastroenterol. 52(3), 385–395.
Gentles R.G. (2019) Discovery of beclabuvir: a potent allosteric inhibitor of the hepatitis C virus polymerase. In: HCV: The Journey from Discovery to a Cure, vol. 1. Ed. Sofia M.J. Topics in Medicinal Chemistry. Springer, Cham. 193‒228.
Balzarini J., Pérez-Pérez M.J., San-Félix A., Schols D., Perno, C.F., Vandamme, A.-M., Camarasa, M.-J., De Clercq E. (1992) 2',5'-Bis-O-(tert-butyldimethylsilyl)-3'-spiro-5''-(4''-amino-1'',2''-oxathiole-2'',2''-dioxide)pyrimidine (TSAO) nucleoside analogues: highly selective inhibitors of human immunodeficiency virus type 1 that are targeted at the viral reverse transcriptase. Proc. Natl. Acad. Sci. USA. 89(10), 4392‒4396.
Balzarini J., Perez-Perez M.-J., San-Felix A., Velazquez S., Camarasa M.-J., De Clercq E., (1992) [2',5'-Bis-O-(tert-butyldimethylsilyl)]-3'-spiro-5''-(4''-amino-1'',2''-oxathiole-2'',2''-dioxide) (TSAO) derivatives of purine and pyrimidine nucleosides as potent and selective inhibitors of human immunodeficiency virus type 1. Antimicrob. Agents Chemother. 36(5), 1073‒1080.
Balzarini J., Pérez-Pérez M.-J., San-Félix A., Camarasa M.-J., Bathurst I.C., Barr P.J., De Clercq E. (1992) Kinetics of inhibition of human immunodeficiency virus type 1 (HIV-1) reverse transcriptase by the novel HIV-1-specific nucleoside analogue [2',5'-bis-O-(tert-butyldimethylsilyl)-β-D-ribofuranosyl]-3'-spiro-5''-(4''-amino-1'',2''-oxathiole-2'',2''-dioxide)thymine (TSAO-T). J. Biol. Chem. 267(17), 11831‒11838.
Camarasa M.J., Pérez-Pérez M.J., Velázques S., San-Félix A., Alvarez R., Ingate S., Jimeno M.L., Karlsson A., De Clercq E., Balzarini J. (1995) TSAO derivatives: highly specific inhibitors of human immunodeficiency virus type 1 (HIV-1) replication. Nucleosides, Nucleotides, Nucleic Acids. 14(3‒5), 585‒594. https://doi.org/10.1080/15257779508012432
Balzarini J., Karlsson A., Vandamme A.-M., Pérez-Pérez M.-J., Zhang H., Vrang L., Öberg B., Bäckbro K., Unge T., San-Félix A., Velázquez S., Camarasa M.-J., De Clercq E. (1993) Human immunodeficiency virus type 1 (HlV-l) strains selected for resistance against the HlV-l-specific [2',5'-bis-O-(tert-butyldimethylsilyl)-3'-spiro-5''-(4''-amino-1'',2''-oxathiole-2'',2''-dioxide)]-β-D-pentofuranosyl (TSAO) nucleoside analogues retain sensitivity to HlV-l-specific nonnucleoside inhibitors. Proc. Natl. Acad. Sci. USA. 90(15), 6952‒6956.
Alvarez R., Jimeno M.-L., Pérez-Pérez M.-J., De Clercq E., Balzarini J., Camarasa M.-J. (1997) Synthesis and anti-human immunodeficiency virus type 1 activity of novel 3′-spiro nucleoside analogues of TSAO-T. Antiviral Chem. Chemother. 8(6), 507‒517.
Angusti A., Manfredini S., Durini E., Ciliberti N., Vertuani S., Solaroli N., Pricl S., Ferrone M., Fermeglia M., Loddo R., Secci B., Visioli A., Sanna T., Collu G., Pezzullo M., La Colla P. (2008) Design, synthesis and anti Flaviviridae activity of N6-, 5′,3′-O- and 5′,2′-O-substituted adenine nucleoside analogs. Chem. Pharm. Bull. (Tokyo). 56(4), 423‒432.
De Burghgraeve T., Selisko B., Kaptein S., Chatelain G., Leyssen P., Debing, Y., Jacobs M., Van Aerschot A., Canard B., Neyts J. (2013) 3′,5′Di-O-trityluridine inhibits in vitro flavivirus replication. Antiviral Res. 98(2), 242‒247.
Chatelain G., Debing Y., De Burghgraeve T., Zmurko J., Saudi M., Rozenski J., Neyts J., Van Aerschot A. (2013) In search of flavivirus inhibitors: evaluation of different tritylated nucleoside analogues. Eur. J. Med. Chem. 65, 249–255.
Saudi M., Zmurko J., Kaptein S., Rozenski J., Neyts J., Van Aerschot A. (2014) In search of Flavivirus inhibitors part 2: tritylated, diphenylmethylated and other alkylated nucleoside analogues. Eur. J. Med. Chem. 76, 98–109.
Vernekar S.K.V., Qiu L., Zhang J., Kankanala J., Li H., Geraghty R.J., Wang Z. (2015) 5′-Silylated 3′-1,2,3-triazolyl thymidine analogues as inhibitors of West Nile virus and dengue virus. J. Med. Chem. 58(9), 4016‒4028.
McGuigan C., Serpi M., Slusarczyk M., Ferrari V., Pertusati F., Meneghesso S., Derudas M., Farleigh L., Zanetta P., Bugert J. (2016) Anti-flavivirus activity of different tritylated pyrimidine and purine nucleoside analogues. ChemistryOpen. 5(3), 227‒235.
Eyer L., Nencka R., De Clercq E., Seley-Radtke K., Růžek D. (2018) Nucleoside analogs as a rich source of antiviral agents active against arthropod-borne flaviviruses. Antiviral Chem. Chemother. 26, 2040206618761299.
St.Vincent M.R., Colpitts C.C., Ustinov A.V., Muqadas M., Joyce M.A., Barsby N.L., Epand R.F., Epand R.M., Khramyshev S.A., Valueva O.A., Korshun V.A., Tyrrell D.L.J., Schang L.M. (2010) Rigid amphipathic fusion inhibitors, small molecule antiviral compounds against enveloped viruses. Proc. Natl. Acad. Sci. USA. 107(40), 17339‒17344.
Orlov A.A., Chistov A.A., Kozlovskaya L.I., Usti-nov A.V., Korshun V.A., Karganova G.G., Osolodkin D.I. (2016) Rigid amphipathic nucleosides suppress reproduction of the tick-borne encephalitis virus. Med. Chem. Comm. 7(3), 495‒499.
Strasfeld L., Chou S. (2010) Antiviral drug resistance: mechanisms and clinical implications. Infect. Dis. Clin. North Am. 24(2), 413‒437.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология