

Молекулярная биология, 2021, T. 55, № 6, стр. 1030-1044
Расчет энергии формирования дуплексов РНК/РНК и ДНК/РНК на основании метода молекулярной динамики
В. М. Голышев a, b, Д. В. Пышный a, b, А. А. Ломзов a, b, *
a Институт химической биологии и фундаментальной медицины Сибирского отеления Российской академии наук
630090 Новосибирск, Россия
b Новосибирский государственный университет
630090 Новосибирск, Россия
* E-mail: lomzov@niboch.nsc.ru
Поступила в редакцию 07.03.2021
После доработки 21.05.2021
Принята к публикации 21.05.2021
Аннотация
Создание подходов прогностического расчета гибридизационных свойств нуклеиновых кислот (НК), а также различных классов их производных считается фундаментом для рационального дизайна конструкций на их основе. Современные достижения в области методов компьютерного моделирования свидетельствуют о возможности таких вычислений. В работе впервые проанализирована возможность расчета энергии формирования ДНК/РНК- и РНК/РНК-дуплексов на примере представительных наборов комплексов (65 и 75 комплексов соответственно). Мы использовали метод классической молекулярной динамики (МД), методы MMPBSA или MMGBSA для расчета энтальпийной (ΔH°) составляющей и квазигармоническое приближение (Q-Harm) или метод анализа нормальных мод колебаний (NMA) для расчета энтропийной (ΔS°) составляющей энергии Гиббса ($\Delta G_{{37}}^{^\circ }$) образования НК-комплексов. Показано, что, используя метод MMGBSA при анализе МД-траектории только НК-дуплекса с учетом эмпирической линейной аппроксимации, можно рассчитывать энтальпию формирования ДНК, РНК и гибридных комплексов различной длины и GC-состава с точностью 8.6%. В рамках каждого типа комплексов комбинация достаточно производительных методов MMGBSA и Q-Harm, в применении к траектории только бимолекулярного комплекса, позволяет рассчитать величину $\Delta G_{{37}}^{^\circ }$ образования дуплекса с достоверностью 10%. Высокая точность прогностического расчета для разных типов природных комплексов (ДНК/ДНК, ДНК/РНК и РНК/РНК) указывает на большую вероятность распространения рассмотренного подхода на аналоги и производные НК. Это в перспективе даст принципиальную возможность перейти к рациональному дизайну новых типов НК-направленных сиквенсспецифических соединений.
Производные и аналоги нуклеиновых кислот (НК) используют для решения задач таргетной терапии различных заболеваний [1, 2], биосенсорных технологий, молекулярно-биологических исследований [3] и биотехнологии [4]. На данный момент известно множество различных производных НК, которые обладают улучшенными относительно природных аналогов свойствами [4]. Несмотря на это, они не в полной мере удовлетворяют потребностям исследователей, а их использование на практике часто ограничено патентным правом. Таким образом, создание новых производных, обладающих заранее заданными биологическими и физико-химическими свойствами, остается актуальной задачей химии и физической химии НК. К одному из важнейших свойств таких аналогов относится их способность взаимодействовать с комплементарными последовательностями НК и эффективность этого взаимодействия. На сегодняшний день не существует способов оценки гибридизационных свойств производных НК на этапе, предшествующем их химическому синтезу. Для хорошо изученных соединений разработан ряд подходов, основанных на анализе эмпирических зависимостей, позволяющих рассчитывать энергию формирования их комплексов с ДНК [5–11] и/или РНК [12] или температуру плавления [13, 14] в зависимости от длины и нуклеотидного состава взаимодействующих цепей. В случае новых соединений необходимо использовать подходы, которые не опираются на полученную экспериментальным путем информацию по их гибридизационным свойствам. Для достаточно больших соединений, состоящих более чем из 100 атомов, подходят методы, основанные на молекулярной механике и/или молекулярной динамике (МД). За последнее десятилетие наблюдается большой прогресс в области разработки различных подходов для расчета термодинамических параметров межмолекулярных взаимодействий [15]. Среди них наиболее распространены методы MMPBSA (Molecular Mechanics/Poisson Boltzmann Surface Area) [16] и MMGBSA (Molecular Mechanics/ Generalized Born Surface Area) [17, 18], методы анализа потенциала средней силы (PMF): например, метод анализа взвешенных диаграмм (WHAM) [19] или метод анализа вариационного профиля свободной энергии (vFEP) [20]. Также за последнее десятилетие разработан ряд подходов, которые позволяют эффективно покрывать рассматриваемое фазовое пространство для более эффективного расчета свободной энергии. Среди них можно выделить методы алхимических превращений [21]: например, методы возмущения свободной энергии (FEP) [22] и термодинамического интегрирования (TI) [23]. К их недостаткам можно отнести необходимость больших затрат вычислительных мощностей и времени расчета, которое необходимо для получения достоверных результатов моделирования, в результате чего вышеупомянутые подходы не могут быть применены для расчета энергий набора комплексов различной длины и GC-состава.
Для оценки энергии комплексообразования очень популярны такие простые и вычислительно эффективные техники, как MMGBSA и MMPBSA. В этих методах для каждой конформации молекул рассчитывают энергии всего комплекса и его составных частей, полученные при МД-моделировании, а растворитель (который был представлен в моделировании в явном виде) заменяют на непрерывную среду с заданной диэлектрической проницаемостью. В этих подходах энергия связывания (ΔGbind) состоит из изменения суммарной молекулярно-механической энергии (ΔEMM), энергии сольватации (ΔGsolv) и вклада конформационной энтропии (TΔS) [24]:
(1)
$\Delta {{G}_{{{\text{bind}}}}} = {\text{ }}\Delta {{E}_{{{\text{MM}}}}} + {\text{ }}\Delta {{G}_{{{\text{solv}}}}} - T\Delta S.$В свою очередь в величину ΔEMM вносит вклад внутренняя энергия (ΔEint) (деформация связей, углов и двугранных углов), электростатическая энергия (ΔEele) и вандерваальсовые взаимодействия (ΔEvdW):
(2)
$\Delta {{E}_{{{\text{MM}}}}} = {\text{ }}\Delta {{E}_{{{\text{int}}}}} + {\text{ }}\Delta {{E}_{{{\text{ele}}}}} + {\text{ }}\Delta {{E}_{{{\text{vdW}}}}}.$Сольватационная энергия равна сумме вкладов от полярного (ΔGPB/GB) и неполярного (ΔGSA) взаимодействия исследуемой молекулы с растворителем:
(3)
$\Delta {{G}_{{{\text{solv}}}}} = {\text{ }}\Delta {{G}_{{{{{\text{PB}}} \mathord{\left/ {\vphantom {{{\text{PB}}} {{\text{GB}}}}} \right. \kern-0em} {{\text{GB}}}}}}} + {\text{ }}\Delta {{G}_{{{\text{SA}}}}}.$Первый член уравнения, ΔGPB/GB, ‒ электростатическая энергия, которая может быть рассчитана либо при прямом решении уравнения Пуассона‒Больцмана (PB, метод MMPBSA), либо при помощи обобщенного метода Борна (GB, метод MMGBSA). Вклад неполярных взаимодействий, ΔGSA, обычно представляют в виде линейной функции от площади молекулы, доступной для взаимодействия с растворителем (SASA) [25]:
где γ и b ‒ коэффициенты линейного уравнения.Для расчета свободной энергии Гиббса энтропийный вклад, учитывающий конформационную составляющую, обычно рассчитывают методами анализа нормальных мод колебаний (NMA) [26] или квазигармонического приближения (Q-Harm) [27]. Первый характеризуется высокой сложностью вычислений и позволяет проанализировать энтропийный вклад только для небольшого числа снимков МД-траектории (до нескольких тысяч), в то время как второй работает существенно быстрее и эффективнее.
Такие методы оценки энергии межмолекулярных взаимодействий широко используют при исследовании взаимодействий биополимеров с малыми молекулами: например, в молекулярно-биологических исследованиях [28] или при разработке новых лекарственных препаратов [29]. Кроме того, продемонстрировано успешное применение этих подходов для расчета энергии формирования комплексов как нативных [30, 31], так и химически модифицированных олигонуклеотидов с ДНК и РНК [32–36]. Однако в последнем случае исследования не носят систематического характера ‒ скорее, это иллюстрация возможностей методов.
Создание подхода прогностического расчета гибридизационных свойств разных классов производных НК может стать фундаментальной основой для рационального дизайна таких соединений и последующего направленного поиска способов их химического синтеза. Для этого на первом этапе требуется проверка применимости расчетов на уже хорошо исследованных немодифицированных комплексах НК с последующим распространением на новые, в том числе еще не синтезированные, аналоги НК. Ранее нами продемонстрировано успешное применение подходов MMPB(GB)SA для расчета энергии и оценки термостабильности набора более 300 ДНК/ДНК-дуплексов [30]. В рамках развития этих исследований мы проанализировали возможность расчета гибридизационных свойств ДНК/РНК- и РНК/РНК-комплексов, используя классический метод МД.
Целью работы была проверка применимости методов MMPBSA и MMGBSA для расчета энтальпийной составляющей и Q-Harm и NMA для расчета энтропийной составляющей энергии Гиббса формирования ДНК/РНК- и РНК/РНК-дуплексов. В ходе исследования проведено моделирование методом МД представительных наборов олигонуклеотидов и их комплексов. Полученные траектории проанализированы с использованием подходов MMPBSA и MMGBSA, а энтропия рассчитана методами NMA и Q-Harm. Данные проанализировали и сравнили с экспериментально полученными значениями термодинамических параметров формирования комплексов олигонуклеотидов.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
База данных термодинамических параметров. Экспериментальные значения термодинамических параметров формирования НК-комплексов взяты из литературных источников. В случае комплементарных ДНК/РНК-дуплексов использованы данные, опубликованные Sugimoto и др. [7]. Из рассмотрения были исключены дуплексы, содержащие фосфатные остатки на 3′-конце. Таким образом, база данных включала термодинамические параметры комплексообразования для 65 дуплексов длиной от 5 до 12 п.о. (в среднем 8 п.о.) и c GC-составом от 0 до 84% (в среднем 53%). Для комплементарных РНК/РНК-дуплексов использованы данные, полученные Xia и др. [37]. Из рассмотрения были также исключены дуплексы, содержащие концевые фосфатные группы. Таким образом, база данных включала термодинамические параметры комплексообразования для 75 дуплексов длиной от 4 до 11 п.о. (в среднем 7 п.о.) и долей G/C-пар от 22 до 100% (в среднем 58%).
Подготовка молекулярных систем для моделирования. Структуры дуплексов были созданы в программе NAB пакета программ AmberTools14 [38] и при помощи программы Chimera [39]. Стартовые структуры РНК/ДНК- и РНК/РНК-дуплексов имели А-форму двойной спирали. Одноцепочечные олигонуклеотиды были получены из дуплексов путем удаления одной из цепей.
Молекулярно-динамическое моделирование. МД-моделирование проведено с применением пакета программ AMBER 14 с использованием параллельных вычислений на центральных процессорах и графических ускорителях программно-аппаратной архитектуры CUDA. Для ДНК использовали силовое поле ff99bsc0 [40], а для РНК ‒ ff99bsc0+chiOL3 [41]. МД-моделирование проводили в явной водной оболочке при температуре 300 К. Электростатические взаимодействия описывали методом сети частиц Эвальда с шагом решетки 1 Å. Давление в системе было равно 1 бар (баростат Берендсена), температура в системе поддерживалась при помощи масштабирования скоростей с периодом привязки 10 пс. Для увеличения шага интегрирования уравнений движения до 2 фс использовали алгоритм SHAKE. Использовали область расчета нековалентных взаимодействий (nonbonded cutoff) 10 Å. Координаты каждого атома системы записывали каждые 10 пс моделирования. Дуплексы и одноцепочечные олигонуклеотиды были помещены в кубическую ячейку с расстоянием 12 Å от моделируемой молекулы до границ ячейки. Количество воды в моделируемой ячейке было в диапазоне от 4000 до 8000 молекул. Для нейтрализации отрицательного заряда НК в периодической ячейке использовали ионы натрия.
Процедура моделирования состояла из следующих шагов:
1) Создание PDB-файла, содержащего данные о координатах каждого атома в структуре дуплекса или одноцепочечной НК.
2) Создание водного окружения (модель воды TIP3P, кубические периодические условия, 12 Å) и добавление ионов натрия для нейтрализации периодической системы.
3) Минимизация всей системы с фиксированной НК (10 000 шагов минимизации) (PMEMD.MPI).
4) Нагрев системы с фиксированной НК в течении 2.5 нс с шагом интегрирования 0.5 фс от 0 до 300 К (PMEMD.CUDA).
5) Уравновешивание плотности системы при постоянном давлении 1 бар в течении 500 пс (SANDER) с зафиксированной НК.
6) Уравновешивание системы при постоянном давлении 1 бар и температуре 300 К в течении 5 нс (PMEMD.CUDA).
7) МД-моделирование в течение 100 нс в NPT-ансамбле (1 бар, 300 К) (PMEMD.CUDA).
8) Расчет энергии формирования комплексов при помощи методов MMGBSA и MMPBSA, расчет энтропии в Q-Harm и NMA (MMPBSA.MPI). Вычисления проводили при использовании только траектории дуплекса (однотраекторный подход) и при использовании независимо полученных траекторий дуплекса и одноцепочечных олигонуклеотидов (трехтраекторный подход).
Для вычисления методом MMGBSA использовали параметры обобщенного метода Борна, предложенные Tsui & Case [17]. Ионная сила раствора была установлена равной 100 мМ. Для расчета методами MMGBSA и MMPBSA и Q-Harm использовали каждый записанный снимок МД-траектории, в случае NMA ‒ каждую десятую сохраненную конформацию.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Ранее нами показано, что применение компьютерных расчетов для определения гибридизационных свойств ДНК/ДНК-дуплексов может давать точность, сопоставимую с точностью экспериментальных методик [30]. Именно поэтому мы использовали методику МД-моделирования и расчета энергии формирования дуплексов в применении к ДНК/РНК- и РНК/РНК-комплексам. Использована база данных величин термодинамических параметров комплексообразования, включающая экспериментальные значения энтальпии, энтропии и свободной энергии Гиббса формирования НК-дуплексов. Характеристики наборов данных приведены в разделе “Экспериментальная часть”.
На первом этапе исследований были созданы молекулярные структуры и проведен расчет методом МД в явной водной оболочке в течении 100 нс для набора 65 дуплексов ДНК/РНК [7] и 75 дуплексов РНК/РНК [37], а также одноцепочечных олигонуклеотидов, входящих в их состав. В случае самокомплементарных олигонуклеотидов расчеты проводили с двумя различными начальными скоростями атомов. Полученные МД-траектории анализировали.
Анализ структуры комплексов
По результатам моделирования РНК/РНК-дуплексов структура А-формы двойной спирали сохраняется вдоль всей траектории длиной 100 нс для каждого из 75 проанализированных комплексов. Отклонение геометрии спирали от исходной неуравновешенной структуры при моделировании не превышает 2.5 Å ‒ величины среднеквадратичного отклонения (СКО) между атомами. Комплексы после уравновешивания в течение 10 нс незначительно изменяли конформацию в течение последующих 90 нс моделирования. Среднее значение СКО вдоль МД-траектории для всего набора комплексов составило 1.56 Å и не превышало 2.56 Å для каждого снимка траектории. В 11 из 74 исследуемых дуплексов наблюдалось расплетание концевых пар нуклеотидов. В ряде случаев для дуплексов с концевыми A/U- или A/T-парами происходило обратимое разрушение второй пары оснований, при этом концевой аденин и предконцевой нуклеотид этой же цепи сохраняли конформацию, близкую к таковой в двойной спирали.
При моделировании ДНК/РНК-дуплексов отклонение от стартовой структуры не превышало 4.1 Å. В 17 из 64 исследуемых структур наблюдали расплетание концевых пар, аналогично случаю бимолекулярных РНК/РНК-комплексов. Среднее значение СКО вдоль МД-траектории для всего набора комплексов составило 2.26 Å и не превышало 3.86 Å для каждого снимка траектории.
Структуры ДНК/РНК- и РНК/РНК-дуплексов несколько отличались. Анализ РНК/РНК-комплексов с помощью программного обеспечения X3DNA [42, 43] выявил, что такие комплексы имеют А-форму двойной спирали. Структуру ДНК/РНК-дуплексов можно охарактеризовать как промежуточную между А- и В-формами. Такое различие конформаций известно и показано экспериментальными методами: ЯМР-спектроскопией [44], рентгеноструктурным анализом [45] и спектроскопией кругового дихроизма [44, 46, 47]. В связи с особенностями структуры гомонуклеотидных последовательностей в ДНК/РНК- и РНК/РНК-дуплексах [48, 49] мы исключили их из рассмотрения.
При МД-анализе одноцепочечных олигонуклеотидов обнаружены значительные конформационные перестройки как в ДНК-, так и в РНК-цепях. В большинстве случаев наблюдали локальное разрушение одноцепочечного стэкинга между прилегающими основаниями, а в некоторых случаях формирование внутримолекулярных шпилечных структур, которые могут быть достаточно устойчивыми на длине траектории в 100 нс. Это приводит к существенно бóльшим значениям СКО ‒ как усредненным вдоль траектории (5.3 Å для РНК- и 5.7 Å для ДНК-цепи ‒ в среднем для всех исследованных олигонуклеотидов), так и их максимальным отклонениям от начальных структур (8.0 и 9.5 Å для РНК- и ДНК-цепей соответственно). Такое поведение одноцепочечных форм типично для НК, что следует из опубликованных экспериментальных данных [50, 51] и результатов компьютерного моделирования [52–54].
Энергия формирования РНК/РНК-комплексов
Энергию формирования комплексов НК определяли как разницу энергий двухцепочечного и одноцепочечного состояний. С использованием данных МД-моделирования рассчитывали изменение внутренней энергии комплексообразования, используя методы MMPBSA и MMGBSA. Для расчета вклада конфигурационной энтропии применяли методы NMA и Q-Harm. Определение вклада одноцепочечных состояний проводили, используя анализ МД-траекторий двух отдельных олигонуклеотидов (трехтраекторный подход, 3tr). Альтернативно из МД-траектории НК-дуплексов вычленяли отдельные олигонуклеотиды и использовали полученный набор конформаций для расчетов (однотраекторный подход, 1tr). Расчет энергии Гиббса комплексообразования проводили, используя все возможные комбинации полученных значений энтальпии и энтропии формирования комплексов в рамках одно- или трехтраекторного подхода.
Сопоставление экспериментально определенных величин термодинамических параметров и значений, полученных с использованием одно- и трехтраекторных подходов для РНК/РНК-комплексов, приведено на рис. 1 и в табл. 1. Наилучшая корреляция экспериментальных и расчетных значений энтальпии комплексообразования была при использовании метода MMGBSA и трехтраекторного подхода. Коэффициент корреляции R2 (где R ‒ коэффициент корреляции Пирсона) между экспериментальными и рассчитанными величинами составлял 0.72; при этом угловой коэффициент линейной зависимости был близок к единице (0.95), а коэффициент сдвига составлял всего 4.6 ккал/моль. При использовании данной зависимости для расчета энтальпии комплексообразования средняя абсолютная величина ошибки ее предсказания составила 8.1 ккал/моль, или 16.3%. При использовании метода MMPBSA корреляция экспериментальных и расчетных величин была гораздо хуже, чем для MMGBSA ‒ средняя абсолютная ошибка была гораздо выше в случае однотраекторного подхода и существенно ниже при трехтраекторном рассмотрении (табл. 1).
Рис. 1.
Корреляционный анализ значений термодинамических параметров, определенных экспериментально и рассчитанных на основании МД-анализа, для РНК/РНК-дуплексов. а ‒ Значения энтальпии (ΔH0) рассчитаны методами MMGBSA с использованием анализа одной (1tr) и трех (3tr) траекторий. б ‒ Значения энергии Гиббса при 37°С ($\Delta G_{{37}}^{^\circ }$) определены экспериментально и на основании анализа одной МД-траектории комбинацией методов MMGBSA и NMA или Q-Harm. Порядок расположения уравнений соответствует порядку подписей на рисунках.
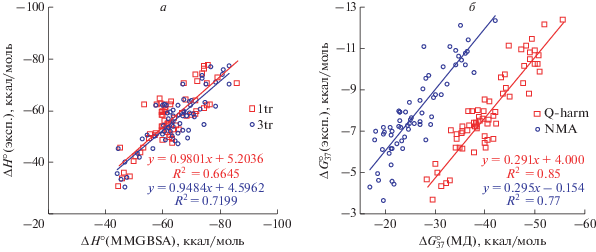
Таблица 1.
Сопоставление значений термодинамических параметров, определенных экспериментально и полученных при анализе МД-траекторий, для РНК/РНК-дуплексовa
| Nb | Метод расчета | a | b | R2 | 〈|ΔE|〉c | 〈|ΔE|/|E|〉, % | 〈|ΔE*|〉c | 〈|ΔE*|/|E*|〉, % |
|---|---|---|---|---|---|---|---|---|
| ΔH° | ||||||||
| 1tr | MMGBSA | 0.98 | 5.20 | 0.66 | 7.2 | 14.5 | 5.1 | 9.29 |
| MMPBSA | 0.68 | ‒33.39 | 0.16 | 21.9 | 38.3 | 7.9 | 14.61 | |
| 3tr | MMGBSA | 0.95 | 4.60 | 0.72 | 8.1 | 16.3 | 3.7 | 8.09 |
| MMPBSA | 1.06 | 5.78 | 0.68 | 5.0 | 10.1 | 4.1 | 8.81 | |
| ΔS° | ||||||||
| 1tr | Q-Harm | 2.40 | 13.64 | 0.67 | 83.4 | 54.0 | 13.5 | 8.73 |
| NMA | 1.47 | 16.48 | 0.60 | 118.4 | 77.2 | 14.6 | 9.47 | |
| 3tr | Q-Harm | 1.68 | ‒15.12 | 0.63 | 70.6 | 45.9 | 12.3 | 9.16 |
| NMA | 1.13 | ‒34.99 | 0.42 | 48.2 | 30.0 | 14.8 | 11.93 | |
| $\Delta G_{{37}}^{^\circ }$ | ||||||||
| 1tr | MMPBSA + Q-Harm | 0.14 | ‒6.22 | 0.18 | 5.5 | 79.3 | 1.40 | 19.22 |
| MMGBSA + Q-Harm | 0.29 | 4.00 | 0.85 | 31.9 | 433.8 | 0.68 | 8.63 | |
| MMPBSA + NMA | 0.07 | ‒8.08 | 0.07 | 10.6 | 145.5 | 1.54 | 20.62 | |
| MMGBSA + NMA | 0.30 | ‒0.15 | 0.77 | 17.7 | 238.2 | 0.74 | 10.92 | |
| 3tr | MMPBSA + Q-Harm | 0.22 | ‒0.48 | 0.50 | 22.9 | 331.2 | 0.89 | 18.34 |
| MMGBSA + Q-Harm | 0.22 | 0.78 | 0.58 | 28.1 | 407.3 | 0.84 | 18.05 | |
| MMPBSA + NMA | 0.22 | ‒2.26 | 0.52 | 16.1 | 226.9 | 0.92 | 18.26 | |
| MMGBSA + NMA | 0.26 | 0.15 | 0.71 | 20.9 | 297.5 | 0.87 | 17.85 | |
a Здесь и далее: коэффициенты a и b определены методом наименьших квадратов из корреляции расчетных и экспериментальных величин термодинамических параметров в рамках каждого метода расчета ‒ например, для величин энтальпии: ΔH°(эксп.) = a ∙ ΔH°(МД) + b; R 2 – коэффициент корреляции этой зависимости. 〈|ΔE|〉 и 〈|ΔE|/|E|〉 ‒ среднее значение абсолютной и относительной ошибки расчета термодинамического параметра. Значения 〈|ΔE*|〉 и 〈|ΔE*|/|E*|〉 рассчитаны после линейной коррекции (коэффициенты a и b) величин термодинамических параметров, полученных методами компьютерного анализа.
Введение линейной эмпирической поправки к рассчитанным величинам энтальпии комплексообразования, полученной из корреляции расчетных и экспериментальных значений (ΔH°(эксп) = = a · ΔH0(МД) + b), приводит к тому, что величина абсолютной ошибки снижается до уровня 3.7 ккал/моль, а относительной – до 8.1% при расчетах методом MMGBSA (3tr), что ниже типичной экспериментальной точности определения энтальпии комплексообразования (10%) [37].
Величину энтропии формирования комплекса рассчитывали методами Q-Harm и NMA при анализе одной или трех независимых траекторий. Сопоставление рассчитанных и экспериментальных значений показывает, что наилучшая корреляция (R2 = 0.67) наблюдается при использовании квазигармонического приближения при анализе одной траектории (табл. 1). Среднее значение величины относительной ошибки расчета энтропии в этом случае составляет 83.4 кал ∙ моль‒1 ∙ К‒1, или 54%. Это в процентном отношении значимо хуже, чем при оптимальном методе расчета энтальпии методом MMPGSA с использованием трехтраекторного анализа. При трехтраекторном анализе ошибки расчета энтропии комплексообразования в 1.2‒2.5 раз ниже, хотя наблюдаемые корреляции существенно хуже. Необходимо отметить, что в линейных зависимостях коэффициент пропорциональности между расчетными и экспериментальными величинами энтропий лежит в диапазоне 1.13–2.40, а свободный член линейной зависимости в диапазоне ‒34.99…+16.48 кал ∙ моль‒1 ∙ К‒1. Это показывает, что конфигурационная энтропия в значительной степени коррелирует с энтропией комплексообразования, но не вносит в нее определяющий вклад. Использование линейной поправки для приближения расчетных величин энтропии комплексообразования к экспериментальным данным дает значимое снижение ошибки расчета до 8.7% при использовании квазигармонического приближения в рамках однотраекторного подхода (табл. 1). Менее достоверная предсказательная способность энтропии при трехтраекторном подходе, скорее всего, связана с неполным покрытием конфигурационного пространства одноцепочечных олигомеров при данной длине траектории в рамках метода классической МД. Кроме того, наблюдаемое значимое различие в энтропии также зависит от ионной силы раствора, которая при моделировании отличается от стандартных условий (1 М NaCl, нейтральные значения pH), в которых были проведены экспериментальные исследования. В одном из классических представлений влияние концентрации одновалентных ионов на термостабильность межмолекулярных НК-комплексов может быть представлено в виде энтропийной поправки, пропорциональной логарифму концентрации катионов в растворе и числу отрицательно заряженных фосфатных остатков в НК-цепи [37, 55, 56]. Такая зависимость соответствует наблюдаемому тренду ‒ линейной зависимости экспериментальных и расчетных значений энтропии формирования дуплексов. Однако в полной мере учесть этот вклад в термостабильность НК-дуплексов в рамках применения метода классической МД не представляется возможным.
Величину свободной энергии Гиббса наиболее часто используют на практике при сопоставлении гибридизационных свойств олигонуклеотидов [55, 56]. Этот параметр определяет величину константы равновесия в соответствии с уравнением Вант-Гоффа и, следовательно, соотношение концентраций одно- и двухцепочечного состояния олигонуклеотидов в растворе при различных температурах. При расчете энергии Гиббса формирования дуплексов при одно- и трехтраекторном подходах рассмотрена прогностическая способность комбинаций методов расчета энтропии и энтальпии. Результаты анализа корреляций расчетных и экспериментальных значений приведены на рис. 1 и в табл. 1. Наилучшая корреляция с R2 = 0.85 наблюдается при использовании однотраекторного анализа методами MMGВSA и Q‑Harm при оценке соответственно энтальпии и энтропии комплексообразования. Наблюдается значительный наклон (a = 0.29) и ненулевой коэффициент сдвига составлял (b = ‒4.0 ккал/моль) данной линейной зависимости (рис. 1б). Это приводит к большим значениям величины абсолютной ошибки. Как видно из данных для прогноза энтальпии и энтропии комплексообразования (табл. 1), оба этих компонента вносят вклад в отличный от единицы наклон корреляции расчетных и экспериментальных значений изменения свободной энергии Гиббса и в пересечение с осью абсцисс не в нулевой точке. Учет линейной корреляции существенно снижает ошибку ‒ до 0.68 ккал/моль, или 8.63%, ‒ что фактически совпадает со средней экспериментальной ошибкой для НК-дуплексов (~7%) [37].
Проведенный анализ показывает достаточно близкие экспериментальные и расчетные значения (без учета линейных поправок) энергии комплексообразования только для изменения энтальпии, рассчитанного методом MMGBSA в рамках трехтраекторного анализа. Сопоставимые значения ошибок дает модель ближайших соседей [37]. Мы сравнили опубликованные ранее инкременты формирования динуклеотидных шагов спирали и инкременты, рассчитанные на основании собранной нами экспериментальной базы данных и соответствующих величин, полученных с помощью компьютерного анализа (рис. 2).
Рис. 2.
Корреляционный анализ значений термодинамических параметров, определенных экспериментально и рассчитанных на основании МД-анализа, для РНК/РНК-дуплексов. а ‒ Прогностическая эффективность метода ближайших соседей (БС; 5'-NN/NN-3') для расчета энтальпии образования РНК/РНК-дуплексов, определенной экспериментально (Эксп.) и с использованием метода MMGBSA при анализе трех траекторий (MMGBSA, 3tr). б ‒ Сопоставление инкрементов модели ближайших соседей для РНК/РНК-комплексов, опубликованных ранее Xia и соавт. [37], определенных на основании экспериментальных данных (Эксп.) и рассчитанных с использованием метода MMPBSA при анализе трех траекторий (MMGBSA, 3tr). Порядок расположения уравнений соответствует порядку подписей на рисунках. Иниц. ‒ фактор инициации формирования двойной спирали.
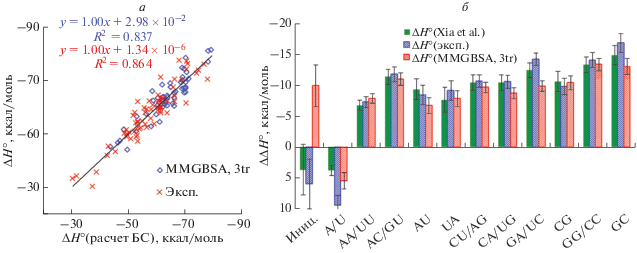
Модель ближайших соседей одинаково хорошо описывает экспериментальные данные и величины, рассчитанные методом MMPBSA с использованием трех траекторий (рис. 2а). Все инкременты значимы (р-value < 4 × 10‒6), за исключением инкремента фактора инициации для экспериментальных данных (р-value = 0.14).
Представленные в литературе и определенные нами на основании экспериментальных и расчетных данных величины формирования динуклеотидных пар близки между собой и одинаковы в рамках погрешностей, за исключением инкрементов для GA/UC и GC/CG, полученных из компьютерных расчетов методом MMPBSA (рис. 2б). В этом случае разница с экспериментально определенными величинами для исследованной базы данных составляет 4.3 и 3.8 ккал/моль соответственно. Наиболее значимое различие наблюдается для фактора инициации (рис. 2б, инкремент “Иниц.”), величина которого составляет ‒9.9 ккал/моль при анализе МД-траекторий и +5.9 ккал/моль при анализе этой же базы данных экспериментальных значений, что, вероятно, связано с низкой значимостью инкремента, определенного на основании экспериментальных данных. Аналогично более отрицательной оказывается поправка, связанная с существованием концевой A/U-пары: +5.4 против +9.4 ккал/моль соответственно. Это может быть связано с не вполне корректным учетом взаимодействия концевых пар оснований, в том числе недостаточно длинной МД-траекторией, чтобы учесть эффект открывания концевых пар с высокой достоверностью. Вместе с тем совокупность полученных данных указывает на адекватность использованного подхода, включающего моделирование методом МД (силовое поле, способ моделирования) и расчет энергии методом MMPBSA для учета взаимодействий внутри дуплексной структуры РНК.
Энергия формирования ДНК/РНК-комплексов
В случае гибридных ДНК/РНК-дуплексов выявлена линейная корреляция расчетных и экспериментальных величин энтальпии комплексообразования (рис. 3, табл. 2). Корреляцию по изменениям энтальпии наблюдали только при анализе одной траектории методом MMGBSA (R2 = 0.71). Наклон корреляционной прямой зависимости близок к единице (а = 0.841), а свободный член близок к нулю (b = ‒0.982 ккал/моль). Для этого способа расчета энтальпии относительная ошибка составляла 23%, а абсолютная – 12.0 ккал/моль. Во всех остальных случаях коэффициент корреляции R2 не превышал значения 0.64. Вместе с тем при трехтраекторном анализе методом MMPBSA значение ошибки расчета оказалось минимальным (16% и 11.4 ккал/моль) при отсутствии корреляции (R2 = 0.13), как и в случае РНК/РНК-комплексов (табл. 1 и 2).
Рис. 3.
Корреляционный анализ значений термодинамических параметров, определенных экспериментально и рассчитанных на основании МД-анализа, для ДНК/РНК-дуплексов. а ‒ Значения энтальпии (ΔH°) рассчитаны методами MMGBSA с использованием анализа одной и трех траекторий. б ‒ Значения энергии Гиббса при 37°С (ΔG°37) определены экспериментально и на основании анализа одной МД-траектории комбинацией методов MMGBSA и NMA или Q-Harm. Порядок расположения уравнений соответствует порядку подписей на рисунках.
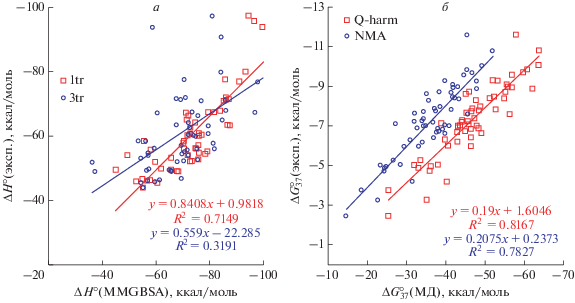
Таблица 2.
Сравнение значений термодинамических параметров, определенных экспериментально и полученных при анализе МД-траекторий, для ДНК/РНК-дуплексова
| N | Метод расчета | a | b | R2 | 〈|ΔE|〉b | 〈|ΔE|/|E|〉, % | 〈|ΔE*||〉b | 〈|ΔE*|/|E*|〉, % |
|---|---|---|---|---|---|---|---|---|
| ΔH° | ||||||||
| 1tr | MMGBSA | 0.84 | 1.0 | 0.71 | 12.0 | 22.9 | 5.1 | 8.2 |
| MMPBSA | 0.49 | ‒44.7 | 0.64 | 25.1 | 44.0 | 8.9 | 10.8 | |
| 3tr | MMGBSA | 0.56 | ‒22.3 | 0.32 | 11.3 | 21.3 | 7.9 | 12.6 |
| MMPBSA | 0.41 | ‒36.9 | 0.13 | 11.4 | 16.0 | 8.7 | 13.8 | |
| ΔS° | ||||||||
| 1tr | Q-Harm | 1.81 | ‒15.3 | 0.55 | 78.2 | 49.0 | 19.0 | 14.3 |
| NMA | 1.77 | 41.1 | 0.74 | 47.5 | 29.2 | 14.3 | 8.4 | |
| 3tr | Q-Harm | 1.33 | ‒31.8 | 0.66 | 60.9 | 38.3 | 16.8 | 9.5 |
| NMA | 1.55 | ‒3.0 | 0.53 | 57.6 | 35.5 | 20.0 | 11.4 | |
| ΔG°37 | ||||||||
| 1tr | MMPBSA + Q-Harm | 0.05 | ‒6.97 | 0.04 | 4.7 | 81.0 | 1.94 | 23.4 |
| MMGBSA + Q-Harm | 0.19 | 1.60 | 0.82 | 35.8 | 561.1 | 0.62 | 9.3 | |
| MMPBSA + NMA | 0.02 | ‒7.38 | 0.00 | 10.5 | 171.6 | 2.01 | 24.2 | |
| MMGBSA + NMA | 0.21 | 0.24 | 0.78 | 26.3 | 407.0 | 0.69 | 9.6 | |
| 3tr | MMPBSA + Q-Harm | ‒0.04 | ‒8.39 | 0.06 | 18.4 | 317.2 | 1.46 | 24.7 |
| MMGBSA + Q-Harm | 0.00 | ‒7.36 | 0.00 | 26.8 | 451.4 | 1.43 | 24.3 | |
| MMPBSA + NMA | ‒0.01 | ‒7.65 | 0.01 | 17.0 | 287.1 | 1.45 | 24.6 | |
| MMGBSA + NMA | 0.03 | ‒6.13 | 0.03 | 25.5 | 423.0 | 1.38 | 23.5 | |
а Все обозначения и размерности см. в табл. 1.
Введение линейной поправки приводит к снижению ошибки до уровня 8.2% (5.1 ккал/моль) в случае метода MMGBSA при анализе траектории только ДНК/РНК-дуплексов, близкой к таковой для РНК/РНК-комплексов.
Значения энтропии формирования ДНК/РНК-комплексов, рассчитанные методом NMA при анализе одной траектории, дают наилучшую корреляцию с экспериментальными величинами (R2 = = 0.74), при этом среднее значение абсолютной ошибки составляет 47.5 кал ∙ моль‒1 ∙ К‒1, или 29% (табл. 2). В случае использования квазигармонического приближения в однотраекторном анализе величина коэффициента корреляции оказалась меньше, R2 = 0.55, а среднее значение абсолютной ошибки составляло 78.2 кал ∙ моль‒1 ∙ К‒1, или 49%. Коэффициент пропорциональности между расчетными и экспериментальными значениями энтропии ДНК/РНК-комплексов лежит в диапазоне 1.3–1.8, а свободный член линейной зависимости ‒ в диапазоне ‒3.0…+41 кал ∙ моль‒1 ∙ К‒1, что указывает на существенные различия значений энтропии формирования комплекса, определенных экспериментально и рассчитанных значений конфигурационной энтропиии. Коэффициент корреляции и наклон близки к таковым в случае РНК/РНК-комплексов (табл. 1 и 2), что указывает на общую природу наблюдаемых закономерностей для разных типов комплексов.
Расчет энтропии формирования РНК-содержащих дуплексов в рамках квазигармонического приближения при анализе одной траектории дает хорошую линейную корреляцию с экспериментальными данными, однако наклон линейной аппроксимации существенно отличаются от единицы, что затрудняет применение такого подхода для оценки полной энтропии комплексообразования. Стоит отметить, что расчет энтропии в квазигармоническом приближении на несколько порядков быстрее, чем при анализе нормальных мод колебаний. Это оправдывает применение Q‑Harm-приближения при оценке энергии Гиббса для большого набора ДНК/РНК- и РНК/РНК-комплексов.
Величины энергии Гиббса формирования гибридных комплексов, рассчитанные на основании метода MMGBSA и конфигурационных энтропий, в высокой степени коррелируют с экспериментальными значениями для ДНК/РНК-дуплексов (рис. 3). Коэффициенты корреляции, R2, составляют 0.82 и 0.78 при расчете энтропии методами Q-Harm и NMA соответственно. В остальных случаях значимой корреляции не наблюдается (R2 < 0.06).
Проведенный анализ применимости модели ближайших соседей для описания величин энтальпии комплексообразования выявил высокую прогностическую эффективность такой модели в применении к данным, полученным экспериментально и методом MMPBSA при анализе одной траектории (рис. 4а). Инкременты модели ближайших соседей, описывающие фактор инициации формирования комплексов, не были статистически значимыми ‒ для экспериментальных и расчетных наборов данных p-value равнялись 0.11 и 0.69 соответственно. При анализе экспериментальной базы данных низкозначимыми оказываются динуклеотидные пары CU/AG и UU/AA. Для величин энтальпий, рассчитанных при однотраекторном анализе методом MMGBSA, все инкременты, кроме фактора инициации, были статистически значимыми (p-value < 4 × 10‒5). Опубликованные ранее и определенные нами величины инкрементов для экспериментальных данных существенно различались для фактора инициации и динуклеотидных пар, у которых в РНК-цепи с 5′-конца находится цитозин (rСХ/dX'G, где Х = A, C, U или G) или гуанин (rGХ/dX'C), за исключением динуклеотидной пары rGU/dAC (рис. 4б). Определенные нами инкременты rСХ/dX'G имеют амплитуды ниже, а rGХ/dX'C – выше, чем представленные в литературе. Также более низкий вклад в стабильность двойной спирали имеют инкременты UG и UU. Сопоставление инкрементов при анализе собранной нами базы данных показывает, что величины инкрементов rСХ/dX'G, rGG/dCC и rGU/dAC экспериментальных энтальпий комплексообразования существенно ниже таковых при их определении методом MMGBSA из анализа одной МД-траектории. Это может быть связано с несогласованностью силовых полей для ДНК и РНК при моделировании гибридных комплексов, которые были оптимизированы для ДНК/ДНК- и РНК/РНК-дуплексов. Кроме того, относительно малая выборка и низкое разнообразие длин и нуклеотидных композиций модельных комплексов приводит к достаточно высокому значению ошибок для отдельных инкрементов модели ближайших соседей (рис. 4б). Расширение выборки в перспективе позволит получить более надежные результаты.
Рис. 4.
Корреляционный анализ значений термодинамических параметров, определенных экспериментально и рассчитанных на основании МД-анализа, для ДНК/РНК-дуплексов. а ‒ Прогностическая эффективность метода ближайших соседей (БС) для расчета энтальпии образования ДНК/РНК-дуплексов, определенной экспериментально (Эксп.) и с использованием метода MMGBSA при анализе трех траекторий (MMGBSA, 1tr). б ‒ Сопоставление инкрементов модели ближайших соседей для ДНК/РНК-комплексов, опубликованных ранее Sugimoto и соавт. [7], определенных на основании экспериментальных данных (Эксп.) и с использованием метода MMPBSA при анализе одной траектории (MMGBSA, 1tr). Порядок расположения уравнений соответсвует порядку подписей на рисунках.
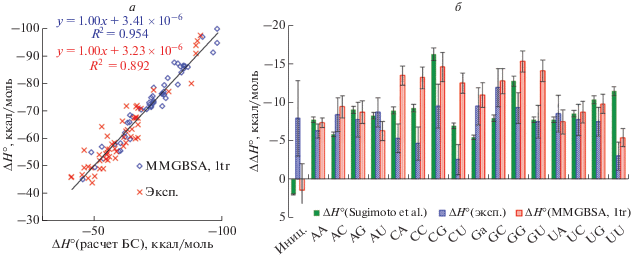
Отрицательные значения факторов инициации и концевых пар, полученные при МД-анализе данных с помощью модели ближайших соседей, превышают таковые для данных из экспериментального анализа для комплексов ДНК/ДНК [30], РНК/РНК и ДНК/РНК. Это указывает на систематическую разницу при моделировании методом МД или расчетах методом MMGBSA. Методом ЯМР показано, что концевые пары оснований находятся в постоянном равновесии между открытым и закрытым состояниями [57], к тому же открытие концевых пар наблюдается не для всех комплексов. Такие события не относятся к многократно повторяющимся в рамках рассматриваемой траектории для каждого моделируемого комплекса, что не позволяет провести достоверный учет вкладов от концевых пар оснований при расчете термодинамических параметров. Несмотря на это, метод MMGBSA дает наилучшую корреляцию экспериментальных и расчетных величин для всех трех типов комплексов.
В результате проведенного нами исследования продемонстрирована возможность расчета термодинамических параметров образования НК-комплексов с использованием подходов, основанных на методе классической МД. При анализе наборов дуплексов РНК/РНК, ДНК/РНК и ДНК/ДНК [30] выявлено, что энтальпия формирования комплексов, рассчитанная методом MMGBSA при анализе МД-траектории только комплекса, в среднем обладает наилучшей корреляцией расчетных и экспериментальных величин для каждого типа комплексов (R2 > 0.73). Ошибка расчета энтальпии формирования комплексов (15‒30%) значительно выше типичной экспериментальной величины (~10%). Высокая корреляция также наблюдается при расчете энтропии в рамках квазигармонического приближения однотраекторного анализа. Однако в этом случае наклон и свободный член линейной зависимости существенно отличались соответственно от единицы и нуля. Как видно из графиков, представленных на рис. 5, по анализу корреляций для экспериментальных и рассчитанных значений энтальпии комплексообразования дуплексов ДНК/ДНК [30], РНК/РНК и ДНК/РНК углы наклона и свободные члены всех линейных уравнений для этих трех пар близки. Мы объединили все полученные в ходе работы данные для определения общей универсальной зависимости, использование которой позволит пересчитывать значения, рассчитанные методом MMGBSA в однотраекторном приближении. Такая линейная функция имеет следующий вид: ΔH°(эксп.) = 0.727 ∙ ΔH°(MMBBSA, 1tr) ‒ 5.32.
Рис. 5.
Корреляционный анализ значений энтальпии (а) и свободной энергии Гиббса (б), определенных экспериментально и рассчитанных методом MMGBSA с использованием одной траектории, для разных типов комплексов. Данные по ДНК/ДНК-комплексам взяты из работы Lomzov и др. [30]. Порядок расположения уравнений соответствует порядку подписей на рисунках.
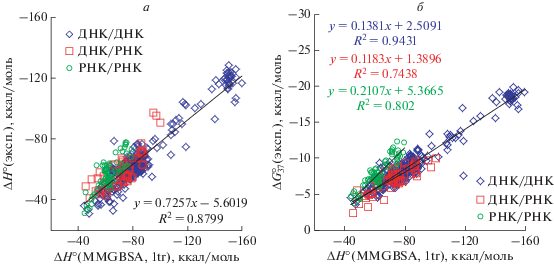
Использование данной линейной коррекции позволяет значительно снизить относительную ошибку расчета энтальпии комплексообразования до 9.3 (7.8), 12.8 (9.9) и 7.5% (6.3%) соответственно для РНК/РНК, ДНК/РНК и ДНК/ДНК (в скобках указана дисперсия для приведенных величин). В среднем для всех типов комплексов ошибка расчета составляла 8.6%, что ниже типичного значения экспериментальной ошибки (10%).
Значительно более высокий уровень корреляции наблюдается между величиной энергии комплексообразования, рассчитанной методом MMGBSA в однотраекторном приближении, и экспериментальной величиной свободной энергии Гиббса при одновременном рассмотрении всех типов комплексов (R2 = 0.897). Однако при более углубленном анализе выявлено, что для каждого из рассмотренных типов комплексов встречаются существенно различающиеся между собой линейные зависимости, а наибольшее значение R2 найдено для выборки из 304 ДНК/ДНК-комплексов. Различия в коэффициентах линейной зависимости связаны, в том числе, с энтропией, вклад которой различен для разных типов комплексов ‒ как по конфигурационной, так и сольватационной составляющей, учитывающей взаимодействие с ионами. Эта гипотеза подтверждается различными линейными зависимостями для экспериментально определенной свободной энергии Гиббса и для рассчитанной на основании энтальпии, полученной методом MMGBSA (1tr), и энтропии в квазигармоническом приближении (табл. 1 и 2). Таким образом, приведенные на рис. 5б линейные зависимости могут быть применены для расчета свободной энергии Гиббса на основании значений энергии комплексообразования, рассчитанных методом MMGBSA в однотраекторном приближении, только в рамках конкретного типа комплексов. Величины ошибки таких расчетов с учетом поправок в виде линейных функций со своими коэффициентами для каждого типа комплексов дают $\Delta G_{{34}}^{^\circ }$ 9.9 (6.7), 10.3 (9.0) и 8.0% (6.9%) соответственно для РНК/РНК, ДНК/РНК и ДНК/ДНК (в скобках указана дисперсия для приведенных величин).
Использование величин изменения внутренней энергии, рассчитанной методом MMGBSA, и конфигурационной энтропии в квазигармоническом приближении для расчета энергии Гиббса формирования дуплекса дает результаты, которые с высоким коэффициентом корреляции линейно связаны с экспериментально определяемыми значениями. Такая зависимость наблюдается для наборов РНК/РНК, ДНК/РНК и ДНК/ДНК различной длины и GC-состава. Вместе с тем высокая дисперсия ошибки расчета указывает на то, что необходимо либо использовать методы более полного покрытия конформационного пространства (sampling-методы), либо проводить анализ для большого числа комплексов различной длины и нуклеотидного состава ‒ с целью достоверного сопоставления рассчитанных данных для разных типов комплексов.
Использование методов компьютерного моделирования с более полным покрытием конформационного пространства при анализе траекторий позволяет определять величину свободной энергии Гиббса. В опубликованных ранее работах показана принципиальная возможность достижения экспериментальной точности (~1 ккал/моль). Применяя метод зонтичной выборки (umbrella sampling) при использовании в качестве координаты расстояния между концами шпилечной структуры РНК, а также метод анализа взвешенных гистограмм (WHAM), L. Smith и др. [58] получили высокую согласованность экспериментальных и расчетных величин свободной энергии. Главный недостаток такого подхода заключается в необходимости получения чрезвычайно длительной МД-траектории ‒ около 200 мкс. Более экономичной может быть комбинация методов алхимических превращений и обмена репликами. В этом случае установлено, что при замене одного основания на другое, того же класса (пуринового или пиримидинового), в РНК-дуплексе размером 6 п.о. точность расчета энергии, связанной с однонуклеотидной заменой, составляет 0.55 ккал/моль [22]; при этом продолжительность МД-траектории небольшая ‒ порядка 1 мкс, что на порядок больше, чем в предлагаемом нами подходе.
Методы МД применяют не только для анализа структуры и гибридизационных свойств нативных олигонуклеотидов, но и для их аналогов и производных ‒ с целью установления их возможной пространственной структуры, влияния модификаций на термостабильность комплексов с НК и причин наблюдаемых физико-химических и молекулярно-биологических эффектов. Для расчета энергии формирования модифицированных комплексов рассматривали замкнутые НК (LNA) [59], пептидил-НК (PNA) [35], 2′-O-Me-производные РНК [34], глицин-морфолиновые аналоги [32] и фосфорилгуанидиновые олигонуклеотиды [33]. Применяли различные подходы, среди которых наиболее распространен использованный и нами метод MMPB(GB)SA. В большинстве случаев показана корреляция рассчитанных энергий с экспериментальными характеристиками термостабильности: энтальпией, свободной энергией Гиббса комплексообразования или температурой плавления. Однако количественного согласования расчетных значений энергий с наблюдаемыми экспериментально при использовании большинства подходов не наблюдается. Это дополнительно усугубляется тем, что обычно анализируют небольшое число комплексов близкой длины и/или GC-состава. В совокупности полученные данные свидетельствует о невозможности количественного сопоставления энергии Гиббса формирования разных типов комплексов, например олигонуклеотидов с измененной химической структурой, при использовании методов классической МД и методов MMPB(GB)SA для расчета энтальпии и Q-Harm или NMA для расчета энтропии комплексообразования. Проведенные нами исследования показывают возможность количественного сопоставления энтальпии формирования РНК/РНК-, ДНК/РНК- и ДНК/ДНК-комплексов. Справедливо ли это утверждение для производных НК, можно будет понять по результатам анализа набора их комплексов, различной длины и GC-состава, с применением предложенного нами подхода.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
В проведенном исследовании проанализирована возможность использования метода МД для прогнозирования величины энергии формирования НК-дуплексов. Для расчета энтальпии комплексообразования использованы методы MMPBSA и MMGBSA, а энтропии – Q-Harm и NMA. Анализ проводили, используя только МД-траекторию НК-дуплекса или три траектории (двухцепоченого состояния и одноцепочечных олигонуклеотидов). Величину энергии Гиббса формирования дуплексов оценивали, используя комбинации величин энтропии и энтальпии, определенных разными методами в рамках одно- или трехтраекторного подходов. В ряде случаев полученные данные характеризовались высоким значением коэффициента корреляции экспериментальных и рассчитанных величин.
На основании полученных результатов можно сделать вывод, что количественный расчет энтальпии комплексообразования допустимо проводить для РНК/РНК-, ДНК/РНК- и ДНК/ДНК-комплексов, используя метод MMGBSA в однотраекторном приближении с применением линейной эмпирической корректировки. Получаемая таким способом величина энтальпии с точностью 8.6% совпадает с экспериментальной. Значения энтропии и свободной энергии Гиббса можно сравнивать количественно только в рамках одного типа комплексов. В этом случае однотраекторный подход и комбинация методов MMGBSA и Q-Harm дают наибольшую точность: в среднем 8.2 и 9.3% соответственно для РНК/РНК- и ДНК/РНК-дуплексов ‒ с учетом линейной поправки для каждого типа комплекса. Использование двух этих способов расчета наиболее вычислительно эффективно и дает точную количественную оценку термодинамических параметров комплексообразования.
Для распространения предлагаемого подхода на аналоги или производные НК необходимо достоверно рассчитать структуру олигомеров и их комплексов с НК. Как только такие структуры или ансамбли структур будут найдены, метод можно использовать для быстрой оценки гибридизационной способности производных НК. Ранее, на глицин-морфолиновых олигомерах [32] и отдельных фосфорилгуанидиновых олигонуклеотидах [33], нами показано, что рассчитанные предлагаемым методом энтальпии комплексообразования близки экспериментально определенным значениям. Однако для количественной оценки достоверности предлагаемого метода необходимы дополнительные исследования на наборе олигомеров различной длины и GC-состава.
Для количественного расчета энтальпии формирования дуплексов можно рекомендовать метод MMGBSA с анализом траектории дуплексной структуры ‒ как эффективный и не слишком времязатратный. В перспективе предложенные нами подходы могут быть также распространены на аналоги и производные нуклеиновых кислот, что даст принципиальную возможность перейти к рациональному дизайну новых типов соединений.
Авторы выражают благодарность Центру коллективного пользования “Сибирский суперкомпьютерный центр СО РАН” (Новосибирск) за предоставленную возможность использования вычислительных кластеров, на которых были проведены расчеты.
Исследование выполнено при финансовой поддержке РФФИ и Правительства Новосибирской области в рамках научного проекта № 20-43-543029, а также при финансовой поддержке базового бюджетного финансирования ПФНИ ГАН № AAAA-A17-117020210021-7.
Статья не содержит экспериментов с привлечением животных или людей.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Bennett C.F. (2019) Therapeutic antisense oligonucleotides are coming of age. Annu. Rev. Med. 70, 307–321.
Smith C.I E., Zain R. (2019) Therapeutic oligonucleotides: State of the art. Annu. Rev. Pharmacol. Toxicol. 59, 605–630.
Benizri S., Gissot A., Martin A., Vialet B., Grinstaff M.W., Barthélémy P. (2019) Bioconjugated oligonucleotides: recent developments and therapeutic applications. Bioconjug. Chem. 30, 366–383.
Glazier D.A., Glazier D.A., Liao J., Roberts B.L., Li X., Yang K., Stevens C. M., Tang W., Tang W. (2020) Chemical synthesis and biological application of modified oligonucleotides. Bioconjug. Chem. 31, 1213–1233.
Pyshnyi D.V., Lomzov A.A., Pyshnaya I.A., Ivanova E.M. (2006) Hybridization of the bridged oligonucleotides with DNA: thermodynamic and kinetic studies. J. Biomol. Struct. Dyn. 23, 567–579.
Banerjee D., Tateishi-Karimata H., Ohyama T., Ghosh S., Endoh T., Takahashi S., Sugimoto N. (2020) Improved nearest-neighbor parameters for the stability of RNA/DNA hybrids under a physiological condition. Nucleic Acids Res. 48(21), 12042–12054.
Sugimoto N., Nakano S., Katoh M., Matsumura A., Nakamuta H., Ohmichi T., Yoneyama M., Sasaki M. (1995) Thermodynamic parameters to predict stability of RNA/DNA hybrid duplexes. Biochemistry. 34, 11211–11216.
Hoshika S., Leal N. A., Kim M.-J., Kim M.-S., Karalkar N. B., Kim H.-J., Bates A. M., Watkins N.E., SantaLucia H.A., Meyer A.J., DasGupta S., Piccirilli J.A., Ellington A.D., SantaLucia J., Georgiadis M.M., Benner S.A. (2019) Hachimoji DNA and RNA: a genetic system with eight building blocks. Science. 363, 884–887.
Watkins N.E., SantaLucia J. (2005) Nearest-neighbor thermodynamics of deoxyinosine pairs in DNA duplexes. Nucleic Acids Res. 33, 6258–6267.
McTigue P. M., Peterson R. J., Kahn J.D. (2004) Sequence-dependent thermodynamic parameters for locked nucleic acid (LNA)-DNA duplex formation. Biochemistry. 43, 5388–5405.
Owczarzy R., You Y., Groth C.L., Tataurov A.V. (2011) Stability and mismatch discrimination of locked nucleic acid–DNA duplexes. Biochemistry. 50, 9352–9367.
Lesnik E.A., Freier S.M. (1995) Relative thermodynamic stability of DNA, RNA, and DNA:RNA hybrid duplexes: relationship with base composition and structure. Biochemistry. 34, 10807–10815.
Giesen U., Kleider W., Berding C., Geiger A., Ørum H., Nielsen P.E. (1998) A formula for thermal stability (T(m)) prediction of PNA/DNA duplexes. Nucleic Acids Res. 26, 5004–5006.
von Ahsen N., Wittwer C.T., Schütz E. (2001) Oligo-nucleotide melting temperatures under PCR conditions: nearest-neighbor corrections for Mg2+, deoxynucleotide triphosphate, and dimethyl sulfoxide concentrations with comparison to alternative empirical formulas. Clin. Chem. 47, 1956–1961.
Armacost K.A., Riniker S., Cournia Z. (2020) Novel directions in free energy methods and applications. J. Chem. Inf. Model. 60, 1–5.
Kollman P.A., Massova I., Reyes C., Kuhn B., Huo S., Chong L., Lee M., Lee T., Duan Y., Wang W., Donini O., Cieplak P., Srinivasan J., Case D.A., Cheatham T.E. (2000) Calculating structures and free energies of complex molecules: combining molecular mechanics and continuum models. Acc. Chem. Res. 33, 889–897
Tsui V., Case D.A. (2000) Molecular dynamics simulations of nucleic acids with a generalized born solvation model. J. Am. Chem. Soc. 122, 2489–2498.
Rastelli G., Rio A.Del., Degliesposti G., Sgobba M. (2009) Fast and accurate predictions of binding free energies using MM-PBSA and MM-GBSA. J. Comput. Chem. 32.
Kumar S., Rosenberg J.M., Bouzida D., Swendsen R.H., Kollman P.A. (1992) The weighted histogram analysis method for free-energy calculations on biomolecules. I. The method. J. Comput. Chem. 13, 1011–1021.
Lee T.-S., Radak B.K., Huang M., Wong K.-Y., York D.M. (2014) Roadmaps through free energy landscapes calculated using the multidimensional vFEP approach. J. Chem. Theory Comput. 10, 24–34.
Williams-Noonan B.J., Yuriev E., Chalmers D.K. (2018) Free energy methods in drug design: prospects of “alchemical perturbation” in medicinal chemistry. J. Med. Chem. 61, 638–649.
Sakuraba S., Asai K., Kameda T. (2015) Predicting RNA duplex dimerization free-energy changes upon mutations using molecular dynamics simulations. J. Phys. Chem. Lett. 6, 4348–4351.
Lee H.-C., Hsu W.-C., Liu A.-L., Hsu C.-J., Sun Y.-C. (2014) Using thermodynamic integration MD simulation to compute relative protein–ligand binding free energy of a GSK3β kinase inhibitor and its analogs. J. Mol. Graph. Model. 51, 37–49.
Wang E., Sun H., Wang J., Wang Z., Liu H., Zhang J.Z.H., Hou T. (2019) End-point binding free energy calculation with MM/PBSA and MM/GBSA: strategies and applications in drug design. Chem. Rev. 119, 9478–9508.
Gilson M.K., Honig B. (1988) Calculation of the total electrostatic energy of a macromolecular system: solvation energies, binding energies, and conformational analysis. Proteins Struct. Funct. Bioinform. 4, 7–18.
Srinivasan J., Cheatham T.E., Cieplak P., Kollman P.A., Case D.A. (1998) Continuum solvent studies of the stability of DNA, RNA, and phosphoramidate-DNA helices. J. Am. Chem. Soc. 120, 9401–9409.
Brooks B.R., Janezic D., Karplus M. (1995) Harmonic analysis of large systems. I. Methodology. J. Comput. Chem. 16, 1522–1542
Choudhary M.I., Shaikh M., Tul-Wahab A., Ur-Rahman A. (2020) In silico identification of potential inhibitors of key SARS-CoV-2 3CL hydrolase (Mpro) via molecular docking, MMGBSA predictive binding energy calculations, and molecular dynamics simulation. PLoS One. 15, e0235030.
Wright D.W., Hall B.A., Kenway O.A., Jha S., Coveney P.V. (2014) Computing clinically relevant binding free energies of HIV-1 protease inhibitors. J. Chem. Theory Comput. 10, 1228–1241.
Lomzov A.A., Vorobjev Y.N., Pyshnyi D.V. (2015) Evaluation of the Gibbs free energy changes and melting temperatures of DNA/DNA duplexes using hybridization enthalpy calculated by molecular dynamics simulation. J. Phys. Chem. B. 119, 15221–15234.
Yesudas J.P., Blinov N., Dew S.K., Kovalenko A. (2015) Calculation of binding free energy of short double stranded oligonucleotides using MM/3D-RISM-KH approach. J. Mol. Liq. 201, 68–76.
Golyshev V.M., Abramova T.V., Pyshnyi D.V., Lom-zov A.A. (2019) Structure and hybridization properties of glycine morpholine oligomers in complexes with DNA and RNA: experimental and molecular dynamics studies. J. Phys. Chem. B. 123, 10571–10581.
Golyshev V.M., Pyshnyi D.V., Lomzov A.A. (2021) Effects of phosphoryl guanidine modification of phosphate residues on the structure and hybridization of oligodeoxyribonucleotides. J. Phys. Chem. B. 125, 2841–2855.
Suresh G., Priyakumar U.D. (2015) Inclusion of methoxy groups inverts the thermodynamic stabilities of DNA-RNA hybrid duplexes: a molecular dynamics simulation study. J. Mol. Graph. Model. 61, 150–159.
Siriwong K., Chuichay P., Saen-oon S., Suparpprom C., Vilaivan T., Hannongbua S. (2008) Insight into why pyrrolidinyl peptide nucleic acid binding to DNA is more stable than the DNA·DNA duplex. Biochem. Biophys. Res. Commun. 372, 765–771.
Lomzov A.A., Gorelov V.V., Golyshev V.M., Abra-mova T.V., Pyshnyi D.V. (2015) 139 Analysis of structure and thermodynamics of modified DNA duplexes using molecular dynamics simulation. J. Biomol. Struct. Dyn. 33, 90–91.
Xia T., SantaLucia J., Burkard M.E., Kierzek R., Schroeder S. J., Jiao X., Cox C., Turner D.H. (1998) Thermodynamic parameters for an expanded nearest-neighbor model for formation of RNA duplexes with Watson‒Crick base pairs. Biochemistry. 37, 14719–14735.
Case D.A., Babin V., Berryman J.T., Betz R.M., Cai Q., Cerutti D.S., Cheatham T.E., Darden T.A., Duke R.E., Gohlke H., Goetz A.W., Gusarov S., Homeyer N., Janowski P., Kaus J., Kolossváry I., Kovalenko A., Lee T.S., LeGrand S., Luchko T., Luo R., Madej B., Merz K.M., Paesani F., Roe D.R., Roitberg A., Sagui C., Salomon-Ferrer R., Seabra G., Simmerling C.L., Smith W., Swails J., Walker R.C., Wang J., Wolf R.M., Wu X., Kollman P.A. (2014) AMBER 14, University of California, San Francisco. https://doi.org/10.13140/rg.2.2.17892.37766
Pettersen E.F., Goddard T.D., Huang C.C., Couch G.S., Greenblatt D.M., Meng E.C., Ferrin T.E. (2004) UCSF Chimera ‒ a visualization system for exploratory research and analysis. J. Comput. Chem. 25, 1605–1612.
Pérez A., Marchán I., Svozil D., Sponer J., Cheat-ham T.E., Laughton C.A., Orozco M. (2007) Refinement of the AMBER force field for nucleic acids: improving the description of α/γ conformers. Biophys. J. 92, 3817–3829.
Zgarbová M., Otyepka M., Šponer J., Mládek A., Banáš P., Cheatham T.E., Jurečka P. (2011) Refinement of the Cornell et al. nucleic acids force field based on reference quantum chemical calculations of glycosidic torsion profiles. J. Chem. Theory Comput. 7, 2886–2902.
Lu X.-J. (2003) 3DNA: a software package for the analysis, rebuilding and visualization of three-dimensional nucleic acid structures. Nucleic Acids Res. 31, 5108–5121.
Lu X.-J., Olson W.K. (2008) 3DNA: a versatile, integrated software system for the analysis, rebuilding and visualization of three-dimensional nucleic-acid structures. Nat. Protoc. 3, 1213–1227.
Gyi J.I., Conn G.L., Lane A.N., Brown T. (1996) Comparison of the thermodynamic stabilities and solution conformations of DNA∙RNA hybrids containing purine-rich and pyrimidine-rich strands with DNA and RNA duplexes. Biochemistry. 35, 12538–12548.
Arnott S., Chandrasekaran R., Millane R.P., Park H.S. (1986) DNA-RNA hybrid secondary structures. J. Mol. Biol. 188, 631–640.
Hall K.B., McLaughlin L.W. (1991) Thermodynamic and structural properties of pentamer DNA-DNA, RNA-RNA and DNA-RNA duplexes of identical sequence. Biochemistry. 30, 10606–10613.
Ломзов А.А., Купрюшкин М.С., Дюдеева Е.С., Пышный Д.В. (2021) Сравнительное исследование гибридизационных свойств фосфорилгуанидиновых олигонуклеотидов с ДНК и РНК. Биоорг. химия. 47(2), 250–258.
Cross C.W., Rice J.S., Gao X. (1997) Solution structure of an RNA∙DNA hybrid duplex containing a 3'-thioformacetal linker and an RNA A-tract. Biochemistry. 36, 4096–4107.
Zimmerman S.B., Pheiffer B.H. (1981) A RNA.DNA hybrid that can adopt two conformations: an X-ray diffraction study of poly(rA).poly(dT) in concentrated solution or in fibers. Proc. Natl. Acad. Sci. USA. 78, 78–82.
Zhou J., Gregurick S.K., Krueger S., Schwarz F.P. (2006) Conformational changes in single-strand DNA as a function of temperature by SANS. Biophys. J. 90, 544–551.
Chen H., Meisburger S.P., Pabit S.A., Sutton J.L., Webb W.W., Pollack L. (2012) Ionic strength-dependent persistence lengths of single-stranded RNA and DNA. Proc. Natl. Acad. Sci. USA. 109, 799–804.
Isaksson J., Acharya S., Barman J., Cheruku P., Chattopadhyaya J. (2004) Single-stranded adenine-rich DNA and RNA retain structural characteristics of their respective double-stranded conformations and show directional differences in stacking pattern. Biochemistry. 43, 15996–16010.
Chakraborty K., Mantha S., Bandyopadhyay S. (2013) Molecular dynamics simulation of a single-stranded DNA with heterogeneous distribution of nucleobases in aqueous medium. J. Chem. Phys. 139(7), 075103.
Sen S., Nilsson L. (2001) MD Simulations of homomorphous PNA, DNA, and RNA single strands: cha-racterization and comparison of conformations and dynamics. J. Am. Chem. Soc. 123, 7414–7422.
Owczarzy R., You Y., Moreira B.G., Manthey J.A., Huang L., Behlke M.A., Walder J.A. (2004) Effects of sodium ions on DNA duplex oligomers: improved predictions of melting temperatures. Biochemistry. 43, 3537–3554.
SantaLucia J. (1998) A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA. 95, 1460–1465.
Nonin S., Leroy J.-L., Gueron M. (1995) Terminal base pairs of oligodeoxynucleotides: imino proton exchange and fraying. Biochemistry. 34, 10652–10659.
Smith L.G., Tan Z., Spasic A., Dutta D., Salas-Estrada L.A., Grossfield A., Mathews D.H. (2018) Chemically accurate relative folding stability of RNA hairpins from molecular simulations. J. Chem. Theory Comput. 14, 6598–6612.
Shen L., Johnson T.L., Clugston S., Huang H., Butenhof K.J., Stanton R.V. (2011) Molecular dynamics simulation and binding energy calculation for estimation of oligonucleotide duplex thermostability in RNA-based therapeutics. J. Chem. Inf. Model. 51, 1957–1965.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология