

Молекулярная биология, 2022, T. 56, № 2, стр. 244-258
Роль ремоделирующего комплекса SWI/SNF в регуляции экспрессии генов воспаления
А. В. Феоктистов a, b, *, С. Г. Георгиева a, c, Н. В. Сошникова a, b
a Институт биологии гена Российской академии наук
119334 Москва, Россия
b Центр точного геномного редактирования и генетических технологий для биомедицины
Института молекулярной биологии им. В.А. Энгельгардта
119991 Москва, Россия
c Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
* E-mail: a.feo95@mail.ru
Поступила в редакцию 22.06.2021
После доработки 07.09.2021
Принята к публикации 07.09.2021
- EDN: HUXFXS
- DOI: 10.31857/S0026898422020070
Аннотация
Процесс воспаления – естественный защитный ответ организма на проникновение чужеродных веществ и молекул. В активации генов воспаления участвует множество белков, сигнальных каскадов и факторов транскрипции. Их координированная работа приводит к изменению экспрессии провоспалительных генов. Состояние хроматина генов, отвечающих на воспалительные стимулы, считается основным фактором, определяющим связывание активаторов транскрипции с регуляторными элементами, и ключевым механизмом в индукции воспалительных генов. Быстрое изменение состояния хроматина, создание открытой структуры и снятие “нуклеосомного барьера” облегчает связывание транскрипционных факторов и инициацию транскрипции. Этот процесс реализуется путем привлечения на хроматин комплексов, модифицирующих и ремоделирующих хроматин. Один из важнейших комплексов, реструктурирующих структуру хроматина в процессе активации генов, – мультисубъединичный комплекс SWI/SNF. SWI/SNF регулирует экспрессию генов воспаления через взаимодействие с факторами транскрипции, в том числе с компонентами сигнального пути NF-κB. Вариабельность субъединиц этого комплекса определяет специфичность связывания с хроматином и активаторами транскрипции. В данном обзоре рассмотрена роль SWI/SNF в регуляции генов воспаления, описано его взаимодействие с хроматином и молекулярные механизмы рекрутирования комплекса SWI/SNF на промоторы.
ВВЕДЕНИЕ
Воспаление – это ответ иммунной системы организма, обеспечивающий выживание тканей. Воспаление провоцируется множеством внешних и внутренних факторов, включая патогены, токсичные соединения и поврежденные клетки. Важную роль в воспалительных реакциях играют клетки врожденной иммунной системы: макрофаги, дендритные и тучные клетки, нейтрофилы и лимфоциты. Реализации воспалительных процессов способствуют и неиммунные клетки – эпителиальные и эндотелиальные, фибробласты.
На поверхности всех этих клеток находятся рецепторы, распознающие сигналы инфекционных и неинфекционных агентов, которые вместе с повреждением клеток запускают сигнальные пути воспаления. Основным сигнальным путем воспаления считается NF-κB, однако огромное количество белков других сигнальных каскадов также принимают участие в модуляции воспалительного ответа. В результате активации этих сигнальных каскадов специфические факторы транскрипции взаимодействуют с регуляторными элементами генов-мишеней и привлекают мультибелковые коактиваторные комплексы и комплексы общего аппарата транскрипции на их промоторы.
Подробное изучение механизмов активации транскрипции провоспалительных генов выявило их значительное разнообразие, которое отражает потребность в высокой специфичности регуляции каждого гена в различных физиологических условиях.
Важнейшую роль в активации провоспалительных генов играет состояние хроматина в их регуляторных элементах. Организация хроматина регулирует доступность ДНК для тех или иных регуляторных факторов. Хроматин также служит платформой, которая влияет на набор белковых факторов, привлекаемых в ходе активации транскрипции. Таким образом, организация и структура хроматина играют решающую роль в модуляции активности факторов транскрипции, связывающихся с данным элементом.
Провоспалительные гены обычно находятся в неактивном состоянии и отличаются высокой плотностью нуклеосом на их регуляторных элементах. Для эффективной активации генов необходимо изменение хроматина и снятие “нуклеосомного барьера”, которое осуществляют специальные белковые комплексы. Комплексы, реорганизующие (ремоделирующие) структуру хроматина, содержат в своем составе АТРазу и реструктурируют хроматин, меняя расположение нуклеосом на молекуле ДНК за счет энергии гидролиза АТР. Эти комплексы привлекаются на хроматин в результате взаимодействия либо с активаторами транскрипции, либо с определенными модификациями С-концевых последовательностей гистонов (табл. 1).
Таблица 1.
Комплексы SWI/SNF и их особенности
| Субъединица | Ген | Аннотированные домены | Взаимодействие с факторами NF-κB | Ген воспаления |
|---|---|---|---|---|
| Общие субъединицы комплексов SWI/SNF | ||||
| BRG1 | SMARCA4 | QLQ, HSA, BRK, SNF2-N, Helicase-C, SnAC, Bromo | Rel-B | Il12b, Ifnb1, Saa3, Ccl5, Il6, Il12b, Nos2 |
| BRM | SMARCA2 | Rel-A, Rel-B | ||
| BAF155 | SMARCC1 | Chromo, SANT, SWIRM, SWIRM-associated | Rel-B | |
| BAF170 | SMARCC2 | |||
| BAF60A | SMARCD1 | SWIB | Rel-B | |
| BAF60B | SMARCD2 | |||
| BAF60C | SMARCD3 | |||
| BAF53A | ACTL6A | Actin | Rel-B | |
| BAF53B | ACTL6B | |||
| β-actin | ACTB | |||
| PBAF-специфичные | ||||
| BAF200 | ARID2 | ARID, RFX-DBD, ZnF-C2H2 | ||
| BRD7 | Bromo | |||
| PHF10 | BAF45A | SAY, DPF | Rel-A, p50 | IL6 |
| BAF180 | PBRM1 | PHD, BAH, HMG-box | ||
| BAF-специфичные | ||||
| BAF250a | ARID1A | ARID, BAF250-C | Il6, CXCL1, IL1A, IL1B, IL6 и IL8 | |
| BAF250b | ARID1B | |||
| BAF45B | DPF1 | Requiem-N, PHD | p50 | IL6 |
| BAF45D | DPF2 | Requiem-N, ZnF-C2H2 | ||
| BAF45C | DPF3 | p50 | ||
| ncBAF-специфичные | ||||
| BRD9 | Bromo | |||
| GLTSCR1 | GLTSCR1 | |||
| GLTSCRL1 | ||||
| Общие для BAF и PBAF | ||||
| BAF47 | SMARCB1 | |||
| BAF57 | SMARCE1 | HMG-box | Rel-B | |
| Общие для BAF и ncBAF | ||||
| SSXT, SYT | SS18 | |||
| CREST | SS18L1 | |||
СИГНАЛЬНЫЕ ПУТИ, АКТИВИРУЮЩИЕ ГЕНЫ ВОСПАЛЕНИЯ (NF-κB)
В ответ на различные стимулы провоспалительные гены могут быстро переходить из репрессированного состояния в транскрипционно активное. Воспалительные стимулы идентифицируются паттернраспознающими рецепторами (PRR, Pattern Recognition Receptors), которые присутствуют в клетках врожденной и адаптивной иммунной системы, например, мембранными Toll-подобные рецепторами (TLR, Toll-like receptors) или цитоплазматическими Nod-подобными рецепторами (NLR, Nod-like receptors) и RLR-рецепторами (RIG-I-like receptors). PRR-рецепторы отвечают за обнаружение микроорганизмов и любых клеточных повреждений [1] путем распознавания консервативных структур микроорганизмов, называемых патоген-ассоциированными молекулярными паттернами (PAMP – Pathogen-associated molecular patterns), а также эндогенных молекул, образующихся в результате внутренних повреждений, называемых молекулярными паттернами, связанными с повреждениями (DAMP, Damage-associated molecular patterns).
После распознавания антигена рецепторы димеризуются и происходит последовательная активация ряда киназ разных сигнальных каскадов. В результате передачи сигнала активируются факторы транскрипции, которые рекрутируются на хроматин и стимулируют экспрессию генов воспаления. Сигнальные каскады, приводящие к индукции генов воспаления, многочисленны, как и факторы транскрипции, рекрутирующиеся на провоспалительные гены в ответ на стимулы.
Наиболее интенсивно гены воспаления индуцируются в ответ на активацию пути NF-κB. Этот путь впервые был описан при обработке экстрактов В-клеток мембранными липополисахаридами (LPS) бактерий Escherichia сoli. Эта обработка приводила к последовательному фосфорилированию семейства факторов NF-κB и их связыванию с энхансером гена легкой цепи IgK [2].
В передаче сигнала участвуют многочисленные ветви разных сигнальных каскадов. Однако общим моментом этого процесса является активация семейства факторов NF-κB, в состав которого входят пять белков: p65 (RelA), c-Rel, RelB, p100/p50 и p105/p52 [3], высококонсервативных у млекопитающих. Каждый белок семейства NF-κB способен образовывать гомодимеры и гетеродимеры. Возможно образование 15 комбинаций димеров [4].
До стимуляции воспалительного ответа большинство белков NF-κB удерживается в цитоплазме иммунных клеток белками IκB, содержащими анкириновые повторы. Белки p50 и p52 первоначально синтезируются в виде предшественников p105 и p100 с IκB-подобным доменом анкиринового повтора на С-конце. Анкириновые повторы IkB взаимодействуют с анкириновыми повторами белков NF-κB. При стимуляции димеры NF-κB высвобождаются из IκB в цитоплазме и перемещаются в ядро, чтобы активировать транскрипцию. Димеры NF-κB могут отделяться от IκB либо за счет фосфорилирования IκB, что ведет к его убиквитинированию и последующей деградации протеасомами, либо путем индукции протеолитического расщепления домена анкириновых повторов гетеродимера p100: RelB [5, 6].
В ядре активаторы NF-κB с помощью домена трансактивации взаимодействуют со многими кофакторами, включая общие факторы транскрипции и комплексы, модифицирующие гистоны [7]. Вносимые ими модификации, в свою очередь, служат платформой для привлечения дополнительных активаторов, таких как комплексы, ремоделирующие хроматин.
КОМПЛЕКСЫ SWI/SNF
Переход к активации транскрипции генов воспаления осуществляется посредством двух механизмов: рекрутирования активаторов транскрипции на регуляторные последовательности и изменения состояния хроматина. У млекопитающих комплексы, связанные с изменением структуры хроматина (ремоделирующие), представлены несколькими семействами: SWI/SNF, ISWI, CHD и INO80. Их классификация основана на структуре основной субъединицы комплекса, обладающей АТРазной активностью [8, 9]. Ремоделирующие комплексы регулируют транскрипцию, влияют на работу промоторов, энхансеров и инсуляторов, участвуют в репликации и репарации ДНК [10].
Основные комплексы, изменяющие топологию хроматина при активации транскрипции и влияющие таким образом на экспрессию генов –комплексы семейства SWI/SNF. Эти комплексы связываются с такими цис-регуляторными элементами, как промоторы и энхансеры, и способствуют установлению и поддержанию открытой структуры хроматина в сайтах связывания факторов транскрипции [11–17]. АТРазная субъединица комплексов SWI/SNF представлена белком BRG1 или его гомологом BRM, поэтому комплексы SWI/SNF часто называют BAF (BRG1/BRM associated factors). Комплексы SWI/SNF – это ключевые регуляторы позиционирования нуклеосом на промоторах [18]. Они также могут сдвигать нуклеосомы вдоль цепи ДНК, удалять нуклеосому и гистоны H2A и H2B из октамера [8, 19].
Выделяют три типа мультибелковых комплексов SWI/SNF млекопитающих, которые содержат как общие, так и специфичные для каждого комплекса субъединицы: канонический BAF (BAF) [20, 21]; полибромассоциированный BAF (PBAF) [22, 23] и неканонический BAF (ncBAF) [24–27]. Каждый из трех комплексов SWI/SNF содержит общие основные субъединицы – BAF155, BAF170, BAF60A, АТРазу BRG1 или BRM и ряд специфических субъединиц, характерных для каждого из комплексов (табл. 1) [28]. Вариабельность белкового состава SWI/SNF увеличивается также за счет того, что некоторые его субъединицы кодируются семействами генов, т.е. могут быть представлены различными гомологичными вариантами.
Существование специфических и вариабельных субъединиц в составе комплекса приводит к его гетерогенности, что обуславливает высокую избирательность взаимодействия комплекса с определенными геномными локусами. Неоднородный состав субъединиц также позволяет комплексу SWI/SNF вносить вклад в регуляцию транскрипции динамическим образом в зависимости от типа клеток, стадии развития и стимулов, которые приводят к изменению экспрессии генов [29–34].
Комплекс семейства BAF был кристаллизован, а его структура определена с помощью одночастичной криоэлектронной микроскопии и масс-спектрометрии (single-particle cryo-EM, cross-linking mass-spectrometry) [35]. В результате в составе комплекса BAF идентифицировали следующие модули: коровый, каталитический “АТРазный” и ARP.
Изучена архитектура и порядок сборки комплексов всех трех семейств BAF, PBAF и ncBAF, организованных по одинаковому принципу [28]. Комплекс SWI/SNF формируется последовательно: сначала cобирается коровый модуль – образуются димеры субъединиц BAF170/BAF155, после этого присоединяется субъединица BAF60, затем встраиваются BAF57 и BAF47. В случае комплекса ncBAF вместо BAF57 и BAF47 могут встраиваться субъединицы GLTSCR1 и GLTSCR1L. Формируется коровый модуль, который связывает BRD9 и взаимодействует с BAF250 (комплекс BAF) или BAF200 (комплекс PBAF). Комплекс BAF связывает DPF2, а затем встраивает АТРазный модуль, содержащий SS18. Комплекс PBAF связывает BRD7 и PHF10, а затем рекрутирует АТРазный модуль без SS18. Формирование комплекса заканчивается встраиванием АТРазного модуля.
Субъединицы комплекса SWI/SNF содержат разнообразные домены, позволяющие комплексам специфически связываться с хроматином, с ДНК, с модификациями N-концевых последовательностей “хвостов” гистонов и опосредовать различные белок-белковые взаимодействия (табл. 1). В число этих доменов входят бромодомены, представленные в субъединицах BRG1/BRM1, BAF180, BRD7, BRD9, которые узнают ацетилированные хвосты гистонов, DPF-домены белков DPF1-3 и PHF10, связывающие модификации H3K14ac и H4ac [36]. Chromo- и BAH-домены с высокой специфичностью связывают метил-лизин-содержащие хвосты гистонов. В связывании ДНК участвуют ARID-домены (AT-rich interaction domain) и HMG-бокс. SANT, SWIRM и QLQ опосредуют белок-белковые взаимодействия при сборке хроматин-белковых комплексов. Домен цинковые пальцы C2H2 (ZNF–C2H2) может связывать РНК и опосредовать белок-белковые взаимодействия, но он наиболее известен своими функциями в составе специфичных к последовательности ДНК-связывающих белков (табл. 1).
Впервые участие комплекса SWI/SNF в регуляции экспрессии генов воспаления показано при активации Т-клеток LPS и иономицином: SWI/SNF млекопитающих немедленно транспортируется в хроматин, подтверждая существование связи между ремоделированием хроматина и индукцией транскрипции в иммунных клетках [37].
ХРОМАТИН ПРИ АКТИВАЦИИ ГЕНОВ ВОСПАЛЕНИЯ И SWI/SNF
Еще десятилетия назад установили, что от состояния хроматина, влияющего на доступность ДНК для факторов транскрипции, сильно зависит избирательность транскрипции генов [38, 39]. Хроматин состоит из нуклеосом, линкерных гистонов, а также многих негистоновых белков [40]. Гистоновые и негистоновые белки хроматина поддерживают целостность ДНК, обеспечивают корректность пространственной организации генома и регуляцию экспрессии генов. Интенсивная транскрипция генов сопровождается открытым состоянием хроматина, сниженным количеством нуклеосом, доступностью ДНК для нуклеаз и большой концентрацией белков-активаторов транскрипции и коактиваторных комплексов. Неактивные гены находятся в “закрытом” хроматине, который характеризуется большой плотностью нуклеосом, низкой доступностью ДНК для нуклеаз, присутствием белков-репрессоров транскрипции и репрессорных комплексов. В нормальных условиях (без стимуляции) хроматин не позволяет генам воспаления транскрибироваться. Для преодоления ингибирующего состояния хроматина необходимо согласованное действие факторов транскрипции и коактиваторных комплексов.
Факторы семейства NF-κB не могут взаимодействовать с хроматинизированной матрицей и требуют связывания с геном комплексов ремоделирования хроматина. В структурных исследованиях показано прямое связывание NF-κB с голой ДНК-матрицей [41, 42]. С гораздо меньшей эффективностью гомодимеры NF-κB p50 связывают сайты κB в составе нуклеосомы in vitro. Расположение сайта связывания относительно нуклеосомы сильно влияет на аффинность связывания p50: предпочтительными являются сайты связывания вблизи края нуклеосомы [43, 44]. В этих же экспериментах связывание p50 c нуклеосомами in vitro зависело от активности ремоделирующего комплекса SWI/SNF [45].
Результаты этих экспериментов указывают на возможность существования in vivo “нуклеосомного барьера”, который закрывает ДНК от связывания активаторов. Несмотря на то, что привлечение активаторных факторов транскрипции на промоторы является ключевым событием в индукции экспрессии генов воспаления в рамках сигнального пути NF-κB, существование “нуклеосомного барьера” обеспечивает рекрутирование белков семейства NF-κB к мишеням с переменной кинетикой в зависимости от гена [46].
Il12b – один из хорошо изученных генов, на котором установлено, что непермиссивная конфигурация хроматина действует как регуляторный барьер, который должен быть устранен для правильной экспрессии гена. Для активации Il12b требуется ремоделирование хроматина и привлечение комплексов SWI/SNF. В ранних работах показано, что для активации Il12b необходимы факторы C/EBP, AP-1 и NFAT, а также c-Rel, входящий в семейство NF-κB [47–49]. Критический сайт связывания с-Rel занят нуклеосомой в положении –30…–175 п.н. выше сайта инициации транскрипции. Эта нуклеосома избирательно ремоделируется при стимуляции LPS. Более того, ремоделирование промотора зависело от синтеза белка de novo, но не зависело от связывания c-Rel [50–52]. Таким образом, при активации гена Il12b ремоделирование хроматина должно происходить раньше, чем связывание c-Rel с ДНК. Чтобы проверить важность нуклеосомного барьера и потребность в специфических ремоделирующих комплексах для его разрешения при экспрессии NF-kB-зависимых генов, использовали нокдаун субъединиц комплекса SWI/SNF, обладающих АТРазной активностью – Brg1 и Brm – в клеточной линии макрофагов [53]. При этом ремоделирование хроматина гена воспаления Il12b сильно ингибировалось одновременно со снижением его экспрессии. Однако нокдаун Brg1 и Brm не влиял на индуцируемую экспрессию гена хемокина Cxcl2. Дальнейшее сравнение профилей экспрессии этих двух генов показало, что, во-первых, Cxcl2 сильно индуцировался через 30 мин после обработки LPS, в то время как наиболее сильная индукция гена Il12b наблюдалась только через 2 ч после обработки и, во-вторых, ингибиторы синтеза белков не подавляли экспрессию Cxcl2. С использованием таких критериев, как кинетика индукции, потребность в синтезе белка и зависимость от комплекса SWI/SNF, выделены три класса индуцируемых воспалительных генов: ранние первичные, поздние первичные и вторичные. Гены раннего первичного ответа индуцируются быстро и не требуют ни Brg1/Brm, ни синтеза нового белка, к ним относятся такие гены, как Cxcl2, Tnf и Ptgs2. Однако некоторые гены первичного ответа (включавшие Ifnb1, Ccl5 и Saa3) нуждались в субъединицах Brg1/Brm для активации, а кинетика их индукции была более медленной, чем у генов раннего первичного ответа. Эти гены получили название генов позднего первичного ответа. Наконец, гены вторичного ответа (Il6, Il12b и Nos2) нуждались в Brg1/Brm и синтезе нового белка [50, 53]. (Под синтезом нового белка подразумевается транскрипция и трансляция генов первичного ответа, необходимых для активации вторичных генов вместе с комплексами SWI/SNF). В соответствии с этой точкой зрения экспрессия значительной части SWI/SNF-зависимых генов, которые индуцируются LPS, зависит от активаторов транскрипции, синтезированных сразу в ответ на стимул.
Другой важный параметр состояния хроматина – чувствительность к нуклеазам. При индукции воспаления комплекс SWI/SNF влияет на чувствительность к нуклеазам генов позднего первичного и вторичного ответа. Нокдаун Brg1/Brm приводит к потере чувствительности генов позднего первичного и вторичного ответа к нуклеазам. Напротив, доступность промоторов генов раннего первичного ответа не изменяется при нокдауне Brg1/Brm [53, 54].
Воспаление контролируется множеством эпигенетических механизмов. Модификации хвостов гистонов влияют на структуру хроматина, а также участвуют в рекрутировании коактиваторных комплексов, которые еще больше реструктурируют хроматин. Ацетилирование гистонов связано с “расслабленными” структурами хроматина, что облегчает транскрипцию и поэтому имеет решающее значение для индукции многих воспалительных генов: ацетилирование гистона H4 в промоторной области воспалительного гена (CXCL8) инициирует повышение привлечения NF-κB к этим промоторам [55, 56]. Гены воспаления раннего ответа имели высокий уровень ацетилирования гистонов, в то время как гены позднего ответа обогащены гистонами с низким уровнем базального ацетилирования, который увеличивался при стимуляции [52].
Установлено, что субъединицы RelA фактора NF-κB быстро проникают в ядро после стимуляции и первоначально связываются только с частью генов [52]. С другими генами RelA связывается в более поздние моменты времени, что предполагает недоступность их сайтов связывания и согласуется с результатами наблюдений, сделанных на промоторе Il12b. В результате проверки модификаций хроматина в генах с ранним и поздним связыванием выявлена следующая закономерность: гены с ранним рекрутированием имели более высокие уровни ацетилирования гистонов, в то время как гены с поздним связыванием имели низкие уровни базального ацетилирования, которое увеличивалось при стимуляции [52]. Таким образом, связывание NF-κB ингибировалось на определенных промоторах в ранней фазе стимуляции из-за недоступной структуры хроматина, а NF-κB мог связываться с этими промоторами поздних генов только после важных событий ремоделирования хроматина.
Изучение активации генов воспаления позволило предложить модель, которая объединяет базальное состояние хроматина с кинетикой индукции транскрипции и селективной потребностью в ремоделировании хроматина. В рамках этой модели кинетика экспрессии конкретного гена коррелирует с базальным состоянием хроматина, а также с синтезом и индукцией факторов транскрипции, способствующих ремоделированию “нуклеосомных барьеров” для рекрутирования димера NF-κB. Важнейшую роль в этой модели играют ремоделирующие комплексы, которые разрушают “нуклеосомные барьеры” для активации транскрипции.
МОЛЕКУЛЯРНЫЕ МЕХАНИЗМЫ РЕКРУТИРОВАНИЯ SWI/SNF НА ГЕНЫ ВОСПАЛЕНИЯ
В процессе активации генов воспаления на их промотор и регуляторные элементы привлекается набор коактиваторов, необходимых для инициации или увеличения базальной транскрипции. Ремоделирующие комплексы SWI/SNF отвечают за позиционирование нуклеосом на промоторах и регуляторных элементах, однако по-разному влияют на разные провоспалительные гены. Функции комплексов SWI/SNF изучают в основном с помощью нокдауна той или иной субъединицы комплекса. Однако несмотря на то, что субъединицы в целом образуют единый функциональный комплекс SWI/SNF, нокдаун разных субъединиц может приводить к увеличению или уменьшению активации одних и тех же генов. Эти наблюдения отражают сложную и специфичную роль комплекса SWI/SNF в регуляции активации генов воспаления. Привлечение комплексов может происходить с помощью разнообразных механизмов, рассмотренных ниже.
Привлечение комплекса SWI/SNF путем взаимодействия с активатором транскрипции
Привлечение SWI/SNF на промоторы генов воспаления может происходить с помощью прямых и опосредованных белок-белковых взаимодействий между первичными или вторичными активаторами транскрипции и субъединицами SWI/SNF. В этом случае структура хроматина позволяет активатору связаться с регуляторным элементом гена, после чего активатор привлекает SWI/SNF, который убирает нуклеосомный барьер (рис. 1а).
Рис. 1.
Способы привлечения SWI/SNF на промоторы провоспалительных генов. а – Привлечение SWI/SNF за счет активатора транскрипции. б – Привлечение SWI/SNF за счет модификаций N-концов гистонов. в – Привлечение SWI/SNF за счет комплекса MEDIATOR. г – Привлечение SWI/SNF за счет длинной некодирующей РНК (lncRNA).
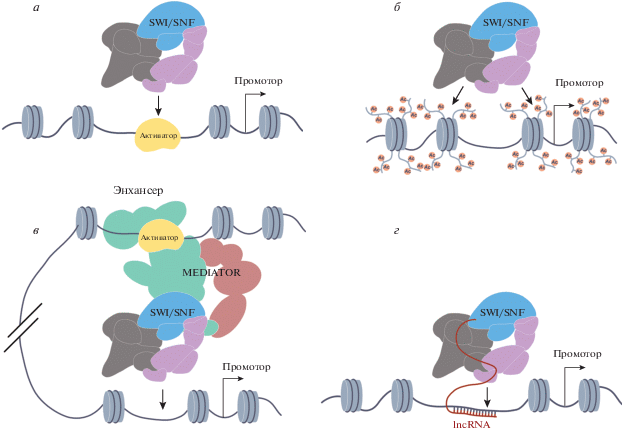
Например, в макрофагах мыши, стимулированых LPS, фактор транскрипции IκBζ, кодируемый геном Nfkbiz, рекрутирует комплекс, содержащий BRG1, на промоторы Il6 и Il12 через белок акирин 2 (Akirin2). Комплекс, состоящий из трех белков – IκB-ζ-Akirin2-BAF60, физически связывает NF-κB и SWI/SNF при активации клеток врожденного иммунитета. Привлекая SWI/SNF к IκB-ζ, консервативный ядерный белок Akirin2 стимулирует промоторы провоспалительных генов в макрофагах мыши во время врожденных иммунных ответов на вирусную или бактериальную инфекцию [57–59]. Это консервативный механизм, так как в культуре клеток S2 дрозофилы акирин необходим для индукции генов, активируемых LPS и IL-1, посредством прямой ассоциации c BAP60, гомологом субъединицы BAF60 SWI/SNF [60].
Показано также, что гетеродимер RelB/p52 из семейства факторов NF-κB взаимодействует с субъединицей BAF комплекса DPF2 (Requiem/REQ), что приводит к активации неканонического пути NF-κB [61]. Сверхэкспрессия DPF2 и его гомологов DPF1, DPF3a, DPF3b, а также субъединицы PHF10 комплекса PBAF значительно усиливают трансактивирующую способность типичных NF-κB-димеров. Используя тандемную аффинную очистку с последующей жидкостной хроматографией и тандемной масс-спектрометрией показали, что Rel-B взаимодействует и с другими субъединицами комплекса SWI/SNF: BRG1, BRM, BAF155, BAF170, BAF60(A/B/C), BAF53A [62].
Для активации транскрипции репортерного гена гетеродимерами RelA/p50 необходимы субъединицы DPF3a и DPF3b, продукты альтернативного сплайсинга гена DPF3. DPF3a и DPF3b прямо взаимодействуют с RelA/p50, но для активации нужен полностью собранный комплекс SWI/SNF [63]. Rel-A взаимодействует также с субъединицами BRM и PHF10 комплекса SWI/SNF, а p50 – с PHF10 и BAF45B/C [63].
Привлечение комплекса SWI/SNF путем взаимодействия с модификациями N-концевых последовательностей гистонов
Структура и свойства хроматина значительно изменяются в процессе активации генов воспаления, в том числе, за счет модификаций N-концов гистонов. Эти модификации привлекают регуляторные комплексы, которые активируют или ингибируют транскрипцию. Примером модификаций, активирующих транскрипцию, может быть ацетилирование лизинов в молекулах гистонов H3 и H4 и триметилирование H3K4. Ковалентные модификации хвостов гистонов осуществляют комплексы, обладающие соответствующими активностями: гистонацетилазной (семейства GNAT, MYST, CBP/P30), гистонметилазной (семейство SET, MLL, EZH), убиквитинлигазной (NEDD4, UHRF1, комплекс CUL4-DDB-ROC1), киназной (PIM1, RPS6KA5/4). Субъединицы SWI/SNF, особенно специфические, содержат различные домены, распознающие модификации N-концов гистонов (табл. 1). Связываясь с ними, субъединицы привлекают весь комплекс SWI/SNF на промоторы генов воспаления (рис. 1б).
Гиперацетилирование гистонов на промоторе генов воспаления коррелирует с их активацией [64, 65]. Ацетилазы могут прямо взаимодействовать с факторами транскрипции NF-κB и привлекаться на гены в момент их активации, как это показано, например, для Rel-A или c-Rel, которые взаимодействуют с p300/CBP [66–71].
Рекрутирование SWI/SNF на ацетилированный хроматин может происходить за счет многочисленных бромодоменов, которые содержатся в субъединицах BRG1, BRD7, BRD9, BAF180 (табл. 1). Например, рекрутирование комплекса SWI/SNF к ацетилированным гистонам происходит в процессе индукции гена интерферона-β (IFNB) человека в ответ на вирусную инфекцию. В экстрактах клеток, инфицированных вирусом Сендай, на регуляторной области промотора посредством кооперативного связывания индуцибельных факторов транскрипции NF-κB, IRF3/IRF7 и ATF-2/c-Jun собирается высокостабильный мультибелковый комплекс, называемый энхансосомой. NF-κB первоначально связывается с консервативным элементом PRDII в промоторе. Это, в свою очередь, облегчает привлечение IRF и ATF-2/c-Jun. Эти факторы транскрипции служат платформой для последовательного рекрутирования комплекса PCAF, модифицирующего хроматин, ацетилтрансфераз p300/CBP и GCN5, а затем комплекса ремоделирования хроматина SWI/SNF. SWI/SNF ремоделирует нижележащие нуклеосомы, которые окружают TATA-бокс [72, 73]. Смещение нуклеосомы позволяет рекрутировать общий фактор транскрипции TFIID вместе с другими факторами транскрипции и РНК-полимеразой II [72, 74, 75]. Рекрутированию ремоделирующих комплексов на ацетилированные гистоны может также способствовать взаимодействие субъединиц SWI/SNF с субъединицами комплексов ацетилаз. Например, субъединица BAF53A (входящая в Arp-модуль комплекса BAF) может взаимодействовать с GCN5 и p300 [76, 77].
Привлечение комплекса SWI/SNF посредством взаимодействия с комплексом MEDIATOR
MEDIATOR – большой многосубъединичный комплекс, необходимый для транскрипции генов РНК-полимеразой II. Основной функцией комплекса MEDIATOR является передача сигнала от активаторов транскрипции, локализованных на энхансерах, на промотор, где происходит сборка преинициаторного комплекса транскрипции. При этом происходит пространственное сближение участка энхансерной ДНК с промотором. Ремоделирующие комплексы SWI/SNF локализуются на энхансерах, а затем могут рекрутироваться на промотор в процессе активации гена (рис. 1в) [78, 79].
Изучение роли комплекса MEDIATOR в активации генов воспаления показало, что RelA взаимодействует с субъединицей Med17 комплекса MEDIATOR [80], что необходимо для активации части генов воспаления, в том числе Ptgs2, Il6 и Cxcl10.
Известно, что субъединицы комплекса MEDIATOR взаимодействуют с комплексом SWI/SNF. С помощью двухгибридного скрининга установлено, что две альтернативные субъединицы киназного модуля – CDK19 и CDK8 – взаимодействуют с АТРазной субъединицей Brg1 комплекса SWI/SNF [81]. Почти весь комплекс SWI/SNF (субъединицы BRM, BAF47, BAF155, BAF170) был аффинно очищен с помощью антител к CDK8-киназе в комплексе с РНК-полимеразой II [82].
Привлечение комплекса SWI/SNF на хроматин с помощью длинных некодирующих РНК
Длинные некодирующие РНК это, как следует из их названия, некодирующие РНК, длина которых превышает 200 н. К ним относятся мРНК-подобные межгенные транскрипты, антисмысловые транскрипты генов, кодирующих белок [83]. Длинные некодирующие РНК могут связываться с многосубъединичными белковыми комплексами и направленно рекрутировать их на определенные комплементарные локусы в геноме (рис. 1г) [84]. Недавно обнаружили, что длинные некодирующие РНК связываются с комплексами SWI/SNF [85, 86], они важны для регуляции врожденного иммунитета и воспалительного ответа [87–89].
Одна из некодирующих РНК – длинная некодирующая РНК Cox2 – наиболее интенсивно индуцируется в ответ на стимуляцию рецепторов TLR [89]. РНК Cox2 взаимодействует с комплексом SWI/SNF в ответ на стимуляцию LPS и рекрутирует комплекс на промоторы генов позднего первичного ответа Ccl5 и Saa3. Нокдаун РНК Cox2 отменяет активацию этих генов, а также рекрутирование BRG1 на промоторы Ccl5 и Saa3 и, как следствие, ремоделирование хроматина [90].
SWI/SNF, ЭНХАНСЕРЫ И СУПЕРЭНХАНСЕРЫ
В активации генов воспаления важную роль играют энхансеры – расположенные дистально от старта транскрипции цис-регуляторные элементы, часто обогащенные сайтами связывания факторов транскрипции. Считается, что энхансеры депонируют коактиваторные комплексы транскрипции. В момент активации генов энхансеры пространственно сближаются с промоторами, что приводит к увеличению концентрации коактиваторов на промоторах и многократному усилению транскрипции. Энхансеры имеют ряд характеристик, которые отличают их от других регуляторных элементов в геноме: обогащение нуклеосом вариантами гистонов H2A.Z и H3.3, модификации гистонов H3K4me1/2 H3K27ac, локализация комплекса MEDIATOR, транскрипция особой энхансерной РНК (eRNA – enhancer RNA), обогащение факторами комплекса АР1 [91]. От промоторов энхансеры отличаются низким содержанием CGI (CpG Island – CpG-островки), низкой чувствительностью к ДНКазам, низкой эволюционной консервативностью [92].
В качестве примера энхансеров воспаления можно привести два энхансера локуса хемокинов на хромосоме 4 человека, регулирующих экспрессию генов IL8 и CXCL1-3 при стимуляции IL-1α [93], и NF-κB-зависимый дистальный энхансер гена MCP-1 (monocyte chemoattractant protein 1 gene), функционирующий при стимуляции фактором некроза опухолей [94].
С помощью антител, распознающих BAF250a, SS18, BAF200, BAF45D, BRD9, в экспериментах по иммунопреципитации хроматина с последующим секвенированием нового поколения показано, что все комплексы SWI/SNF локализуются на энхансерах: комплексы семейства BAF обогащены на активных суперэнхансерах, тогда как комплексы семейств ncBAF и PBAF реже встречаются на энхансерах и чаще на промоторах [95, 96].
Комплексы SWI/SNF не только локализуются на энхансерах для последующего рекрутирования на промоторы воспалительных генов, но также активно ремоделируют нуклеосомы энхансерных элементов, обеспечивая их правильное функционирование. С использованием нокдауна субъединиц BAF250a, BRG1 или BAF47 с последующим ATAC-секвенированием (технология ATAC-seq основана на работе транспозазы, которая встраивает специфичные последовательности – баркоды – в открытый хроматин; по результатам секвенирования такой библиотеки можно определить доступность хроматина на уровне единичных клеток) показано, что хроматин энхансерных элементов утрачивает чувствительность к транспозазе. Эти эксперименты свидетельствуют о блокировании нуклеосомами энхансерных сайтов связывания и потере их доступности для факторов транскрипции (OCT4, AP-1, E2A, EBF1, CEBPβ) в клетках мыши и человека [13, 14, 17, 97–100].
Комплексы семейства BAF влияют и на другие аспекты архитектуры энхансера. В эмбриональных фибробластах мыши и линиях опухолевых клеток человека разрушение комплекса приводит к снижению уровня H3K4me и H3K27ac. BAF прямо связывается с гистон-ацетилтрансферазой p300, а его недостаток ведет к гипоацетилированию энхансеров [101]. В совокупности эти данные показывают, что ключевая функция комплекса BAF состоит в создании и поддержании архитектуры хроматина энхансера в ответ на рекрутирование факторов транскрипции.
Факторы транскрипции семейства AP-1 (FOS/ JUN), которые часто локализуются на энхансерных элементах, вместе с факторами NF-kB участвуют в активации генов воспаления, модулируя индукцию транскрипции [102]. При этом факторы AP-1, кооперируясь со специфическими активаторами, могут создавать энхансеры, специфичные для данного типа клеток [103, 104], и действовать в ответ на стимул [105–107]. Комплекс SWI/SNF взаимодействует непосредственно с АР-1 [103], он необходим для ацетилирования H3K27 в сайтах связывания АР-1 [14, 99, 100]. Таким образом, посредством взаимодействия с факторами АР-1 SWI/SNF может реализовываться еще один механизм регуляции активации транскрипции воспалительных генов.
SWI/SNF-КОМПЛЕКСЫ КАК РЕПРЕССОРЫ ГЕНОВ ВОСПАЛЕНИЯ
Помимо активации транскрипции генов воспаления, комплексы SWI/SNF могут также участвовать в их репрессии посредством ассоциации с такими корепрессорами, как NCoR, mSin3a и/или HDAC1/3.
Деацетилазы гистонов (HDAC) являются антагонистами гистонацетилаз. Снимая ацетилирование с гистонов, HDAC вызывают снижение транскрипционной активности генов. BAF250a связывает промоторы генов IL6 и IL8, где он действует, в первую очередь, как репрессор посредством взаимодействий с HDAC [108, 109].
Ген воспаления Il6 обычно репрессируется, когда BAF250a связывается с промотором Il6. Мутации, инактивирующие функции гена BAF250a, приводят к активации экспрессии генов цитокинов при раке яичников. В частности, в генно-инженерной модели рака яичников у мышей одновременная делеция BAF250a и активация мутантного аллеля фосфатидилинозитол-3-киназы-α p110 (PIK3CA H1047R) приводила к развитию опухоли яичников с повышенной экспрессией Il6 по сравнению с контролем [108]. Сходным образом нокдаун BAF250a в линии иммортализованных клеток нормального эпителия яичников человека, усиленно экспрессирующих PIK3CA, приводил к индукции таких генов цитокинов, как CXCL1, IL1A, IL1B, IL6 и IL8 [109]. Изучение механизма репрессии BAF250a показало, что нокдаун BAF250a приводит к потере связывания корепрессоров mSin3A (комплекс, ассоциированный с деацетилазами) и HDAC1 с промоторами генов IL6 и IL8 [109].
В нестимулированных макрофагах мыши многие гены воспаления репрессированы комплексами NCoR, ассоциированными с HDAC1 и HDAC3 [64, 110]. Субъединицы BRG1, BAF170, BAF155 и BAF47 комплекса SWI/SNF также связаны с корепрессорным комплексом NCoR и необходимы для поддержания генов в нестимулированном состоянии вместе гистондеацетилазами [111].
Выяснение SWI/SNF-зависимого механизма репрессии генов может много сказать о базальном контроле транскрипции генов воспаления, а также о разрешении зависимых от стимулов воспалительных реакций. Кроме того, универсальный характер этого механизма в клетках разного типа имеет важное значение в случае болезней, при которых аберрантно повышается уровень воспалительных цитокинов, например, при раке или аутоиммунных заболеваниях.
SWI/SNF-КОМПЛЕКС В РЕГУЛЯЦИИ TAD ГЕНОВ ВОСПАЛЕНИЯ
Топологически ассоциированными доменами (TAD) называют фрагменты генома в пределах одной хромосомы, участки которого могут взаимодействовать между собой. Участки между двумя разными TAD между собой практически не взаимодействуют. Границы TAD обычно консервативны между видами и активно связывают белки CTCF и когезины, которые блокируют распространение TAD. Считается, что TAD модулируют экспрессию генов, ограничивая взаимодействие между энхансерами и промоторами внутри одного TAD, однако окончательно функции TAD не установлены.
При иммунопреципитации хроматина антителами, узнающими субъединицу BRD9 комплекса nсBAF, с последующим полногеномным секвенированием было показано, что комплексы ncBAF предпочитают связываться с сайтами CTCF и когезина и локализуются на границе TAD [95, 112]. Таким образом, комплекс ncBAF может взаимодействовать с этими структурными белками для образования и/или поддержания петель хроматина, что имеет решающее значение для развития воспалительной реакции макрофагов в ответ на стимуляцию [113]. Индуцируемая делеция Brd9 у мыши и/или быстрая химическая деградация BRD9 у человека приводит к снижению экспрессии воспалительных генов из-за неправильной активации энхансеров и локального образования петель хроматина в ответ на стимуляцию клеток LPS [113]. Результаты этих экспериментов указывают на важную роль семейства ncBAF комплексов SWI/SNF в регуляции генов воспаления посредством поддержания границ TAD.
ЗАКЛЮЧЕНИЕ
Регуляция экспрессии генов воспаления происходит на многих уровнях. Большую роль в этом процессе играет комплекс SWI/SNF, который меняет структуру хроматина регуляторных последовательностей генов воспаления, делая ее открытой для взаимодействия с факторами транскрипции. Биохимическая гетерогенность семейства SWI/SNF обусловливает функционально специфические свойства индивидуальных комплексов и обеспечивает тонкий контроль регуляции индукции генов воспаления.
В настоящее время роль комплекса SWI/SNF в контроле экспрессии воспалительных генов изучена далеко не полностью. Подробное исследование взаимодействий и взаимного влияния комплексов SWI/SNF, активаторов транскрипции и коактиваторных комплексов, а также взаимодействия с РНК-полимеразой II и другими эпигенетическими регуляторами будет способствовать пониманию специфичных для генов воспаления требований к ремоделированию комплекса SWI/SNF в ответ на стимуляцию.
Роль SWI/SNF в возникновении онкологических заболеваний изучена достаточно хорошо. Мутации в генах, кодирующих субъединицы SWI/SNF, встречаются в 20–25% случаев рака [10, 114–116]. SWI/SNF активно участвует в регуляции острого воспаления, которое может переходить в хроническое и, соответственно, приводить к онкотрансформации клеток [117]. Поэтому понимание роли комплексов SWI/SNF в регуляции генов воспаления важно как для фундаментальной науки, так и для практической медицины.
Исследование выполнено при финансовой поддержке Российского фонда фундаментальных исследований в рамках научного проекта № 20-14-50525.
В настоящей работе людей и животных не использовали в качестве объектов исследования.
Авторы сообщают об отсутствии конфликта интересов.
Список литературы
Akira S., Uematsu S., Takeuchi O. (2006) Pathogen recognition and innate immunity. Cell. 124, 783–801.
Sen R., Baltimore D. (1986) Inducibility of κ immunoglobulin enhancer-binding protein NF-κB by a posttranslational mechanism. Cell. 47, 921–928.
Ghosh S., May M.J., Kopp E.B. (1998) NF-κB and rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 16, 225–260.
Hoffmann A., Baltimore D. (2006) Circuitry of nuclear factor κB signaling. Immunol. Rev. 210, 171–186.
Vallabhapurapu S., Karin M. (2009) Regulation and function of NF-κB transcription factors in the immune system. Annu. Rev. Immunol. 27, 693–733.
Hayden M.S., Ghosh S. (2008) Shared principles in NF-κB signaling. Cell. 132, 344–362.
Fenouil R., Cauchy P., Koch F., Descostes N., Cabeza J.Z., Innocenti C., Ferrier P., Spicuglia S., Gut M., Gut I., Andrau J.C. (2012) CpG islands and GC content dictate nucleosome depletion in a transcription-independent manner at mammalian promoters. Genome Res. 22, 2399–2408.
Clapier C.R., Iwasa J., Cairns B.R., Peterson C.L. (2017) Mechanisms of action and regulation of ATP-dependent chromatin-remodelling complexes. Nat. Rev. Mol. Cell. Biol. 18, 407–422.
Hargreaves D.C., Crabtree G.R. (2011) ATP-dependent chromatin remodeling: genetics, genomics and mechanisms. Cell Res. 21, 396–420.
Mittal P., Roberts C.W.M. (2020) The SWI/SNF complex in cancer – biology, biomarkers and therapy. Nat. Rev. Clin. Oncol. 17, 435–448.
Han L., Madan V., Mayakonda A., Dakle P., Woon T.W., Shyamsunder P., Nordin H.B.M., Cao Z., Sundaresan J., Lei I., Wang Z., Koeffler H.P. (2019) Chromatin remodeling mediated by ARID1A is indispensable for normal hematopoiesis in mice. Leukemia. 33, 2291–2305.
Gao F., Elliott N.J., Ho J., Sharp A., Shokhirev M.N., Hargreaves D.C. (2019) Heterozygous mutations in SMARCA2 reprogram the enhancer landscape by global retargeting of SMARCA4. Mol. Cell. 75, 891–904.
King H.W., Klose R.J. (2017) The pioneer factor OCT4 requires the chromatin remodeller BRG1 to support gene regulatory element function in mouse embryonic stem cells. eLife. 6, e22631.
Kelso T.W.R., Porter D.K., Amaral M.L., Shokhirev M.N., Benner C., Hargreaves D.C. (2017) Chromatin accessibility underlies synthetic lethality of SWI/ SNF subunits in ARID1A-mutant cancers. eLife. 6, e30506.
Miller E.L., Hargreaves D.C., Kadoch C., Chang C.Y., Calarco J.P., Hodges C., Buenrostro J.D., Cui K., Greenleaf W.J., Zhao K., Crabtree G.R. (2017) TOP2 synergizes with BAF chromatin remodeling for both resolution and formation of facultative heterochromatin. Nat. Struct. Mol. Biol. 24, 344–352.
Bao X., Rubin A.J., Qu K., Zhang J., Giresi P.G., Chang H.Y., Khavari P.A. (2015) A novel ATAC-seq approach reveals lineage-specific reinforcement of the open chromatin landscape via cooperation between BAF and p63. Genome Biol. 16, 1–17.
Bossen C., Murre C.S., Chang A.N., Mansson R., Rodewald H.R., Murre C. (2015) The chromatin remodeler Brg1 activates enhancer repertoires to establish B cell identity and modulate cell growth. Nat. Immunol. 16, 775–784.
Euskirchen G., Auerbach R.K., Snyder M. (2012) SWI/SNF chromatin-remodeling factors: multiscale analyses and diverse functions. J. Biol. Chem. 287, 30897–30905.
Kwon H., Imbalzano A.N., Khavari P.A., Kingston R.E., Green M.R. (1994) Nucleosome disruption and enhancement of activator binding by a human SW1/SNF complex. Nature. 370, 477–481.
Clapier C.R., Cairns B.R. (2009) The biology of chromatin remodeling complexes. Annu. Rev. Biochem. 78, 273–304.
Wang W., Côté J., Xue Y., Zhou S., Khavari P.A., Biggar S.R., Muchardt C., Kalpana G.V., Goff S.P., Yaniv M., Workman J.L., Crabtree G.R. (1996) Purification and biochemical heterogeneity of the mammalian SWI-SNF complex. EMBO J. 15, 5370–5382.
Lemon B., Inouye C., King D.S., Tjian R. (2001) Selectivity of chromatin-remodelling cofactors for ligand-activated transcription. Nature. 414, 924–928.
Raab J.R., Resnick S., Magnuson T. (2015) Genome-Wide transcriptional regulation mediated by biochemically distinct SWI/SNF complexes. PLoS Genet. 11, 1–26.
Alpsoy A., Dykhuizen E.C. (2018) Glioma tumor suppressor candidate region gene 1 (GLTSCR1) and its paralog GLTSCR1-like form SWI/SNF chromatin remodeling subcomplexes. J. Biol. Chem. 293, 3892–3903.
Michel B.C., D’Avino A.R., Cassel S.H., Mashtalir N., McKenzie Z.M., McBride M.J., Valencia A.M., Zhou Q., Bocker M., Soares L.M.M., Pan J., Remillard D.I., Lareau C.A., Zullow H.J., Fortoul N., Gray N.S., Bradner J.E., Chan H.M., Kadoch C. (2018) A non-canonical SWI/SNF complex is a synthetic lethal target in cancers driven by BAF complex perturbation. Nat. Cell Biol. 20, 1410–1420.
Wang X., Wang S., Troisi E.C., Howard T.P., Haswell J.R., Wolf B.K., Hawk W.H., Ramos P., Oberlick E.M., Tzvetkov E.P., Ross A., Vazquez F., Hahn W.C., Park P.J., Roberts C. W.M. (2019) BRD9 defines a SWI/SNF sub-complex and constitutes a specific vulnerability in malignant rhabdoid tumors. Nat. Commun. 10, 1–11.
Brien G.L., Remillard D., Shi J., Hemming M.L., Chabon J., Wynne K., Dillon E.T., Cagney G., Van Mierlo G., Baltissen M.P., Vermeulen M., Qi J., Fröhling S., Gray N.S., Bradner J.E., Vakoc C.R., Armstrong S.A. (2018) Targeted degradation of BRD9 reverses oncogenic gene expression in synovial sarcoma. eLife. 7, 1–26.
Mashtalir N., D’Avino A.R., Michel B.C., Luo J., Pan J., Otto J.E., Zullow H.J., McKenzie Z.M., Kubiak R.L., St Pierre R., Valencia A.M., Poynter S.J., Cassel S.H., Ranish J.A., Kadoch C. (2018) Modular organization and assembly of SWI/SNF family chromatin remodeling complexes. Cell. 175, 1272–1288.
Lessard J., Wu J.I., Ranish J., Wan M., Winslow M.M., Staahl B.T., Wu H., Aebersold R., Graef I.A., Crabtree G.R. (2007) An essential switch in subunit composition of a chromatin remodeling complex during neural development. Neuron. 55, 201–215.
Ho L., Ronan J.L., Wu J., Staahl B.T., Chen L., Kuo A., Lessard J., Nesvizhskii A.I., Ranish J., Crabtree G.R. (2009) An embryonic stem cell chromatin remodeling complex, esBAF, is essential for embryonic stem cell self-renewal and pluripotency. Proc. Natl. Acad. Sci. USA. 106, 5181–5186.
Priam P., Krasteva V., Rousseau P., D’Angelo G., Gaboury L., Sauvageau G., Lessard J.A. (2017) SMARCD2 subunit of SWI/SNF chromatin-remodeling complexes mediates granulopoiesis through a CEBPϵ dependent mechanism. Nat. Genet. 49, 753–764.
Witzel M., Petersheim D., Fan Y., Bahrami E., Racek T., Rohlfs M., Puchałka J., Mertes C., Gagneur J., Ziegenhain C., Enard W., Stray-Pedersen A., Arkwright P.D., Abboud M.R., Pazhakh V., Lieschke G.J., Krawitz P.M., Dahlhoff M., Schneider M.R., Wolf E., Horny H.P., Schmidt H., Schäffer A.A., Klein C. (2017) Chromatin-remodeling factor SMARCD2 regulates transcriptional networks controlling differentiation of neutrophil granulocytes. Nat. Genet. 49, 742–752.
Forcales S.V., Albini S., Giordani L., Malecova B., Cignolo L., Chernov A., Coutinho P., Saccone V., Consalvi S., Williams R., Wang K., Wu Z., Baranovskaya S., Miller A., Dilworth F.J., Puri P.L. (2012) Signal-dependent incorporation of MyoD-BAF60c into Brg1-based SWI/SNF chromatin-remodelling complex. EMBO J. 31, 301–316.
Lickert H., Takeuchi J.K., Von Both I., Walls J.R., McAuliffe F., Adamson S.L., Henkelman R.M., Wrana J.L., Rossant J., Bruneau B.G. (2004) Baf60c is essential for function of BAF chromatin remodelling complexes in heart development. Nature. 432, 107–112.
Mashtalir N., Suzuki H., Farrell D.P., Sankar A., Luo J., Filipovski M., D’Avino A.R., St Pierre R., Valencia A.M., Onikubo T., Roeder R.G., Han Y., He Y., Ranish J.A., DiMaio F., Walz T., Kadoch C. (2020) A structural model of the endogenous human BAF complex informs disease mechanisms. Cell. 183, 802–817.
Сошникова Н.В., Шейнов А.А., Татарский Е.В., Георгиева С.Г. (2020) DPF-домен как уникальная структурная единица в активации транскрипции, дифференцировке и онкотрансформации. Acta Naturae. 12, 57–65.
Zhao K., Wang W., Rando O.J., Xue Y., Swiderek K., Kuo A., Crabtree G.R. (1998) Rapid and phosphoinositol-dependent binding of the SWI/SNF-like BAF complex to chromatin after T lymphocyte receptor signaling. Cell. 95, 625–636.
Weintraub H., Groudine M. (1976) Chromosomal subunits in active genes have an altered conformation. Science. 193, 848–856.
Wu C., Bingham P.M., Livak K.J., Holmgren R., Elgin S.C.R. (1979) The chromatin structure of specific genes: I. Evidence for higher order domains of defined DNA sequence. Cell. 16, 797–806.
Luger K., Mäder A.W., Richmond R.K., Sargent D.F., Richmond T.J. (1997) Crystal structure of the nucleosome core particle at 2.8 Å resolution. Nature. 389, 251–260.
Rye R. (1995) Structure of the NF-kappa B p50 homodimer bound to DNA. Nature. 378, 603–605.
Wang V.Y. (2012) NF-κB regulation: lessons from structures. Immunol. Rev. 246, 36–58.
Steger D.J., Workman J.L. (1997) Stable co-occupancy of transcription factors and histones at the HIV-1 enhancer. EMBO J. 16, 2463–2472.
Angelov D., Lenouvel F., Hans F., Müller C.W., Bouvet P., Bednar J., Moudrianakis E.N., Cadet J., Dimitrov S. (2004) The histone octamer is invisible when NF-κB binds to the nucleosome. J. Biol. Chem. 279, 42374–42382.
Lone I.N., Shukla M.S., Charles Richard J.L., Peshev Z.Y., Dimitrov S., Angelov D. (2013) Binding of NF-κB to nucleosomes: effect of translational positioning, nucleosome remodeling and linker histone H1. PLoS Genet. 9(9), e1003830.
Saccani S., Pantano S., Natoli G. (2001) Two waves of nuclear factor κB recruitment to target promoters. J. Exp. Med. 193, 1351–1359.
Plevy S.E., Gemberling J.H., Hsu S., Dorner A.J., Smale S.T. (1997) Multiple control elements mediate activation of the murine and human interleukin 12 p40 promoters: evidence of functional synergy between C/EBP and Rel proteins. Mol. Cell. Biol. 17, 4572–4588.
Zhu C., Gagnidze K., Gemberling J.H.M., Plevy S.E. (2001) Characterization of an activation protein-1-binding site in the murine interleukin-12 p40 promoter: demonstration of novel functional elements by a reductionist approach. J. Biol. Chem. 276, 18519–18528.
Zhu C., Rao K., Xiong H., Gagnidze K., Li F., Horvath C., Plevy S. (2003) Activation of the murine interleukin-12 p40 promoter by functional interactions between NFAT and ICSBP. J. Biol. Chem. 278, 39372–39382.
Weinmann A.S., Plevy S.E., Smale S.T. (1999) Rapid and selective remodeling of a positioned nucleosome during the induction of IL-12 p40 transcription. Immunity. 11, 665–675.
Weinmann A.S., Mitchell D.M., Sanjabi S., Bradley M.N., Hoffmann A., Liou H.C., Smale S.T. (2001) Nucleosome remodeling at the IL-12 p40 promoter is a TLR-dependent, Rel-independent event. Nat. Immunol. 2, 51–57.
Zhou L., Nazarian A.A., Xu J., Tantin D., Corcoran L.M., Smale S.T. (2007) An inducible enhancer required for Il12b promoter activity in an insulated chromatin environment. Mol. Cell. Biol. 27, 2698–2712.
Ramirez-Carrozzi V.R., Nazarian A.A., Li C.C., Gore S.L., Sridharan R., Imbalzano A.N., Smale S.T. (2006) Selective and antagonistic functions of SWI/SNF and Mi-2β nucleosome remodeling complexes during an inflammatory response. Genes Dev. 20, 282–296.
Ramirez-Carrozzi V.R., Braas D., Bhatt D.M., Cheng C.S., Hong C., Doty K.R., Black J.C., Hoffmann A., Carey M., Smale S.T. (2009) A unifying model for the selective regulation of inducible transcription by CpG islands and nucleosome remodeling. Cell. 138, 114–128.
Barnes P.J. (2009) Targeting the epigenome in the treatment of asthma and chronic obstructive pulmonary disease. Proc. Am. Thorac. Soc. 6, 693–696.
Ito K., Ito M., Elliott W.M., Cosio B., Caramori G., Kon O.M., Barczyk A., Hayashi S., Adcock I.M., Hogg J.C., Barnes P.J. (2005) Decreased histone deacetylase activity in chronic obstructive pulmonary disease. N. Engl. J. Med. 352, 1967–1976.
Yamamoto M., Yamazaki S., Uematsu S., Sato S., Hemmi H., Hoshino K., Kaisho T., Kuwata H., Takeuchi O., Takeshige K., Saitoh T., Yamaoka S., Yamamoto N., Yamamoto S., Muta T., Takeda K., Akira S. (2004) Regulation of Toll/IL-1-receptor-mediated gene expression by the inducible nuclear protein IκBζ. Nature. 430, 218–222.
Yamazaki S., Matsuo S., Muta T., Yamamoto M., Akira S., Takeshige K. (2008) Gene-specific requirement of a nuclear protein, IκB-ζ, for promoter association of inflammatory transcription regulators. J. Biol. Chem. 283, 32404–32411.
Tartey S., Matsushita K., Vandenbon A., Ori D., Imamura T., Mino T., Standley D.M., Hoffmann J.A., Reichhart J.M., Akira S., Takeuchi O. (2014) Akirin2 is critical for inducing inflammatory genes by bridging IκB-ζ and the SWI/SNF complex. EMBO J. 33, 2332–2348.
Bonnay F., Nguyen X., Cohen-Berros E., Troxler L., Batsche E., Camonis J., Takeuchi O., Reichhart J., Matt N. (2014) Akirin specifies NF-κB selectivity of Drosophila innate immune response via chromatin remodeling. EMBO J. 33, 2349–2362.
Tando T., Ishizaka A., Watanabe H., Ito T., Iida S., Haraguchi T., Mizutani T., Izumi T., Isobe T., Akiyama T., Inoue J., Iba H. (2010) Requiem protein links RelB/p52 and the Brm-type SWI/SNF complex in a noncanonical NF-κB pathway. J. Biol. Chem. 285, 21951–21960.
Bouwmeester T., Bauch A., Ruffner H., Angrand P.O., Bergamini G., Croughton K., Cruciat C., Eberhard D., Gagneur J., Ghidelli S., Hopf C., Huhse B., Mangano R., Michon A.M., Schirle M., Schlegl J., Schwab M., Stein M.A., Bauer A., Casari G., Drewes G., Gavin A.C., Jackson D.B., Joberty G., Neubauer G., Rick J., Kuster B., Superti-Furga G. (2004) A physical and functional map of the human TNF-α/NF-κB signal transduction pathway. Nat. Cell. Biol. 6, 97–105.
Ishizaka A., Mizutani T., Kobayashi K., Tando T., Sakurai K., Fujiwara T., Iba H. (2012) Double plant homeodomain (PHD) finger proteins DPF3a and -3b are required as transcriptional co-activators in SWI/SNF complex-dependent activation of NF-κB RelA/p50 heterodimer. J. Biol. Chem. 287, 11924–11933.
Hargreaves D.C., Horng T., Medzhitov R. (2009) Control of inducible gene expression by signal-dependent transcriptional elongation. Cell. 138, 129–145.
Bayarsaihan D. (2011) Epigenetic mechanisms in inflammation. J. Dent. Res. 90, 9–17.
Zhong H., Voll R.E., Ghosh S. (1998) Phosphorylation of NF-κB p65 by PKA stimulates transcriptional activity by promoting a novel bivalent interaction with the coactivator CBP/p300. Mol. Cell. 1, 661–671.
Zhong H., May M.J., Jimi E., Ghosh S. (2002) The phosphorylation status of nuclear NF-κB determines its association with CBP/p300 or HDAC-1. Mol. Cell. 9, 625–636.
Dong J., Jimi E., Zhong H., Hayden M.S., Ghosh S. (2008) Repression of gene expression by unphosphorylated NF-κB p65 through epigenetic mechanisms. Genes Dev. 22, 1159–1173.
Mukherjee S.P., Behar M., Birnbaum H.A., Hoffmann A., Wright P.E., Ghosh G. (2013) Analysis of the RelA:CBP/p300 interaction reveals its involvement in NF-κB-driven transcription. PLoS Biol. 11(9), e1001647.
Garbati M.R., Alço G., Gilmore T.D. (2010) Histone acetyltransferase p300 is a coactivator for transcription factor REL and is C-terminally truncated in the human diffuse large B-cell lymphoma cell line RC-K8. Cancer Lett. 291, 237–245.
Haery L., Lugo-Picó J.G., Henry R.A., Andrews A.J., Gilmore T.D. (2014) Histone acetyltransferase-deficient p300 mutants in diffuse large B cell lymphoma have altered transcriptional regulatory activities and are required for optimal cell growth. Mol. Cancer. 13, 1–13.
Agalioti T., Lomvardas S., Parekh B., Yie J., Maniatis T., Thanos D. (2000) Ordered recruitment of chromatin modifying and general transcription factors to the IFN-β promoter. Cell. 103, 667–678.
Lomvardas S., Thanos D. (2002) Modifying gene expression programs by altering core promoter chromatin architecture. Cell. 110, 261–271.
Thanos D., Maniatis T. (1995). Virus induction of human IFN-β gene expression requires the assembly of an enhanceosome. Cell. 83, 1091–1100.
Ford E., Thanos D. (2010) The transcriptional code of human IFN-β gene expression. Biochim. Biophys. Acta – Gene Regul. Mech. 1799, 328–336.
Park J., Wood M.A., Cole M.D. (2002) BAF53 forms distinct nuclear complexes and functions as a critical c-Myc-interacting nuclear cofactor for oncogenic transformation. Mol. Cell. Biol. 22, 1307–1316.
Raisner R., Kharbanda S., Jin L., Jeng E., Chan E., Merchant M., Haverty P.M., Bainer R., Cheung T., Arnott D., Flynn E.M., Romero F.A., Magnuson S., Gascoigne K.E. (2018) Enhancer activity requires CBP/P300 bromodomain-dependent histone H3K27 Acetylation. Cell Rep. 24, 1722–1729.
Soutourina J. (2018) Transcription regulation by the Mediator complex. Nat. Rev. Mol. Cell Biol. 19, 262–274.
Malik S., Roeder R.G. (2005) Dynamic regulation of pol II transcription by the mammalian Mediator complex. Trends Biochem. Sci. 30, 256–263.
van Essen D., Engist B., Natoli G., Saccani S. (2009) Two modes of transcriptional activation at native promoters by NF-κB p65. PLoS Biol. 7, 0549–0562.
Fukasawa R., Tsutsui T., Hirose Y., Tanaka A., Ohkuma Y. (2012) Mediator CDK subunits are platforms for interactions with various chromatin regulatory complexes. J. Biochem. (Tokyo). 152, 241–249.
Cho H., Orphanides G., Sun X., Yang X.-J., Ogryzko V., Lees E., Nakatani Y., Reinberg D. (1998) A human RNA polymerase II complex containing factors that modify chromatin structure. Mol. Cell. Biol. 18, 5355–5563.
Yao R.W., Wang Y., Chen L.L. (2019) Cellular functions of long noncoding RNAs. Nat. Cell Biol. 21, 542–551.
Moran V.A., Perera R.J., Khalil A.M. (2012) Emerging functional and mechanistic paradigms of mammalian long non-coding RNAs. Nucl. Acids Res. 40, 6391–6400.
Kawaguchi T., Tanigawa A., Naganuma T., Ohkawa Y., Souquere S., Pierron G., Hirose T., Steitz J.A. (2015) SWI/SNF chromatin-remodeling complexes function in noncoding RNA-dependent assembly of nuclear bodies. Proc. Natl. Acad. Sci. USA. 112, 4304–4309.
Zhu Y., Rowley M.J., Böhmdorfer G., Wierzbicki A.T. (2013) A SWI/SNF chromatin-remodeling complex acts in noncoding RNA-mediated transcriptional silencing. Mol. Cell. 49, 298–309.
Peng X., Gralinski L, Armour C.D., Ferris M.T., Thomas M.J., Proll S., Bradel-Tretheway B.G., Korth M.J., Castle J.C., Biery M.C., Bouzek H.K., Haynor D.R., Frieman M.B., Heise M., Raymond C.K., Baric R.S., Katze M.G. (2010) Unique signatures of long noncoding RNA expression in response to virus infection and altered innate immune signaling. mBio. 1, 206–210.
Rapicavoli N.A., Qu K., Zhang J., Mikhail M., Laberge R.M., Chang H.Y. (2013) A mammalian pseudogene lncRNA at the interface of inflammation and antiinflammatory therapeutics. eLife. 2013, 1–16.
Carpenter S., Aiello D., Atianand M.K., Ricci E.P., Gandhi P., Hall L.L., Byron M., Monks B., Henry-Bezy M., Lawrence J.B., O’Neill L.A., Moore M.J., Caffrey D.R., Fitzgerald K.A. (2013) A long noncoding RNA mediates both activation and repression of immune response genes. Science. 341, 789–792.
Hu G., Gong A.Y., Wang Y., Ma S., Chen X., Chen J., Su C.J., Shibata A., Strauss-Soukup J.K., Drescher K.M., Chen X.M. (2016) LincRNA-Cox2 promotes late inflammatory gene transcription in macrophages through modulating SWI/SNF-mediated chromatin remodeling. J. Immunol. 196, 2799–2808.
Tafessu A., Banaszynski L.A. (2020) Establishment and function of chromatin modification at enhancers: chromatin landscape at enhancers. Open Biol. 10, 200255.
Andersson R., Sandelin A. (2020) Determinants of enhancer and promoter activities of regulatory elements. Nat. Rev. Genet. 21, 71–87.
Weiterer S.S., Meier-Soelch J., Georgomanolis T., Mizi A., Beyerlein A., Weiser H., Brant L., Mayr-Buro C., Jurida L., Beuerlein K., Müller H., Weber A., Tenekeci U., Dittrich-Breiholz O., Bartkuhn M., Nist A., Stiewe T., van IJcken W.F., Riedlinger T., Schmitz M.L., Papantonis A., Kracht M. (2020) Distinct IL-1α-responsive enhancers promote acute and coordinated changes in chromatin topology in a hierarchical manner. EMBO J. 39, 1–22.
Teferedegne B., Green M.R., Guo Z., Boss J.M. (2006) Mechanism of action of a distal NF-κB-dependent enhancer. Mol. Cell. Biol. 26, 5759–5770.
Gatchalian J., Malik S., Ho J., Lee D.S., Kelso T.W.R., Shokhirev M.N., Dixon J.R., Hargreaves D.C. (2018) A non-canonical BRD9-containing BAF chromatin remodeling complex regulates naive pluripotency in mouse embryonic stem cells. Nat. Commun. 9, 5139.
Pan J., McKenzie Z.M., D’Avino A.R., Mashtalir N., Lareau C.A., St. Pierre R., Wang L., Shilatifard A., Kadoch C. (2019) The ATPase module of mammalian SWI/SNF family complexes mediates subcomplex identity and catalytic activity–independent genomic targeting. Nat. Genet. 51, 618–626.
Wang X., Lee R.S., Alver B.H., Haswell J.R., Wang S., Mieczkowski J., Drier Y., Gillespie S.M., Archer T.C., Wu J.N., Tzvetkov E.P., Troisi E.C., Pomeroy S.L., Biegel J.A., Tolstorukov M.Y., Bernstein B.E., Park P.J., Roberts C.W.M. (2017) SMARCB1-mediated SWI/SNF complex function is essential for enhancer regulation. Nat. Genet. 49, 289–295.
Alexander J.M., Hota S.K., He D., Thomas S., Ho L., Pennacchio L.A., Bruneau B.G. (2015) Brg1 modulates enhancer activation in mesoderm lineage commitment. Dev. Camb. 142, 1418–1430.
Mathur R., Alver B.H., San Roman A.K., Wilson B.G., Wang X., Agoston A.T., Park P.J., Shivdasani R.A., Roberts C.W.M. (2017) ARID1A loss impairs enhancer-mediated gene regulation and drives colon cancer in mice. Nat. Genet. 49, 296–302.
Nakayama R.T., Pulice J.L. Valencia A.M., McBride M.J., McKenzie Z.M., Gillespie M.A., Ku W.L., Teng M., Cui K., Williams R.T., Cassel S.H., Qing H., Widmer C.J., Demetri G.D., Irizarry R.A., Zhao K., Ranish J.A., Kadoch C. (2017) SMARCB1 is required for widespread BAF complex-mediated activation of enhancers and bivalent promoters. Nat. Genet. 49, 1613–1623.
Alver B.H., Kim K.H., Lu P., Wang X., Manchester H.E., Wang W., Haswell J.R., Park P.J., Roberts C.W.M. (2017) The SWI/SNF chromatin remodelling complex is required for maintenance of lineage specific enhancers. Nat. Commun. 8, 1–10.
Fujioka S., Niu J., Schmidt C., Sclabas G.M., Peng B., Uwagawa T., Li Z., Evans D.B., Abbruzzese J.L., Chiao P.J. (2004) NF-κB and AP-1 connection: mechanism of NF-κB-dependent regulation of AP-1 activity. Mol. Cell. Biol. 24, 7806–7819.
Vierbuchen T., Ling E., Cowley C.J., Couch C.H., Wang X., Harmin D.A., Roberts C.W.M., Greenberg M.E. (2017) AP-1 transcription factors and the BAF complex mediate signal-dependent enhancer selection. Mol. Cell. 68, 1067–1082.e12.
Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 38, 576–589.
Mancino A., Termanini A., Barozzi I., Ghisletti S., Ostuni R., Prosperini E., Ozato K., Natoli G. (2015) A dual cis-regulatory code links IRF8 to constitutive and inducible gene expression in macrophages. Genes Dev. 29, 394–408.
Fonseca G.J., Tao J., Westin E.M., Duttke S.H., Spann N.J., Strid T., Shen Z., Stender J.D., Sakai M., Link V.M., Benner C., Glass C.K. (2019) Diverse motif ensembles specify non-redundant DNA binding activities of AP-1 family members in macrophages. Nat. Commun. 10, 414–430.
Ghisletti S., Barozzi I., Mietton F., Polletti S., De Santa F., Venturini E., Gregory L., Lonie L., Chew A., Wei C.L., Ragoussis J., Natoli G. (2010) Identification and characterization of enhancers controlling the inflammatory gene expression program in macrophages. Immunity. 32, 317–328.
Chandler R.L., Damrauer J.S., Raab J.R., Schisler J.C., Wilkerson M.D., Didion J.P., Starmer J., Serber D., Yee D., Xiong J., Darr D.B., Pardo-Manuel de Villena F., Kim W.Y., Magnuson T. (2015) Coexistent ARID1A-PIK3CA mutations promote ovarian clear-cell tumorigenesis through pro-tumorigenic inflammatory cytokine signalling. Nat. Commun. 6, 1–14.
Kim M., Lu F., Zhang Y. (2016) Loss of HDAC-mediated repression and gain of NF-κB activation underlie cytokine induction in ARID1A- and PIK3CA-mutation-driven ovarian cancer. Cell Rep. 17, 275–288.
Ogawa S., Lozach J., Jepsen K., Sawka-Verhelle D., Perissi V., Sasik R., Rose D.W., Johnson R.S., Rosenfeld M.G., Glass C.K. (2004) A nuclear receptor corepressor transcriptional checkpoint controlling activator protein 1-dependent gene networks required for macrophage activation. Proc. Natl. Acad. Sci. USA. 101, 14461–14466.
Underhill C., Qutob M.S., Yee S.P., Torchia J. (2000) A novel nuclear receptor corepressor complex, N-CoR, contains components of the mammalian SWI/SNF complex and the corepressor KAP-1. J. Biol. Chem. 275, 40463–40470.
Ong C.T., Corces V.G. (2014) CTCF: an architectural protein bridging genome topology and function. Nat. Rev. Genet. 15, 234–246.
Cuartero S., Weiss F.D., Dharmalingam G., Guo Y., Ing-Simmons E., Masella S., Robles-Rebollo I., Xiao X., Wang Y.F., Barozzi I., Djeghloul D., Amano M.T., Niskanen H., Petretto E., Dowell R.D., Tachibana K., Kaikkonen M.U., Nasmyth K.A., Lenhard B., Natoli G., Fisher A.G., Merkenschlager M. (2018) Control of inducible gene expression links cohesin to hematopoietic progenitor self-renewal and differentiation. Nat. Immunol. 19, 932–941.
Lu C., Allis C.D. (2017) SWI/SNF complex in cancer. Nat. Genet. 49, 178–179.
Kadoch C., Crabtree G.R. (2015) Mammalian SWI/SNF chromatin remodeling complexes and cancer: mechanistic insights gained from human genomics. Sci. Adv. 1, 1–18.
Wanior M., Krämer A., Knapp S., Joerger A.C. (2021) Exploiting vulnerabilities of SWI/SNF chromatin remodelling complexes for cancer therapy. Oncogene. 40, 3637–3654.
Furman D., Campisi J., Verdin E., Carrera-Bastos P., Targ S., Franceschi C., Ferrucci L., Gilroy D.W., Fasano A., Miller G.W., Miller A.H., Mantovani A., Weyand C.M., Barzilai N., Goronzy J.J., Rando T.A., Effros R.B., Lucia A., Kleinstreuer N., Slavich G.M. (2019) Chronic inflammation in the etiology of disease across the life span. Nat. Med. 25, 1822–1832.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология