

Молекулярная биология, 2022, T. 56, № 2, стр. 296-319
Аналоги S-аденозил-L-метионина в изучении метилтрансфераз
А. Ю. Руденко a, b, С. С. Марьясина a, c, П. В. Сергиев c, В. И. Польшаков a, *
a Факультет фундаментальной медицины, Московский государственный университет им. М.В. Ломоносова
119991 Москва, Россия
b Институт органической химии им. Н.Д. Зелинского Российской академии наук
119991 Москва, Россия
c Институт функциональной геномики, Московский государственный университет им. М.В. Ломоносова
119991 Москва, Россия
* E-mail: vpolsha@mail.ru
Поступила в редакцию 30.08.2021
После доработки 18.10.2021
Принята к публикации 20.10.2021
- EDN: ZTQQLX
- DOI: 10.31857/S0026898422020148
Аннотация
Метилтрансферазы (МТазы) играют важную роль в функционировании живых систем, катализируя реакции метилирования ДНК, РНК, белков и малых молекул, включая эндогенные соединения и лекарственные препараты. Многие заболевания человека ассоциированы с нарушениями в работе этих ферментов, поэтому изучение МТаз представляется актуальной задачей. Большинство МТаз используют в качестве донора метильной группы кофактор S-аденозил-L-метионин (SAM). Аналоги SAM нашли широкое применение: они используются в исследовании каталитической активности МТаз, идентификации субстратов новых МТаз, для модификации субстратов метилирования или их связывания с МТазами. В настоящем обзоре рассмотрены синтетические аналоги SAM и задачи, которые удается решать с их помощью.
ВВЕДЕНИЕ
Метилтрансферазы (МТазы) играют важную роль в функционировании живых систем, катализируя реакции метилирования ДНК, РНК, белков и малых молекул, включая эндогенные соединения и лекарственные препараты. Так, метилирование ДНК определяет эпигенетическую регуляцию экспрессии генов [1]. Метилирование РНК необходимо для правильного протекания процессов с участием этих молекул, прежде всего синтеза белка и сплайсинга [2]. Метилирование расширяет функциональное разнообразие белков [3]. Оно участвует в большом количестве внутриклеточных процессов, например, метилирование гистонов обеспечивает регуляцию экспрессии генов [4]. Метилирование малых молекул – один из способов изменения их свойств и участия в биохимических процессах [5].
Нарушение работы МТаз изменяет характер внутриклеточных процессов метилирования, что, в свою очередь, часто приводит к возникновению различных заболеваний [2, 6], поэтому изучение МТаз представляется важной задачей. Разработано множество методов и подходов к изучению этих ферментов, связанных с использованием аналогов кофактора метилирования – S-аденозил-L-метионина (SAM, или AdoMet). С помощью аналогов SAM можно влиять на активность МТаз, определять субстраты новых белков этого класса, химически модифицировать субстраты метилирования.
Значительная часть обзора посвящена синтетическим аналогам SAM, полученным за последние два десятилетия, и задачам, которые удается решать с их помощью.
МЕТИЛТРАНСФЕРАЗЫ
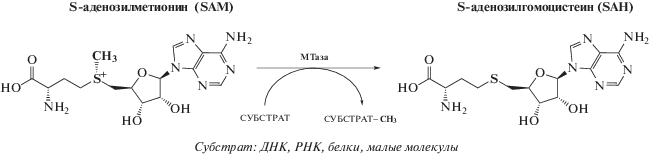
Метилтрансферазы [Е.С. 2.1.1] – обширная группа ферментов, катализирующих реакцию метилирования различных субстратов и регулирующих такие биологические процессы, как метаболизм, биосинтез, функционирование и деградацию нуклеиновых кислот, активность белков, детоксикацию экзогенных соединений [7]. Подавляющее большинство известных МТаз используют в качестве донора метильной группы SAM, который после отщепления CH3-группы превращается в S-аденозил-L-гомоцистеин (SAH) (рис. 1) [8].
Рис. 1.
Перенос метильной группы SAM-зависимой МТазой с кофактора на субстрат. В ходе этой реакции SAM превращается в SAH.
На данный момент в протеоме человека известно около 300 SAM-зависимых МТаз, из которых примерно для 120 известен тип субстрата (малые молекулы, липиды, белки или нуклеиновые кислоты) и тип метилируемого атома (как правило, азот, кислород, углерод или сера) [9, 10].
Роль МТаз в биологических процессах
В зависимости от типа субстрата метилирование выполняет различные функции в организме. Метилирование ДНК отвечает за эпигенетическую регуляцию экспрессии генов эукариот, играет важную роль в эмбриональном развитии многоклеточных, участвует в импринтинге генома и канцерогенезе. В прокариотических клетках метилирование используется для контроля репарации и времени репликации ДНК [11], а также в системах рестрикции-модификации для защиты от чужеродных генетических элементов [12].
Метилирование РНК дополняет четырехбуквенный генетический код нуклеотидами, содержащими модифицированные основания или 2'-метилгидроксильные группы. Оно обеспечивает биохимическую, биофизическую и метаболическую стабилизацию РНК, необходимые для выполнения этими молекулами специфических функций, а также контроль качества РНК. Метилирование присуще большинству РНК, выполняющих каталитическую функцию, в том числе участвующих в синтезе белка и сплайсинге [13]. Также оно играет роль в возникновении резистентности бактерий к антибиотикам [14].
Метилирование белков позволяет существенно расширить 20-аминокислотный “алфавит”, внося большее разнообразие в аминокислотный состав белков и изменяя их функциональные свойства. Чаще всего метилированию подвергаются аминогруппа лизина и гуанидиновый фрагмент аргинина, реже – гидроксильные группы серина и тирозина, и карбоксильные группы боковых цепей глутаминовой и аспарагиновой кислот [15]. Также метилированию могут подвергаться N- и С-концевые группы любых аминокислотных остатков в белках, включая амидные фрагменты боковых цепей глутамина и аспарагина [16]. Наиболее изучено и представлено в клетках метилирование лизина и аргинина в молекулах гистонов. Эти модификации регулируют транскрипцию генов [17].
Метилирование малых молекул – один из процессов биосинтеза многих эндогенных соединений. В число продуктов метилирования с участием МТаз входят кофакторы белков, сигнальные и защитные молекулы, компоненты мембран, пигменты и многие другие соединения [18, 19].
Кофактор метилирования SAM
SAM – кофактор почти всех известных МТаз [8], выступает в качестве донора метильной группы в реакции метилирования N, C, O и S-нуклеофильных центров. Это второй по распространенности кофактор органической природы после АТР [20]. Главная особенность молекулы SAM – трехвалентный атом серы, который обеспечивает ее высокую реакционную способность в реакции метилирования различных субстратов (ДНК, РНК, белки, малые молекулы), обычно протекающей по SN2-механизму.
Асимметрически замещенный ион сульфония в молекуле SAM представляет собой хиральный центр. S- и R-эпимеры оптически стабильны и могут быть разделены. Показано, что реакция метилирования стереоспецифична, причем предпочтительно в реакцию вступает только природный S-эпимер, тогда как R-эпимер, связываясь в том же активом центре, не переносит метильную группу на субстрат [21, 22].
SAM также используется в качестве источника метиленовых групп (в биосинтезе циклопропилжирных кислот), аминогрупп (в синтезе 7,8‑диаминоперлагоновой кислоты, предшественника биотина), рибозильных фрагментов (в биосинтезе эпоксикевозина, модифицированного нуклеозида тРНК) и аминопропильных групп (при синтезе этилена и полиаминов) [23].
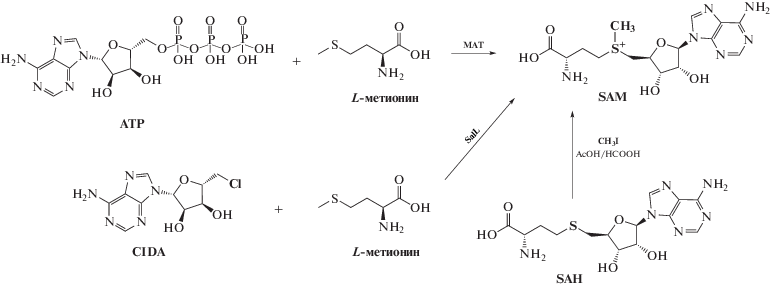
В живых системах SAM как правило образуется из АТР и L‑метионина при каталитическом участии метионин-аденозилтрансферазы (MAT, S‑adenosylmethionine synthetase) [24, 25]. Эту ферментативную реакцию используют и для синтеза SАМ в препаративных количествах из АТР и метионина [26, 27]. Для увеличения конверсии в реакционную смесь добавляют неорганическую пирофосфатазу, утилизирующую образующийся пирофосфат [28]. Другой вариант реакции получения SAM основан на использовании фермента SalL, который катализирует взаимодействие 5‑хлораденозина (CIDA) с метионином (рис. 2) [29]. Еще один вариант получения SAM – выделение этого продукта из дрожжей [30]. Также применяется синтетический способ получения SAM, основанный на взаимодействии метилгалогенида с SAH в кислой среде [22, 31]. В последнем случае, однако, образуется смесь S- и R-эпимеров.
В нейтральной и щелочной среде SAM подвергается самопроизвольному разложению по двум параллельным направлениям: расщеплению до метилтиоаденозина (МТА) и гомосеринлактона, а также гидролизу до аденина и S-рибозилметионина [32].
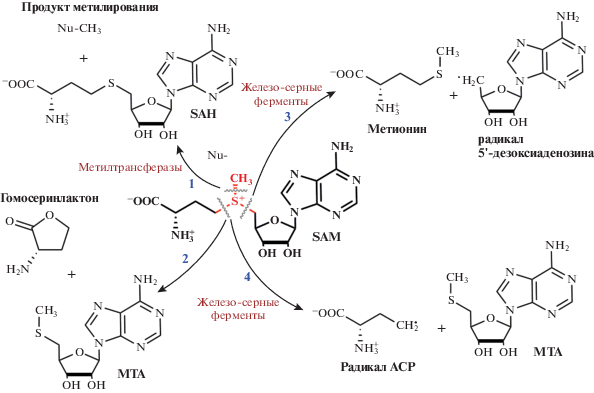
Присутствие трехвалентного атома серы определяет четыре основных пути метаболизма SAM в клетках: (1) удаление метильной группы в реакции метилирования с образованием SAH; (2) разрыв связи сера-углерод метиониновой цепи с образованием МТА и гомосеринлактона; (3) радикальное, катализируемое железосодержащими ферментами, расщепление связи между атомом серы и остатком рибозы с образованием реакционноспособного 5'-дезоксиаденозильного радикала; (4) радикальное расщепление с образованием радикала ACP и MTA (рис. 3) [33–35].
Рис. 3.
Схема разложения SAM. Путь 1 – катализируемый МТазами перенос метильной группы; путь 2 – неферментативное отщепление гомосеринолактона; пути 3 и 4 – радикальное расщепление, катализируемое железо-серными ферментами. Перепечатано (адаптировано) из [33] с разрешения издательства.
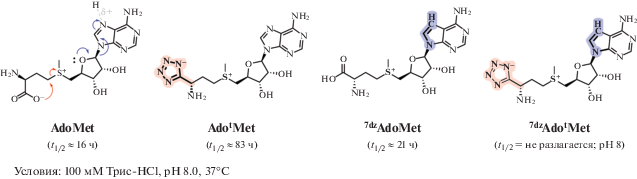
Показано, что синтез функционально активных изостерических аналогов SAМ увеличивает их устойчивость в основных реакциях разложения (депуринизация, внутримолекулярная циклизация и эпимеризация сульфония). При этом связывание таких аналогов SAМ с МТазами протекает почти идентично исходному кофактору [36–38]. Сотрудниками Thorson J.S. синтезированы аналоги SAM (рис. 4), в которых атом азота в положении 7 аденинового фрагмента заменен на углерод, а карбоксильная группа – на тетразольный цикл, что привело к значительному увеличению стабильности аналога SAM при pH 8. Биохимические и структурные исследования показали, что эти аналоги SAM связываются с МТазой DnrK (карминомицин 4-O-метилтрансфераза), при этом ферментативная активность белка полностью сохраняется [36].
Рис. 4.
Изостерические аналоги SAM. Указано время полуразложения соединений в условиях, приближенных к физиологическим. tMet – тетразол-L-метионин, 7dzAdo – S-7-деазааденозил. Перепечатано из [36] с разрешения издательства.
Еще одна изостерическая замена в молекуле SAM – замена карбоксильной группы на фосфониевую или фосфоновую (рис. 5), в результате которой SAM лишается возможности лактонизации, становясь более устойчивым. Показано, что такие молекулы способны эффективно связываться с МТазами [39, 40].
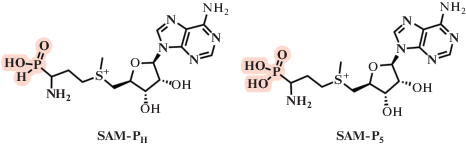
Копродукт метилирования SAH
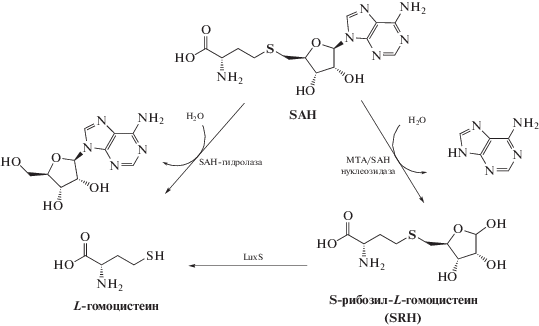
В результате переноса МТазами метильной группы с SAM на субстрат образуется молекула SAH. В большинстве случаев SAH является конкурентным ингибитором МТаз, и часто прочность связывания SAH с МТазами оказывается даже выше, чем у кофактора SAM [33, 41]. Поэтому очень важен клеточный гомеостаз SAM и SAH, который должен точно регулировать деградацию SAH после реакции метилирования. В клетках эукариот SAH распадается с образованием гомоцистеина и аденозина в реакции, катализируемой SAH-гидролазой (рис. 6) [41]. В клетках прокариот SAH расщепляется до аденина и S‑рибозилгомоцистеина преимущественно в реакции, катализируемой MTA/SAH-нуклеозидазой [42]. Все известные прокариотические MTA/SAH-нуклеозидазы относятся к трехсубстратным ферментам, способным отщеплять аденин не только от SAH, но также от MTA и 5'dAdo [43].
СИНТЕТИЧЕСКИЕ АНАЛОГИ SAM
Синтетические аналоги SAM создают и используют для решения нескольких типов задач, связанных с изучением или управлением активностью SAM-зависимых МТаз. В настоящем обзоре синтетические аналоги SAM рассмотрены исходя из задач, решаемых с их помощью.
Первую группу составляют ингибиторы МТаз. Эти соединения связываются с ферментом и блокируют его каталитическую активность. Во вторую группу мы выделяем аналоги SAM, которые содержат различные функциональные группы вместо метильной. Они узнаются МТазой, связываются с ферментом и вступают в каталитические реакции, что позволяет модифицировать субстраты новыми молекулярными фрагментами. Третья группа синтетических аналогов SAM – соединения, узнаваемые МТазой в качестве кофактора, которые под действием МТазы образуют аддукт с субстратом фермента. Наконец, к четвертой группе мы отнесли аналоги SAM, способные связываться с МТазами, но в дальнейшие каталитические реакции не вступают. Они используются для введения функциональных фрагментов, которые позволяют изучать МТазы.
Ингибиторы МТаз
Существует множество ингибиторов МТаз, различающихся механизмом действия. Они были получены с помощью различных стратегий, включая виртуальный и высокопроизводительный скрининг с последующей оптимизацией соединений-лидеров [44–46].
Некоторые соединения природного происхождения (рис. 7) действуют как конкурентные ингибиторы ДНК-МТаз (DNMT) [47]. К их числу относятся полифенолы, флавоноиды, антрахиноны и соединения некоторых других классов. Одними из первых описанных ингибиторов МТаз стали куркумин, (–)-эпигаллокатехин-3-галлат (EGCG), маханин, генистеин, кверцетин, силибинин, лютеолин, казинол Q, гиперицин, босвеллиевая кислота и ликопин [47, 48].
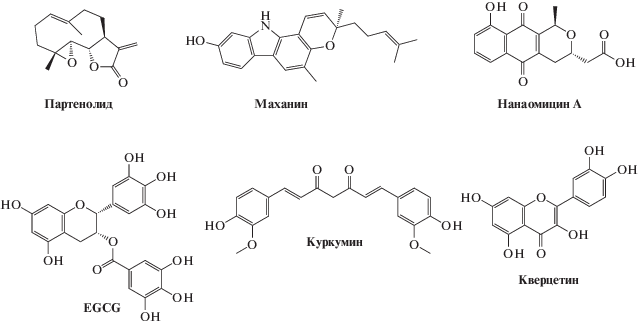
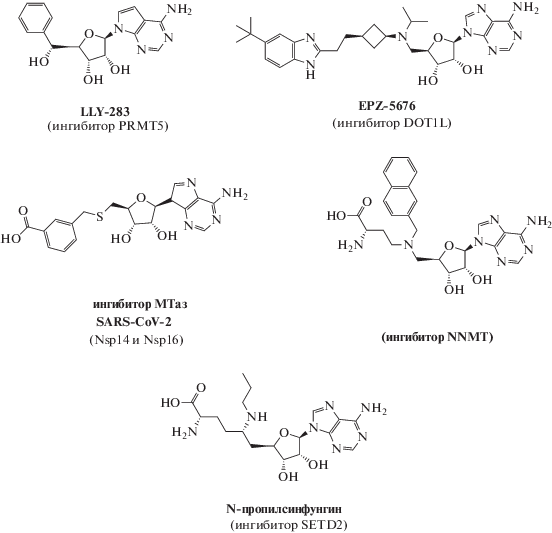
Другой класс ингибиторов – аналоги кофактора SAM, участвующие в конкурентном связывании с МТазами и не обладающие специфичностью в отношении конкретной МТазы, т.е. это так называемые PAN-ингибиторы. К этой группе можно отнести копродукт метилирования SAH, продукт разложения SAM – метилтиоаденозин, а также синфунгин – антигрибковый агент бактериального происхождения. Модификация заместителей в молекуле ингибитора в соответствии с особенностями активного центра МТазы позволяет увеличить селективность его связывания с конкретным ферментом (рис. 8) [49–56].
Рис. 8.
Примеры синтетических ингибиторов МТаз. LLY-283 – селективный ингибитор МТазы PRMT5, отвечающей за образование большинства моно- и диметилированных остатков аргинина в белках [57]; соединение EPZ-5676 – ингибитор МТазы DOT1L, отвечающей за метилирование лизина-79 в гистоне H3 [49, 51, 52]; потенциальный ингибитор МТаз Nsp14 и Nsp16 вируса SARS-Cov-2, отвечающих за метилирование кепа мРНК [56]; бисубстратный ингибитор никотинамид-N-МТазы (NNMT) – фермента, отвечающего за образование N-метилникотинамида из никотинамида [50]; N-пропилсинфунгин – производное синфунгина – селективный ингибитор МТазы SETD2, метилирующей остатки лизина в белках [53, 54].
Препараты нуклеозидной природы – азацитидин (5‑азацитидин) и децитабин (5‑аза‑2′‑дезоксицитидин), действие которых основано на ингибировании ДНК-МТаз, в частности DNMT3A, используют в терапии миелодиспластического синдрома [58]. Однако эти препараты нельзя отнести к аналогам кофактора. Это классические нуклеозидные ингибиторы – аналоги субстрата (ДНК). Метилирование этих фрагментов в составе цепи ДНК приводит к образованию ковалентной сшивки с ферментом, приводящей к необратимому блокированию его дальнейшей активности [59].
Аналоги SAM, используемые для переноса новых функциональных групп на субстрат
В этом разделе рассмотрены аналоги и производные SAM, которые могут служить синтетическими кофакторами для введения новых функциональных групп, включая различные метки, в субстраты МТаз. Они используются для поиска субстратов новых МТаз, а также в изучении функциональной активности ферментов. В качестве переносимой метки может выступать, например, изотопно-замещенная метильная группа. Кроме того, МТазы склонны “ошибаться” и переносить на субстраты более сложные функциональные группы.
ПЕРЕНОС ИЗОТОПНО-ЗАМЕЩЕННОЙ МЕТИЛЬНОЙ ГРУППЫ
С помощью изотопно-замещенной по метильной группе молекулы SAM в субстрат МТаз можно внедрять радиоактивные или стабильные изотопы углерода и водорода в реакциях in vitro метилирования.
А) Перенос радиоактивной метки. Для введения радиоактивной метки в субстрат МТазы проводят реакцию in vitro метилирования рекомбинантной МТазой или клеточным экстрактом [60] в присутствии молекулы SAM, содержащей атомы 14С или 3H в метильной группе.
Протекание реакции метилирования под действием SAM-зависимых МТаз сопровождается образованием SAH, ингибирующего реакцию. Поэтому для увеличения числа оборотов МТазы в реакционную смесь часто добавляют SAH-нуклеозидазу – фермент, расщепляющий SAH [61].
Количество введенной в макромолекулы радиоактивной метильной группы оценивают с помощью сцинцилляционного счетчика [62] или измеряют радиоавтографически после разделения в полиакриламидном геле [63]. Перед подсчетом радиоактивности необходимо отделить непрореагировавший радиоактивный SAM и низкомолекулярные радиоактивные продукты его распада от меченого субстрата. При изучении ДНК-МТаз классическим считается использование фильтровальной бумаги DE-81 для связывания ДНК после окончания реакции [64]. В случае метилирования белков предложено выделять белки и пептиды из реакционной смеси с помощью ZipTip-С4 – наконечников для пипеток, содержащих сорбент С4, селективно связывающий пептиды [65]. При проведении in vitro метилирования РНК продукт отделяют, осаждая РНК из реакционной смеси этанолом после фенол-хлороформенной экстракции белков [63]. Еще один вариант детекции меченого продукта реакции в присутствии избытка радиоактивного SAM получил название SPA (scintillation proximity assay). В этом случае используют предварительно биотинилированные субстраты, например, модифицированные пептиды, взаимодействующие с шариками, содержащими сцинтиллятор [66].
Изучение процессов метилирования с использованием радиоактивных меток in vitro позволяет идентифицировать и подтверждать макромолекулярные субстраты МТаз. Например, этим способом установлено, что МТаза YbeA метилирует псевдоуридин в позиции 1915 23S рРНК Escherichia coli [67], а МТаза YfiC E. coli отвечает за модификацию m6A37 в валиновой тРНК [62].
Б) Перенос метильной группы, содержащей стабильные изотопы. С помощью изотопнозамещенной молекулы SAM можно ввести в субстрат МТазы стабильные изотопы 2H и/или 13С. Такая изотопная метка позволяет проводить количественную оценку содержания продукта метилирования с помощью масс-спектрометрических методов. Например, in vitro метилирование РНК с помощью S‑(5'‑аденозил)‑L‑метионина‑d3 с последующим анализом РНК в экспериментах LC‑MS/MS было использовано для определения активности метилтрансферазы METTL14. Этот белок связывает МТазу-партнер METTL3 и в составе прочного гетеродимерного комплекса METTL3-METTL14 осуществляет модификацию m6A в мРНК млекопитающих [68].
Кроме того, введение изотопа 13С открывает возможности изучения метилирования с помощью метода ядерного магнитного резонанса (ЯМР), поскольку этот изотоп углерода имеет ненулевой спиновый магнитный момент и может быть детектирован методами ЯМР. Например, методом твердотельного ЯМР анализировали опухолевые ткани, выращенные с добавлением 13C-метионина [69]. Попадая в клетки, меченый метионин превращается в 13C-меченный SAM, который далее служит субстратом МТаз. В указанной работе с помощью измерения спектров ЯМР 13C,1H-HSQC метильных групп изучали различия в распределении 13CH3-групп в опухолях и нормальных тканях, которые могут быть связаны с нетипичным метаболизмом метионина, в частности, вследствие некорректной работы МТаз.
ПЕРЕНОС ДВАЖДЫ-АКТИВИРОВАННОЙ ФУНКЦИОНАЛЬНОЙ ГРУППЫ
Сульфониевый центр SAM является ключевым фрагментом в реакции трансметилирования, поэтому чаще всего модификации подвергают именно эту часть молекулы. Разработки аналогов SAM привели к эффективным синтетическим кофакторам для мечения субстратов путем переноса активированных функциональных групп с SAM на субстрат, которые в дальнейшем могут быть лигированы с более сложными фрагментами. Такие функциональные группы были названы “дважды активированными”, а метод получил название “метилтрансферазно-направленный перенос активированных групп” (methyltransferase-directed transfer of activated groups, mTAG) [70]. В качестве переносимых групп выступают аминосодержащие фрагменты, которые могут быть модифицированы в реакции с эфиром N-гидроксисукцинимида, фрагменты с концевым алкином или азидом, которые могут быть функционализированы в реакции азид-алкинового циклоприсоединения (“клик”‑химия) или фотоактивируемые фрагменты.
Механизм переноса метильной группы с SAM на молекулу субстрата детально исследован [71]. Одним из факторов, определяющих скорость переноса, является малый размер переносимой метильной группы. Ее замена на этильную или пропильную у трехвалентного атома серы SAM приводит к значительному уменьшению скорости переноса вследствие стерических эффектов [72, 73]. В 2006 году группа Elmar Weinhold сообщила о новом типе аналогов SAM, в котором метильная группа заменена на аллил или пропаргил (рис. 9) [74, 75]. Скорость реакции (SN2) переноса функциональной группы с таких аналогов SAM на субстрат оказывается лишь на порядок медленнее, несмотря на стерический фактор. Реакция протекает вследствие стабилизации переходного состояния за счет участия π-электронов двойной или тройной связи. Показано, что при использовании ДНК-МТазы из Thermus aquaticus (M.TaqI, метилирует остаток аденина (N6) в TCGA-сайте) перенос пропаргила с аналога SAM на ДНК осуществляется достаточно эффективно [74].
Рис. 9.
Схема синтеза аналогов SAM, содержащих пропаргильную и аллильную группы. Перепечатано (адаптировано) из работы [75] с разрешения издательства.
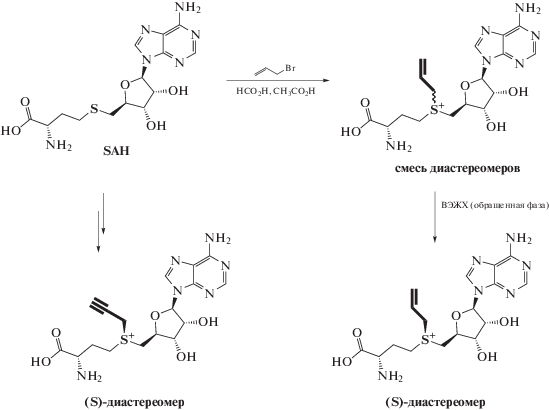
Подобные производные SAM получают реакцией нуклеофильного замещения активированного радикала (аллил или пропаргил) с SAH (рис. 9). Реакцию проводят в кислых условиях, что стабилизирует образующийся продукт и влияет на региоселективность за счет протонирования аминогрупп [75]. В ходе реакции образуются два диастереомера, которые могут быть разделены методом ВЭЖХ.
Пропаргил-содержащие аналоги представляют большой интерес как репортеры метилирования, поскольку перенесенная на субстрат тройная связь может быть в дальнейшем модифицирована функциональной меткой с помощью реакции азид-алкинового циклоприсоединения (AAC) [76–79].
Невысокая стабильность пропаргил-SAM, связанная с быстрым гидролизом алкинильной группы в физиологических условиях (период полупревращения составляет несколько минут), затрудняет работу с ним (рис. 10) [80]. Так, для некоторых МТаз реакция трансметилирования требует длительного время инкубирования. За это время пропаргил-SAM претерпевает гидролиз до keto-AdoMet, что приводит к низкой конверсии переноса пропаргильного фрагмента на субстрат [71, 81].
Рис. 10.
Пропаргильные аналоги SAM и ProSeAM. Показаны пути разложения соединений и их стабильность. Перепечатано (адаптировано) из работы [81] с разрешения издательства.
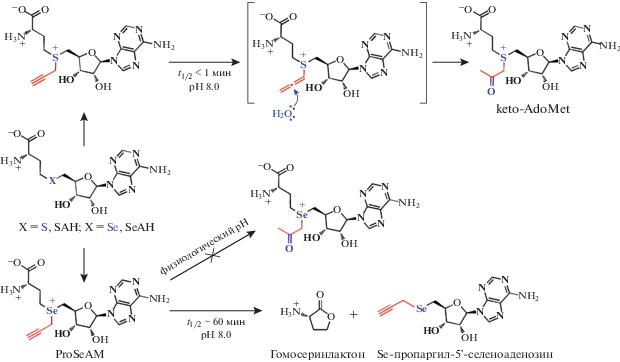
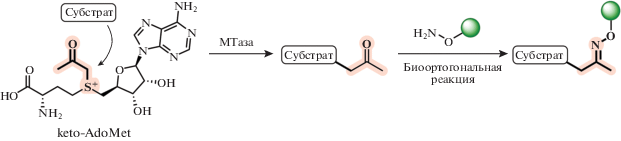
Позже было показано, что кетоновый аналог keto-AdoMet (рис. 11) также может использоваться для мечения субстратов, поскольку высокая реакционная способность и отсутствие кето-групп в белках и ДНК позволяет производить специфичное мечение. Данный подход впервые был использован в работе Zhaohui Sunny Zhou. После переноса кето-группы с keto-AdoMet на субстрат в присутствии тиопурин-S-метилтрансферазы (TPMT) она была введена в реакцию с гидроксиламиновым производным красителя Alexa Fluor 647. Это позволило выделить продукт реакции in vivo метилирования методом ВЭЖХ из клеточного лизата [82].
Рис. 11.
Использование keto-AdoMet в реакции трансметилирования. Перепечатано (адаптировано) из работы [82] с разрешения издательства.
Модифицированные аналоги пропаргил-SAM, обладающие повышенной стабильностью, описаны в работе группы E. Weinhold. Был показан перенос радикала пент‑2-ен-4-инила с аналога SAM (AdoEnYn, рис. 12) на гистон H3 в присутствии лизинметилтрансферазы гистона H3 Dim-5 при pH 9. Гистон H3, модифицированный концевым алкином, может дополнительно связываться с азид-ПЭГ-биотином с помощью CuAAC реакции [83].
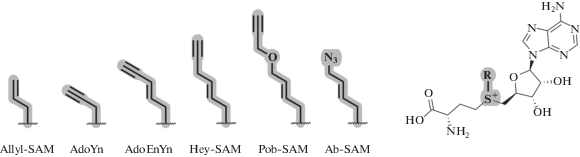
Группой Minkui Luo синтезирован аналог SAM, содержащий 4‑пропаргилоксибут-2-енильный радикал (Pob-SAM, рис. 12). Обнаружено, что при использовании нативной формы аргининметилтрансферазы (PRMT1) перенос функционального фрагмента на субстрат не происходит. Однако методом рациональной белковой инженерии получены мутантные формы фермента с изменениями в SAM-связывающем кармане. Так, фермент Y39F/M48G-PRMT1 показал высокую эффективность модификации гистона H4 в присутствии Pob-SAM, которые на следующем шаге были модифицированы флуоресцентным зондом в реакции CuAAC [85]. Аналоги SAM с громоздким функциональным фрагментом зачастую оказываются малоактивными при использовании нативных форм МТаз, но эту проблему часто удается решить путем создания модифицированных форм ферментов [85–87]. У некоторых ДНК- и РНК-МТаз не наблюдается существенного влияния размера переносимого фрагмента на ферментативную активность. Так, нативные формы МТаз Hen1, Ecm1, M. TaqI, M. HhaI и Dam эффективно катализируют перенос большого фрагмента содержащего флуорофор или биотиновый остаток [88–93].
В работе группы Klimašauskas Saulius исследована устойчивость различных алкинил-содержащих аналогов SAM. Показано, что они имеют период полуразложения в интервале от 3 мин до 5 ч при pH 7.4 [80] и деградируют по пути гидратации алкина. В группе Minkui Luo предложили заменить атом серы в SAM на селен. Для синтезированных ими селен-содержащих производных SAM (SeAM) не наблюдается гидратирования алкинильного фрагмента при pH 7.5. Обнаружено, что разложение селеновых аналогов идет по пути образования пропаргил-Se-MTA и гомосеринлактона (рис. 10) [94]. Реакционная способность селен-производного ProSeAM выше по сравнению с его серным аналогом, что увеличивает эффективность ферментативного переноса функциональной группы из-за повышенной активации селенониевого центра по сравнению с сульфониевым аналогом [95]. Преимущество ProSeAM продемонстрировано эффективным мечением серии субстратов PKMT (Protein lysine methyltransferases) [81, 96]. Высокая стабильность ProSeAM и совместимость с большинством МТаз, позволила провести протеомный анализ активности эндогенных белковых МТаз в различных клеточных линиях [81].
Команда J. Thorson изучала возможность ферментативного синтеза серии производных SAM и SeAM из АТР и соответствующих производных метионина в присутствии различных ферментов MAT. Показано, что каталитическая субъединица SAM-синтазы человека MATII (hMAT2A) и SAM-синтаза метаногенных термофильных архей Methanocaldococcus jannaschii (mMAT) проявляют высокую субстратную пластичность. Известно, что экспрессия гена hMAT2A повышена в раковых клетках. Это может открыть путь к синтезу аналогов S/Se – AdoMet в качестве метаболических зондов для исследования роли метилирования в развитии опухолевых процессов [97, 98].
Использование природных ферментов MAT в синтезе аналогов SAM ограничено алифатическими группами у атома серы аналога SAM [99, 100]. Поиск универсальной SAM-синтазы, способной конвертировать производные метионина, содержащие S-бензильный фрагмент, привели к созданию формы I122A/I330A фермента MAT из Cryptosporidium hominis (ChMAT). В 2020 году команда Andrea Rentmeister путем точечных замен аминокислотных остатков в гидрофобном связывающем кармане ChMAT, в котором располагается метильная группа SAM, определила, что замена аминокислотных остатков на менее объемные влияет на активность фермента по отношению к аналогам метионина, содержащим крупные концевые фрагменты. Выяснено, что ChMAT-I122A и ChMAT-I330A катализируют реакцию взаимодействия АТР с S-бензильным производным метионина с удовлетворительной конверсией 5–25%. Фермент, содержащий двойную замену I122A/I330A (PC-ChMAT) показал конверсию уже 65–70%. Сравнение рентгеноструктурных данных для ChMAT и PC-ChMAT показало, что указанные замены аминокислотных остатков не изменяют общую структуру активного сайта, но при этом уменьшают пространственное затруднение в активном центре. Аналогичным образом была сконструирована форма L147A/I351A термостабильной SAM-синтазы из M. jannaschii (PC-MjMAT), которая катализирует конверсию стерически нагруженных производных метионина и АТР в аналоги SAM при 65°C, что позволило их использовать в каскадных реакциях с МТазой M.TaqI, у которой оптимальная активность наблюдается при повышенных температурах [101]. Параллельно команда J. Thorson исследовала 38 мутантных форм SAM-синтаз hMAT2A, аминокислотные замены в которых влияли на связывание фермента с кофактором в области карбоксильной группы SAM. Установлено, что у формы K289L hMAT2A наблюдается изменение селективности в сторону аналога метионина – L-метионинола. Полученный аналог SAM обладал повышенной стабильностью [102].
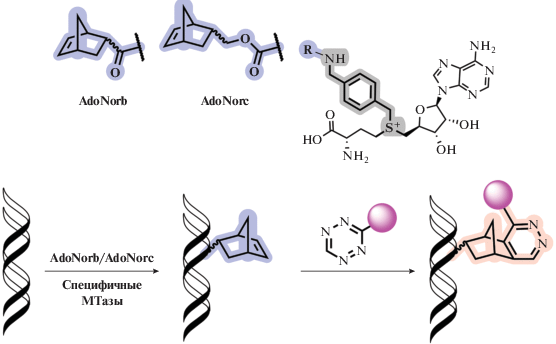
Дважды активированные аналоги SAM, не содержащие тройную связь, нашли применение в изучении МТаз [70, 84, 103]. A. Rentmeister и ее коллеги на примере двух МТаз (Ecm1 и M. TaqI) показали перенос норборненового фрагмента (рис. 13), присоединенного через п-ксилиленовый линкер к атому серы аналога SAM (AdoNorb и AdoNorc), на нуклеиновые кислоты различного размера. Последующее биоконъюгирование с флуорофорами или биотином проводили в отсутствие солей меди (I), что может быть полезным в богатой тиолами клеточной среде из-за малой устойчивости в ней азидов [104]. Показано, что бензильные фрагменты переносятся ДНК-МТазой Ecm1 с более высокой каталитической эффективностью, чем метильная группа из SAM. Это демонстрирует широкую применимость соответствующих аналогов SAM [105].
Рис. 13.
Вверху норборненовые аналоги SAM, используемые для введения меток в ДНК, внизу – общая схема процесса мечения. Перепечатано (адаптировано) из работы [105] (лицензия Creative Commons Attribution-NonCommercial 3.0).
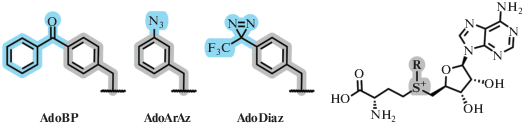
В работе A. Rentmeister и коллег получены новые производные SAM, несущие наиболее распространенные фотосшивающие фрагменты (арилазид, диазирин и бензофенон), присоединенные через бензильный линкер (рис. 14). Показано, что полученные фото-кросс-линкеры могут ферментативно переноситься на N7-кеп-мРНК – мишень МТазы Ecm1. При фотоактивации модифицированного субстрата УФ-излучением происходит сшивка меченой РНК с взаимодействующим с ней кеп-связывающим белком eIF4E [91].
Рис. 14.
Аналоги SAM c фото-сшивающейся группой AdoBP, AdoArAz, AdoDiaz. Перепечатано (адаптировано) из работы [91] с разрешения издательства.
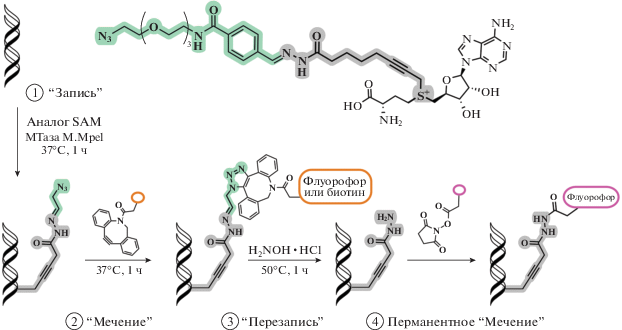
Дальнейшее развитие методов биоконъюгации привело к созданию перезаписывающихся меток. В 2020 году группа Paco Fernandez-Trillo разработала новый аналог SAM, у которого в качестве метки для субстрата имеется ацилгидразоновый линкер и концевой азид (рис. 15). Дизайн метки позволяет модифицировать ДНК-субстраты МТаз (M.TaqI и M.MpeI), а затем удалять метку и снова модифицировать тот же субстрат функциональными фрагментами, перманентно или перезаписываемо. На примере ДНК-МТазы M.MpeI разработан протокол выделения флуоресцентной ДНК с последующей перезаписью метки на биотин для выделения на стрептавидиновых магнитных шариках [106].
Рис. 15.
Вверху аналог SAM, содержащий ацилгидразоновый линкер и концевой азид, внизу показана общая схема процесса последовательной функционализации ДНК. Перепечатано (адаптировано) из работы [106] с разрешения издательства.
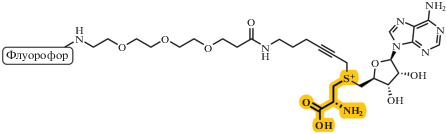
Сравнительно низкая стабильность аналогов SAM накладывает ограничения на их синтез и выделение. Основная проблема классического метода синтеза возникает на последней стадии – S-присоединении высокоэлектрофильных алкилтрифлатов или аллилбромидов к предшественнику аналога SAM. Этот шаг протекает с низким выходом и требует большого избытка (20‒200 eq) дорогостоящего бифункционального линкера. Модификация стратегии синтеза, предложенная Johan Hofkens и коллегами, позволяет получить неприродные аналоги SAM, содержащие цистеин, а не гомоцистеин, в аминокислотной части молекулы, из доступных исходных соединений с высоким выходом (рис. 16). Предложенный метод меняет последовательность присоединения заместителей к атому серы: сначала формируется аналог МТА, модифицированный функциональным линкером или красителем, а затем создается каркас аналога SAM путем присоединения аминокислотной части. Также показано, что MTaза M.TaqI может переносить метку большого размера c неприродного аналога SAM, содержащую PEG-линкер с красителем, на субстрат [107].
Рис. 16.
Гомолог SAM, укороченный на одну СН2-группу, в аминокислотном фрагменте и содержащий флуорофорную группу.
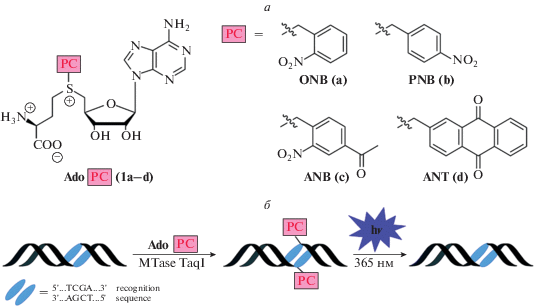
Использование молекулярных систем с фотоактивируемыми группами является важным инструментом в исследовании взаимодействий и функций биомолекул [108–110]. МТазы с использованием кофактора SAM постсинтетически метилируют биомолекулы, тем самым модулируя их активность. Во многих случаях данный процесс является динамическим и обратимым [111]. В 2018 году впервые описаны аналоги SAM (рис. 17), содержащие фотоактивируемые группы, которые могут быть перенесены на субстрат (ДНК) в присутствии МТазы M.TaqI, тем самым блокируя сайт связывания эндонуклеазы рестрикции R.TaqI. Удаление фотоактивируемой группы происходит при облучении УФ-светом (365 нм) в течение 10‒30 мин. Таким образом, использование данных аналогов SAM позволяет имитировать естественные процессы с участием ферментов метилирования и деметилирования, и регулировать эти процессы во времени [112, 113]. Позже был предложен ферментативный каскад, содержащий модифицированную SAM-синтазу (PC-MAT) и МТазы ДНК (MTaqI) или РНК (Ecm1), что позволило вводить фотоактивируемые группы, используя АТР и аналоги метионина [101, 114–118].
Рис. 17.
Аналоги SAM, содержащие фотоактивируемые группы (а); и схема катализируемого МТазой переноса фотоактивируемой группы от AdoPC на ДНК с последующим фоторасщеплением (б). Перепечатано (адаптировано) из работы [113] (лицензия Creative Commons Attribution-NonCommercial 3.0).
Аналоги SAM, ковалентно связывающиеся с субстратом
Другая разновидность аналогов SAM – молекулы, способные под действием МТаз ковалентно связываться с субстратом. В состав таких аналогов SAM чаще всего входит азиридиновое кольцо или акцептор Михаэля.
Азиридиновые производные – это высокоактивные электрофилы с трехчленным циклом, содержащим один атом азота. Производные MTA, содержащие азиридиновую группу вместо атома серы, могут вступать в реакцию SN2 с раскрытием цикла и образованием конъюгата кофактора и субстрата (рис. 18). В работе 1998 года, представленной группой E. Weinhold, показано алкилирование ДНК-субстрата азиридиновым производным MTA в присутствии МТазы M.TaqI (N6-аденин-ДНК-метилтрансфераза), тогда как в отсутствие фермента эта реакция не наблюдалась [119]. Описанный подход открыл возможность создавать бифункциональные структуры, содержащие флуорофорную или аффинную метку, для детектирования положения аналога кофактора после конъюгации [120–123].
Рис. 18.
Введение метки в ДНК с помощью несущего метку производного MTA, ковалентно сшивающегося посредством азиридиновой группы с субстратом. Перепечатано (адаптировано) из работы [120] с разрешения издательства.
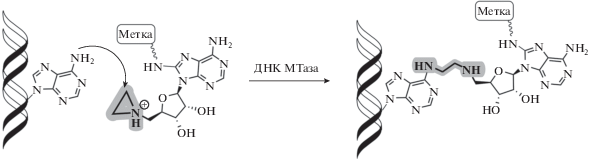
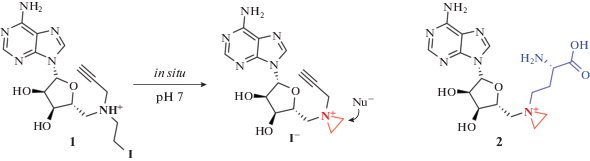
Далее было создано 5'-N-замещенное соединение 1 (рис. 19), которое содержит фрагмент азотистого иприта и функциональную группу [124]. Такие соединения достаточно стабильны и позволяют избежать трудностей, связанных с низкой стабильностью азиридинового фрагмента. Азиридиновый цикл, обладающий высокой реакционной способностью, легко образуется in situ после внутримолекулярной циклизации (рис. 19). Перенесенная функциональная группа делает возможным дальнейшее биортогональное присоединение дополнительных меток.
Рис. 19.
Производное MTA 1 и производное SAM 2, содержащие высокореакционный фрагмент азотистого иприта.
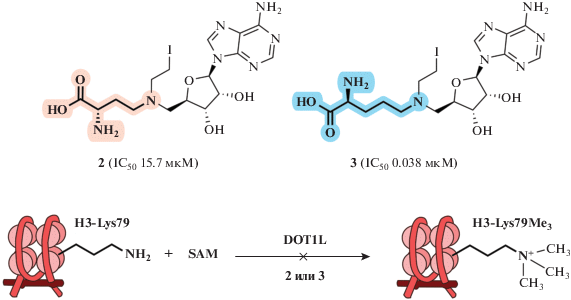
Аналог SAM 2 (рис. 19), содержащий аминокислотную часть, показал более высокую алкилирующую способность в сравнении с соединением 1, что обусловлено увеличением прочности связывания модифицированного кофактора с МТазой [125]. Эффективность данного подхода показана для субстратов МТаз различных классов [126–128].
Влияние аминокислотной части аналогов SAM описано в работе команды Yongcheng Song. Показано, что гомолог соединения 2, в котором длина аминокислотной части кофактора увеличена на одну метиленовую группу (рис. 20), может значительно увеличить ингибирование МТазы DOT1L (метилирует лизин 79 гистона H3). Так, IC50 такого гомолога снизилась до 0.038 мкM в сравнении с 15.7 мкМ для соединения 2. Предполагается, что гомолог, содержащий дополнительную CH2-группу, в большей степени воспроизводит связывание SAM с МТазой, поскольку две связи C–N (~1.47 Å каждая) значительно короче, чем связи C–S (~1.82 Å) в SAM и SAH [51].
Рис. 20.
Соединение 3 (гомолог соединения 2, содержащего фрагмент азотистого иприта) ингибирует каталитическую активность МТазы DOT1L. Перепечатано (адаптировано) из работы [51] с разрешения издательства.
Азиридиновые производные имеют хорошие перспективы использования в качестве зондов для идентификации субстратов МТаз [129, 130], однако ряд недостатков ограничивают их широкое применение. Во-первых, синтез таких молекул трудоемок: низкие выходы на последних стадиях, образование β-галогенида при снятии защитных групп и низкая стабильность продукта усложняют их получение. Во-вторых, образованные бисубстратные аддукты необратимо ингибируют МТазы, что требует стехиометрических количеств фермента для мечения. В-третьих, высокая реакционная способность азиридиновых аналогов может приводить к неспецифическому и неферментативному алкилированию нуклеофильных центров молекул ДНК и белков. На сегодняшний день азиридининовые аналоги SAM можно получить только синтетическим путем.
Несмотря на удобство и универсальность переноса МТазой функциональной группы с аналога SAM на субстрат [101], на практике существует ограничение такого подхода для изучения взаимодействия субстрат-МТаза in situ, поскольку субстраты других МТаз, присутствующих в клетке, также могут быть помечены, что приводит к исчезновению специфичности. В этом случае прямая связь между конкретной парой субстрат–фермент не может быть установлена.
Решение данной проблемы предложено James K. Coward и Anthony E. Pegg. Они разработали концепцию ингибирования фермента мультисубстратными аддуктами [131]. Суть этой концепции основана на том, что прочность связывания субстрата и/или кофактора с ферментом может быть невысокой, однако прочность связывания бисубстрата, состоящего из ковалентно связанных молекул субстрата и кофактора, усиливается многократно из-за потери энтропийного вклада в свободную энергию связывания для одного из фрагментов бисубстрата [132].
В 2016 году группа Zhaohui Sunny Zhou провела ферментативный синтез аналога SAM AdoVin (S-аденозил-L-винтионин, рис. 21), содержащий акцептор Михаэля. На примере тиопуринметилтрансферазы (TPMT) показана возможность выделять из реакционных смесей белок в комплексе с аддуктом субстрат-AdoVin, что позволило идентифицировать новый субстрат фермента [133].
Рис. 21.
Получение AdoVin из L-винтионина и АТР. Далее под действием МТазы TPMT AdoVin образует аддукт субстрат-AdoVin, играющий роль бисубстратного ингибитора. Перепечатано (адаптировано) из работы [133] с разрешения издательства.
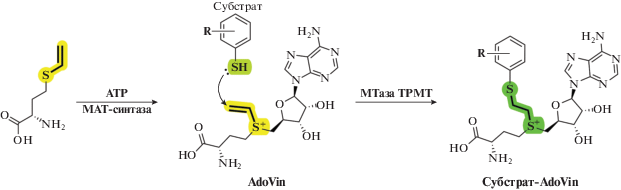
Аналоги SAM, связывающиеся с МТазой
Для большинства МТаз SAH конкурирует за сайт связывания SAM и часто имеет константу связывания более высокую, чем SAM [18, 33]. Аналоги SAH были предложены в качестве агентов для селективного фотоиндуцированного выделения МТаз и дальнейшего изучения аддуктов методами масс-спектрометрии. Впервые соединения-“крючки” для SAM-зависимых МТаз были синтезированы группой E. Weinhold [134]. Такие “крючки” состоят из трех частей: фрагмента SAH для направленного нековалентного связывания с ферментами (рис. 22-1), фотореактивной группы для ковалентной сшивки с ферментом (рис. 22-2) и биотин-содержащего фрагмента, служащего для последующего выделения образующихся аддуктов с помощью магнитных шариков с иммобилизованным стрептавидином (рис. 22-3). SAH был выбран в качестве направляющей группы, к которой через амино-линкеры в положении N-6 или C-8 пуринового остатка прикреплялись остальные фрагменты (рис. 22).
Рис. 22.
Общее строение трифункциональной молекулы-крючка [134]. 1 – фрагмент SAH, 2 – фотореактивная группа, 3 – остаток биотина.
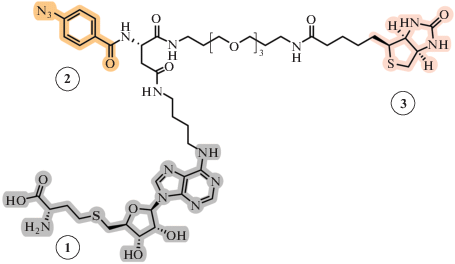
Эффективность этих соединений продемонстрирована на примере различных МТаз, действующих на ДНК, РНК и белки. Показано, что данные соединения, образовавшие ковалентную сшивку в результате облучения, позволяют выделять и идентифицировать SAH-связанные белки из сложных смесей, таких как клеточный лизат. Эффективность выделения МТаз без ковалентной сшивки фотореактивной группой оказалась существенно более низкой [134, 135]. На основе данного каркаса получены также “крючки”, содержащие флуоресцентные метки [136, 137].
Iredia Iyamu и Rong Huang в работе 2020 года получили азо-аналог копродукта SAH (рис. 23). Он представляет собой бисубстратный ингибитор МТаз, оснащенный флуоресцентным красителем, присоединенным через длинный линкер к положению N6 аденинового кольца. Высокая константа связывания с МТазой NNMT позволила разработать метод конкурентного анализа для оценки и поиска новых ингибиторов NNMT с использованием поляризации флуоресценции (FP). Важно отметить, что конкурентный анализ с использованием FP дает возможность идентифицировать ингибиторы, которые напрямую или аллостерически препятствуют связыванию кофактора с активным сайтом NNMT. Высокая воспроизводимость метода позволяет использовать его для высокопроизводительного скрининга ингибиторов NNMT [138].
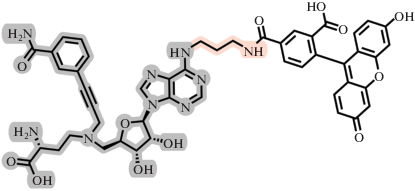
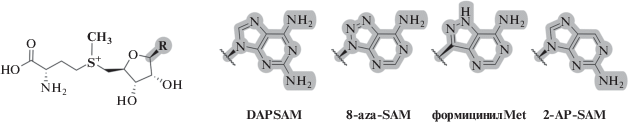
Флуоресцентные аналоги SAM могут быть использованы для определения константы связывания кофактора не только с МТазами, но и с другими SAM-связывающими молекулами. Так, с помощью измерения флуоресценции аналога DAPSM (рис. 24) определена константа связывания SAM с рибопереключателем РНК (III riboswitch RNA) [139–141].
Рис. 24.
Некоторые флуоресцентные производные SAM. Перепечатано (адаптировано) из работы [8] с разрешения издательства.
ЗАКЛЮЧЕНИЕ
Приведенные выше примеры позволяют заключить, что возможности использования синтетических аналогов SAM в изучении МТаз и их субстратов очень велики.
На протяжении последних двух десятилетий разработано огромное количество разнообразных синтетических аналогов SAM, каждый из которых имеет свои особые свойства. Интерес к подобным соединениям возрастает год от года. Этому способствует постоянно расширяющийся круг задач, в которых подобные молекулы могут быть использованы: это и управление активностью МТаз с помощью селективных низкомолекулярных ингибиторов, и установление функций МТаз с помощью уникальных меток, и разнообразная модификация субстратов МТаз.
Тем не менее, все еще остается множество нерешенных вопросов. Например, большинство аналогов SAM обладают низкой устойчивостью в условиях, необходимых для дальнейших реакций с участием МТаз. Другая сложность состоит в том, что структурные отличия аналогов SAM от природного кофактора как правило влияют на прочность их связывания с МТазами. Синтетические молекулы могут недостаточно хорошо узнаваться МТазой; могут связываться, но не вступать в последующую ферментативную реакцию; а могут связываться с ферментом, вступать в реакцию, но, в силу высокой аффинности, не высвобождаться из активного центра МТазы после завершения ферментативной реакции. Также нерешенным остается вопрос неспецифичности МТаз при использовании аналогов SAM в клеточных системах (in vivo или in vitro). Идентификация, модификация или выделение субстрата изучаемой МТазы осложняется наличием большого количества других МТаз в клетке, конкурирующих за кофактор, что приводит к неспецифическому переносу функциональный группы на нецелевые субстраты.
Тем не менее, постоянное расширение синтетической базы, а также непрерывно растущее число работ в области разработки новых молекул, отвечающих определенным свойствам, необходимым для изучения МТаз, позволяют надеяться на то, что в ближайшее время многие из этих проблем будут преодолены.
Работа выполнена при финансовой поддержке РФФИ (грант № 20-04-00318) и Междисциплинарной научно-образовательной школы Московского университета “Молекулярные технологии живых систем и синтетическая биология”.
Настоящая статья не содержит описания каких-либо результатов, полученных с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Moore L.D., Le T., Fan G. (2013) DNA methylation and its basic function. Neuropsychopharmacology. 38(1), 23–38.
Chen X., Sun Y.Z., Liu H., Zhang L., Li J.Q., Meng J. (2017) RNA methylation and diseases: experimental results, databases, Web servers and computational models. Brief. Bioinformatics. 20(3), 896–917.
Clarke S. (1993) Protein methylation. Curr. Opin. Cell Biol. 5(6), 977–983.
Lee D.Y., Teyssier C., Strahl B.D., Stallcup M.R. (2005) Role of protein methylation in regulation of transcription. Endocrine Rev. 26(2), 147–170.
Noel J.P., Dixon R.A., Pichersky E., Zubieta C., Ferrer J.L. (2003) Chapter two. Structural, functional, and evolutionary basis for methylation of plant small molecules. Recent Adv. Phytochem. 37(C), 37–58.
Stojković V., Fujimori D.G. (2017) Mutations in RNA methylating enzymes in disease. Curr. Opin. Chem. Biol. 41, 20–27.
Bateman A., Martin M.J., Orchard S., Magrane M., Agivetova R., Ahmad S., Alpi E., Bowler-Barnett E.H., Britto R., Bursteinas B., Bye-A-Jee H., Coetzee R., Cukura A., Silva A.Da, Denny P., Dogan T., Ebe-nezer T.G., Fan J., Castro L.G., Garmiri P., Georghiou G., Gonzales L., Hatton-Ellis E., Hussein A., Ignatchenko A., Insana G., Ishtiaq R., Jokinen P., Joshi V., Jyothi D., Lock A., Lopez R., Luciani A., Luo J., Lussi Y., MacDougall A., Madeira F., Mahmoudy M., Menchi M., Mishra A., Moulang K., Nightingale A., Oliveira C.S., Pundir S., Qi G., Raj S., Rice D., Lopez M.R., Saidi R., Sampson J., Sawford T., Speretta E., Turner E., Tyagi N., Vasudev P., Volynkin V., Warner K., Watkins X., Zaru R., Zellner H., Bridge A., Poux S., Redaschi N., Aimo L., Argoud-Puy G., Auchincloss A., Axelsen K., Bansal P., Baratin D., Blatter M.C., Bolleman J., Boutet E., Breuza L., Casals-Casas C., de Castro E., Echioukh K.C., Coudert E., Cuche B., Doche M., Dornevil D., Estreicher A., Famiglietti M.L., Feuermann M., Gasteiger E., Gehant S., Gerritsen V., Gos A., Gruaz-Gumowski N., Hinz U., Hulo C., Hyka-Nouspikel N., Jungo F., Keller G., Kerhornou A., Lara V., Le Mercier P., Lieberherr D., Lombardot T., Martin X., Masson P., Morgat A., Neto T.B., Paesano S., Pedruzzi I., Pilbout S., Pourcel L., Pozzato M., Pruess M., Rivoire C., Sigrist C., Sonesson K., Stutz A., Sundaram S., Tognolli M., Verbregue L., Wu C.H., Arighi C.N., Armi-nski L., Chen C., Chen Y., Garavelli J.S., Huang H., Laiho K., McGarvey P., Natale D.A., Ross K., Vinayaka C.R., Wang Q., Wang Y., Yeh L.S., Zhang J. (2021) UniProt: The universal protein knowledgebase in 2021. Nucl. Acids Res. 49(D1), D480–D489.
Zhang J., Zheng Y.G. (2016) SAM/SAH analogs as versatile tools for SAM-dependent methyltransferases. ACS Chem. Biol. 11(3), 583–597.
Martin J.L., McMillan F.M. (2002) SAM (dependent) I AM: the S-adenosylmethionine-dependent methyltransferase fold. Curr. Opin. Struct. Biol. 12(6), 783–793.
Cornelissen N.V., Michailidou F., Muttach F., Rau K., Rentmeister A. (2020) Nucleoside-modified AdoMet analogues for differential methyltransferase targeting. Chem. Commun. 56(14), 2115–2118.
Greenberg M.V.C., Bourc’his D. (2019) The diverse roles of DNA methylation in mammalian development and disease. Nat. Rev. Mol. Cell. Biol. 20(10), 590–607.
Koonin E.V., Makarova K.S., Wolf Y.I. (2017) Evolutionary genomics of defense systems in archaea and bacteria. Annu. Rev. Microbiol. 71(1), 233–261.
Sergiev P.V., Aleksashin N.A., Chugunova A.A., Polikanov Y.S., Dontsova O.A. (2018) Structural and evolutionary insights into ribosomal RNA methylation. Nat. Chem. Biol. 14(3), 226–235.
Wilson D.N. (2014) Ribosome-targeting antibiotics and mechanisms of bacterial resistance. Nat. Rev. Microbiol. 12(1), 35–48.
Boriack-Sjodin P.A., Swinger K.K. (2016) Protein methyltransferases: a distinct, diverse, and dynamic family of enzymes. Biochemistry. 55(11), 1557–1569.
Heurgu V., Champ S. (2002) The hemK gene in Escherichia coli encodes the N5-glutamine methyltransferase that modifies peptide release factors. EMBO J. 21(4), 769–778.
Jambhekar A., Dhall A., Shi Y. (2019) Roles and regulation of histone methylation in animal development. Nat. Rev. Mol. Cell. Biol. 20(10), 625–641.
Liscombe D.K., Louie G.V., Noel J.P. (2012) Architectures, mechanisms and molecular evolution of natural product methyltransferases. Nat. Prod. Rep. 29(10), 1238–1250.
Zhang C., Sultan S.A., Rehka T., Chen X. (2021) Biotechnological applications of S-adenosyl-methionine-dependent methyltransferases for natural products biosynthesis and diversification. Bioresources Bioprocessing. 8 (1), 1–21.
Fauman E.B., Blumenthal R.M., Cheng X. (1999) Structure and evolution of Adomet-dependent methyltransferases. S-Adenosylmethionine-Dependent Methyltransferases. Structure and Functions. Eds Cheng X. and Blumenthal. World Sci. Publ., 1–38.
Borchardt R.T., Wu Y.S. (1976) Potential inhibitors of S-adenosylmethionine-dependent methyltransferases. 5. Role of the asymmetric sulfonium pole in the enzymatic binding of S-adenosyl-L-methionine. J. Med. Chem. 19(9), 1099–1103.
de La Haba G., Jamieson G.A., Mudd S.H., Richards H.H. (1959) S-adenosylmethionine: the relation of configuration at the sulfonium center to enzymatic reactivity. J. Am. Chem. Soc. 81(15), 3975–3980.
Fontecave M., Atta M., Mulliez E. (2004) S-adenosylmethionine: nothing goes to waste. Trends Biochem. Sci. 29(5), 243–249.
Markham G.D., Pajares M.A. (2009) Structure-function relationships in methionine adenosyltransferases. Cell. Mol. Life Sci. 66(4), 636–648.
Popadić D., Mhaindarkar D., Dang Thai M.H.N., Hailes H.C., Mordhorst S., Andexer J.N. (2021) A bicyclic: S-adenosylmethionine regeneration system applicable with different nucleosides or nucleotides as cofactor building blocks. RSC Chem. Biol. 2(3), 883–891.
Park J., Tai J., Roessner C.A., Scott A.I. (1996) Enzymatic synthesis of S-adenosyl-L-methionine on the preparative scale. Bioorg. Med. Chem. 4(12), 2179–2185.
Davis T.D., Kunakom S., Burkart M.D., Eustaquio A.S. (2018) Preparation, assay, and application of chlorinase SalL for the chemoenzymatic synthesis of S-adenosyl-L-methionine and analogs. Meth. Enzymol. 604, 367–388.
Walsby C.J., Hong W., Broderick W.E., Cheek J., Ortillo D., Broderick J.B., Hoffman B.M. (2002) Electron-nuclear double resonance spectroscopic evidence that S-adenosylmethionine binds in contact with the catalytically active [4Fe-4S]+ cluster of pyruvate formate-lyase activating enzyme. J. Am. Chem. Soc. 124(12), 3143–3151.
Sadler J.C., Humphreys L.D., Snajdrova R., Burley G.A. (2017) A tandem enzymatic sp2-C-methylation process: coupling in situ S-adenosyl-L-methionine formation with methyl transfer. ChemBioChem. 18(11), 992–995.
Hayakawa K., Kajihata S., Matsuda F., Shimizu H. (2015) 13C-metabolic flux analysis in S-adenosyl-l-methionine production by Saccharomyces cerevisiae. J. Biosci. Bioengin. 120(5), 532–538.
Moriya S.S., Samejima K., Taira H., Hiramatsu K., Kawakita M. (2020) ESI-Q-TOF-MS determination of polyamines and related enzyme activity for elucidating cellular polyamine metabolism. Anal. Biochem. 607, 113831.
Matos J.R., Wong C.H. (1987) S-adenosylmethionine: stability and stabilization. Bioorg. Chem. 15(1), 71–80.
Mariasina S.S., Chang C.F., Petrova O.A., Efimov S.V., Klochkov V.V., Kechko O.I., Mitkevich V.A., Sergiev P.V., Dontsova O.A., Polshakov V.I. (2020) Williams–Beuren syndrome-related methyltransferase WBSCR27: cofactor binding and cleavage. FEBS J. 287(24), 5375–5393.
Yeates T.O. (2002) Structures of SET domain proteins: protein lysine methyltransferases make their mark. Cell. 111 (1), 5–7.
Dixon M.M., Huang S., Matthews R.G., Ludwig M. (1996) The structure of the C-terminal domain of methionine synthase: presenting S-adenosylmethionine for reductive methylation of B12. Structure. 4(11), 1263–1275.
Huber T.D., Wang F., Singh S., Johnson B.R., Zhang J., Sunkara M., Van Lanen S.G., Morris A.J., Phillips G.N., Thorson J.S. (2016) Functional AdoMet isosteres resistant to classical AdoMet degradation pathways. ACS Chem. Biol. 11(9), 2484–2491.
McKean I.J.W., Sadler J.C., Cuetos A., Frese A., Humphreys L.D., Grogan G., Hoskisson P.A., Burley G.A. (2019) S-adenosylmethionine cofactor modifications enhance the biocatalytic repertoire of small molecule C-alkylation. Angew. Chem. Int. Ed. 58(49), 17583–17588.
Vranken C., Fin A., Tufar P., Hofkens J., Burkart M.D., Tor Y. (2016) Chemoenzymatic synthesis and utilization of a SAM analog with an isomorphic nucleobase. Org. Biomol. Chem. 14(26), 6189–6192.
Alferov K.V., Zhukov Y.N., Khurs E.N., Khomutov R.M. (2003) Stable organophosphorus analogues of S-adenosylmethionine and S-methylmethionine. Mendeleev Commun. 13(6), 243–244.
Жуков Ю.Н., Хомутов А.Р., Осипова Т.И., Хомутов Р.М. (1999) Синтез фосфиновых аналогов серусодержащих аминокислот. Известия РАН, Сер. хим. Вып. № 7, 1360–1363
Ueland P.M. (1982) Pharmacological and biochemical aspects of S-adenosylhomocysteine and S-adenosylhomocysteine hydrolase. Pharmacol. Rev. 34(3), 223–253.
Walker R.D., Duerre J.A. (1975) S-adenosylhomocysteine metabolism in various species. Can. J. Biochem. 53(3), 312–319.
Parveen N., Cornell K.A. (2011) Methylthioadenosine/S-adenosylhomocysteine nucleosidase, a critical enzyme for bacterial metabolism. Mol. Microbiol. 79(1), 7–20.
Medina-Franco J.L., Méndez-Lucio O., Dueñas-González A., Yoo J. (2015) Discovery and development of DNA methyltransferase inhibitors using in silico approaches. This paper is dedicated to the memory of Dr Eugene Athanas Bratoeff Titeff honoring his many contributions to drug discovery. Drug Discov. Today. 20(5), 569–577.
Richart L., Margueron R. (2020) Drugging histone methyltransferases in cancer. Curr. Opin. Chem. Biol. 56, 51–62.
Lin Q., Jiang F., Schultz P.G., Gray N.S. (2001) Design of allele-specific protein methyltransferase inhibitors. J. Am. Chem. Soc. 123(47), 11608–11613.
Zwergel C., Valente S., Mai A. (2016) DNA methyltransferases inhibitors from natural sources. Curr. Top. Med. Chem. 16(7), 680–696.
Saldívar-González F.I., Gómez-García A., Chávez-Ponce De León D.E., Sánchez-Cruz N., Ruiz-Rios J., Pilón-Jiménez B.A., Medina-Franco J.L. (2018) Inhibitors of DNA methyltransferases from natural sources: a computational perspective. Front. Pharmacol. 9(OCT), 1144.
Yu W., Chory E.J., Wernimont A.K., Tempel W., Scopton A., Federation A., Marineau J.J., Qi J., Barsyte-Lovejoy D., Yi J., Marcellus R., Iacob R.E., Engen J.R., Griffin C., Aman A., Wienholds E., Li F., Pineda J., Estiu G., Shatseva T., Hajian T., Al-Awar R., Dick J.E., Vedadi M., Brown P.J., Arrowsmith C.H., Bradner J.E., Schapira M. (2012) Catalytic site remodelling of the DOT1L methyltransferase by selective inhibitors. Nat. Commun. 3(1), 1–12.
Gao Y., Van Haren M.J., Moret E.E., Rood J.J.M., Sartini D., Salvucci A., Emanuelli M., Craveur P., Babault N., Jin J., Martin N.I. (2019) Bisubstrate inhibitors of nicotinamide N-methyltransferase (NNMT) with enhanced activity. J. Med. Chem. 62(14), 6597–6614.
Yao Y., Chen P., Diao J., Cheng G., Deng L., Anglin J.L., Prasad B.V.V., Song Y. (2011) Selective inhibitors of histone methyltransferase DOT1L: design, synthesis, and crystallographic studies. J. Am. Chem. Soc. 133(42), 16746–16749.
Rugo H.S., Jacobs I., Sharma S., Scappaticci F., Paul T.A., Jensen-Pergakes K., Malouf G.G. (2020) The promise for histone methyltransferase inhibitors for epigenetic therapy in clinical oncology: a narrative review. Adv. Therapy. 37(7), 3059–3082.
Zheng W., Ibáñez G., Wu H., Blum G., Zeng H., Dong A., Li F., Hajian T., Allali-Hassani A., Amaya M.F., Siarheyeva A., Yu W., Brown P.J., Schapira M., Vedadi M., Min J., Luo M. (2012) Sinefungin derivatives as inhibitors and structure probes of protein lysine methyltransferase SETD2. J. Am. Chem. Soc. 134(43), 18004–18014.
Tisi D., Chiarparin E., Tamanini E., Pathuri P., Coyle J.E., Hold A., Holding F.P., Amin N., Martin A.C.L., Rich S.J., Berdini V., Yon J., Acklam P., Burke R., Drouin L., Harmer J.E., Jeganathan F., Van Montfort R.L.M., Newbatt Y., Tortorici M., Westlake M., Wood A., Hoelder S., Heightman T.D. (2016) Structure of the epigenetic oncogene MMSET and inhibition by N-alkyl sinefungin derivatives. ACS Chem. Biol. 11(11), 3093–3105.
Chen D., Dong C., Dong G., Srinivasan K., Min J., Noinaj N., Huang R. (2020) Probing the plasticity in the active site of protein N-terminal methyltransferase 1 using bisubstrate analogues. J. Med. Chem. 63(15), 8419–8431.
Bobiļeva O., Bobrovs R., Kaņepe I., Patetko L., Kalniņš G., Šišovs M., Bula A.L., Grī Nberga S., Borodušķis M.R., Ramata-Stunda A., Rostoks N., Jirgensons A., Tā Rs K., Jaudzems K. (2021) Potent SARS-CoV-2 mRNA cap methyltransferase inhibitors by bioisosteric replacement of methionine in SAM cosubstrate. ACS Med. Chem. Lett. 12(7), 1102–1107.
Bonday Z.Q., Cortez G.S., Grogan M.J., Antonysamy S., Weichert K., Bocchinfuso W.P., Li F., Kennedy S., Li B., Mader M.M., Arrowsmith C.H., Brown P.J., Eram M.S., Szewczyk M.M., Barsyte-Lovejoy D., Vedadi M., Guccione E., Campbell R.M. (2018) LLY-283, a potent and selective inhibitor of arginine methyltransferase 5, PRMT5, with antitumor activity. ACS Med. Chem. Lett. 9(7), 612–617.
Leone G., Voso M.T., Teofili L., Lübbert M. (2003) Inhibitors of DNA methylation in the treatment of hematological malignancies and MDS. Clin. Immunol. 109(1), 89–102.
Silverman L.R. (2004) DNA methyltransferase inhibitors in myelodysplastic syndrome. Best Pract. Res. Clin. Haematol. 17(4), 585–594.
Shimba S., Bokar J.A., Rottman F., Reddy R. (1995) Accurate and efficient N-6-adenosine methylation in spliceosomal U6 small nucelar RNA by HeLa cell extract in vitro. Nucl. Acids Res. 23(13), 2421–2426.
Siegrist J., Netzer J., Mordhorst S., Karst L., Gerhardt S., Einsle O., Richter M., Andexer J.N. (2017) Functional and structural characterisation of a bacterial O-methyltransferase and factors determining regioselectivity. FEBS Lett. 591(2), 312–321.
Golovina A.Y., Sergiev P.V., Golovin A.V., Serebryakova M.V., Demina I., Govorun V.M., Dontsova O.A. (2009) The yfiC gene of E. coli encodes an adenine-N6 methyltransferase that specifically modifies A37 of tRNA1Val(cmo5UAC). RNA. 15(6), 1134–1141.
Mendel M., Chen K.M., Homolka D., Gos P., Pandey R.R., McCarthy A.A., Pillai R.S. (2018) Methylation of structured RNA by the m6A writer METTL16 is essential for mouse embryonic development. Mol. Cell. 71(6), 986–1000.e11.
Rubin R.A., Modrich P. (1977) EcoRI methylase. Physical and catalytic properties of the homogeneous enzyme. J. Biol. Chem. 252(20), 7265–7272.
Hevel J.M., Price O.M. (2020) Rapid and direct measurement of methyltransferase activity in about 30 min. Methods 175, 3–9.
Wu J., Xie N., Feng Y., Zheng Y.G. (2012) Scintillation proximity assay of arginine methylation. J. Biomol. Screening. 17(2), 237–244.
Ero R., Peil L., Liiv A., Remme J. (2008) Identification of pseudouridine methyltransferase in Escherichia coli. RNA. 14(10), 2223–2233.
Liu J., Yue Y., Han D., Wang X., Fu Y., Zhang L., Jia G., Yu M., Lu Z., Deng X., Dai Q., Chen W., He C. (2014) A METTL3–METTL14 complex mediates mammalian nuclear RNA N6-adenosine methylation. Nat. Chem. Biol. 10(2), 93–95.
Morvan D., Demidem A., Guenin S., Madelmont J.C. (2006) Methionine-dependence phenotype of tumors: metabolite profiling in a melanoma model using L-[methyl-13C]methionine and high-resolution magic angle spinning 1H-13C nuclear magnetic resonance spectroscopy. Magn. Reson. Med. 55(5), 984–996.
Lukinavičius G., Lapiene V., Staševskij Z., Dalhoff C., Weinhold E., Klimašauskas S. (2007) Targeted labeling of DNA by methyltransferase-directed transfer of activated groups (mTAG). J. Am. Chem. Soc. 129(10), 2758–2759.
Binda O., Boyce M., Rush J.S., Palaniappan K.K., Bertozzi C.R., Gozani O. (2011) A chemical method for labeling lysine methyltransferase substrates. ChemBioChem. 12(2), 330–334.
Dippe M., Brandt W., Rost H., Porzel A., Schmidt J., Wessjohann L.A. (2015) Rationally engineered variants of S-adenosylmethionine (SAM) synthase: reduced product inhibition and synthesis of artificial cofactor homologues. Chem. Commun. 51(17), 3637–3640.
Ho D.K., Wu J.C., Santi D.V., Floss H.G. (1991) Stereochemical studies of the C-methylation of deoxycytidine catalyzed by HhaI methylase and the N-methylation of deoxyadenosine catalyzed by EcoRI methylase. Arch. Biochem. Biophys. 284(2), 264–269.
Dalhoff C., Lukinavičius G., Klimašauskas S., Weinhold E. (2006) Direct transfer of extended groups from synthetic cofactors by DNA methyltransferases. Nat. Chem. Biol. 2(1), 31–32.
Dalhoff C., Lukinavičius G., Klimašauskas S., Weinhold E. (2006) Synthesis of S-adenosyl-L-methionine analogs and their use for sequence-specific transalkylation of DNA by methyltransferases. Nat. Protocols. 1(4), 1879–1886.
Jalali E., Thorson J.S. (2021) Enzyme-mediated bioorthogonal technologies: catalysts, chemoselective reactions and recent methyltransferase applications. Curr. Opin. Biotechnol. 69, 290–298.
Tomkuvienė M., Mickutė M., Vilkaitis G., Klimašauskas S. (2019) Repurposing enzymatic transferase reactions for targeted labeling and analysis of DNA and RNA. Curr. Opin. Biotechnol. 55, 114–123.
Muttach F., Rentmeister A. (2016) One-pot modification of 5′-capped RNA based on methionine analogs. Methods. 107, 3–9.
Deen J., Vranken C., Leen V., Neely R.K., Janssen K.P.F., Hofkens J. (2017) Methyltransferase-directed labeling of biomolecules and its applications. Angew. Chem. Int. Ed. 56(19), 5182–5200.
Lukinavičius G., Tomkuvienè M., Masevičius V., Klimašauskas S. (2013) Enhanced chemical stability of AdoMet analogues for improved methyltransferase-directed labeling of DNA. ACS Chem. Biol. 8(6), 1134–1139.
Bothwell I.R., Islam K., Chen Y., Zheng W., Blum G., Deng H., Luo M. (2012) Se-adenosyl-L-selenomethionine cofactor analogue as a reporter of protein methylation. J. Am. Chem. Soc. 134(36), 14905–14912.
Lee B.W.K., Sun H.G., Zang T., Ju-Kim B., Alfaro J.F., Zhou Z.S. 2010. Enzyme-catalyzed transfer of a ketone group from an S-adenosyl-L-methionine analogue: a tool for the functional analysis of methyltransferases. J. Am. Chem. Soc. 132(11), 3642–3643.
Peters W., Willnow S., Duisken M., Kleine H., Macherey T., Duncan K.E., Litchfield D.W., Lüscher B., Weinhold E. (2010) Enzymatic site-specific functionalization of protein methyltransferase substrates with alkynes for click labeling. Angew. Chem. Int. Ed. 49(30), 5170–5173.
Huber T.D., Johnson B.R., Zhang J., Thorson J.S. (2016) AdoMet analog synthesis and utilization: current state of the art. Curr. Opin. Biotechnol. 42, 189–197.
Wang R., Zheng W., Yu H., Deng H., Luo M. (2011) Labeling substrates of protein arginine methyltransferase with engineered enzymes and matched S-adenosyl-L-methionine analogues. J. Am. Chem. Soc. 133(20), 7648–7651.
Islam K., Bothwell I., Chen Y., Sengelaub C., Wang R., Deng H., Luo M. (2012) Bioorthogonal profiling of protein methylation using azido derivative of S-adenosyl-L-methionine. J. Am. Chem. Soc. 134(13), 5909–5915.
Islam K., Zheng W., Yu H., Deng H., Luo M. (2011) Expanding cofactor repertoire of protein lysine methyltransferase for substrate labeling. ACS Chem. Biol. 6(7), 679–684.
Kunkel F., Lurz R., Weinhold E. (2015) A 7-deazaadenosylaziridine cofactor for sequence-specific labeling of DNA by the DNA cytosine-c5 methyltransferase M.HhaI. Molecules. 20(11), 20805–20822.
Plotnikova A., Osipenko A., Masevičius V., Vilkaitis G., Klimašauskas S. (2014) Selective covalent labeling of miRNA and siRNA duplexes using HEN1 methyltransferase. J. Am. Chem. Soc. 136(39), 13550–13553.
Flade S., Jasper J., Gieß M., Juhasz M., Dankers A., Kubik G., Koch O., Weinhold E., Summerer D. (2017) The N6-position of adenine is a blind spot for TAL-effectors that enables effective binding of methylated and fluorophore-labeled DNA. ACS Chem. Biol. 12(7), 1719–1725.
Muttach F., Mäsing F., Studer A., Rentmeister A. (2017) New AdoMet analogues as tools for enzymatic transfer of photo-cross-linkers and capturing RNA–protein interactions. Chemistry – A Eur. J. 23(25), 5988–5993.
Holstein J.M., Muttach F., Schiefelbein S.H.H., Rentmeister A. (2017) Dual 5′ cap labeling based on regioselective RNA methyltransferases and bioorthogonal reactions. Chemistry – A Eur. J. 23(25), 6165–6173.
Holstein J.M., Anhäuser L., Rentmeister A. (2016) Modifying the 5′-cap for click reactions of eukaryotic mRNA and to tune translation efficiency in living cells. Angew. Chem. Int. Ed. 55(36), 10899–10903.
Bothwell I.R., Luo M. (2014) Large-scale, protection-free synthesis of Se-adenosyl-l-selenomethionine analogues and their application as cofactor surrogates of methyltransferases. Organic Lett. 16(11), 3056–3059.
Iwig D.F., Grippe A.T., McIntyre T.A., Booker S.J. (2004) Isotope and elemental effects indicate a rate-limiting methyl transfer as the initial step in the reaction catalyzed by Escherichia coli cyclopropane fatty acid synthase. Biochemistry. 43(42), 13510–13524.
Willnow S., Martin M., Lüscher B., Weinhold E. (2012) A selenium-based click adomet analogue for versatile substrate labeling with wild-type protein methyltransferases. ChemBioChem. 13(8), 1167–1173.
Singh S., Zhang J., Huber T.D., Sunkara M., Hurley K., Goff R.D., Wang G., Zhang W., Liu C., Rohr J., Van Lanen S.G., Morris A.J., Thorson J.S. (2014) Facile chemoenzymatic strategies for the synthesis and utilization of S-adenosyl-L-methionine analogues. Angew. Chem. Int. Ed. 53(15), 3965–3969.
Niland C.N., Ghosh A., Cahill S.M., Schramm V.L. (2021) Mechanism and inhibition of human methionine adenosyltransferase 2A. Biochemistry. 60(10), 791–801.
Wang R., Islam K., Liu Y., Zheng W., Tang H., Lailler N., Blum G., Deng H., Luo M. (2013) Profiling genome-wide chromatin methylation with engineered posttranslation apparatus within living cells. J. Am. Chem. Soc. 135(3), 1048–1056.
Lukinavičius G., Lapinaite A., Urbanavičiute G., Gerasimaite R., Klimašauskas S. (2012) Engineering the DNA cytosine-5 methyltransferase reaction for sequence-specific labeling of DNA. Nucl. Acids Res. 40(22), 11594–11602.
Michailidou F., Klöcker N., Cornelissen N.V., Singh R.K., Peters A., Ovcharenko A., Kümmel D., Rentmeister A. (2021) Engineered SAM synthetases for enzymatic generation of AdoMet analogs with photocaging groups and reversible DNA modification in cascade reactions. Angew. Chem. Int. Ed. 60(1), 480–485.
Huber T.D., Clinger J.A., Liu Y., Xu W., Miller M.D., Phillips G.N., Thorson J.S. (2020) Methionine adenosyltransferase engineering to enable bioorthogonal platforms for AdoMet-utilizing enzymes. ACS Chem. Biol. 15(3), 695–705.
van Dülmen M., Muthmann N., Rentmeister A. (2021) Chemo-enzymatic modification of the 5′ Cap maintains translation and increases immunogenic properties of mRNA. Angew. Chem. Int. Ed. 60(24), 13280–13286.
Hong V., Presolski S.I., Ma C., Finn M.G. (2009) Analysis and optimization of copper-catalyzed azide-alkyne cycloaddition for bioconjugation. Angew. Chem. Int. Ed. 48(52), 9879–9883.
Muttach F., Muthmann N., Reichert D., Anhäuser L., Rentmeister A. (2017) A benzylic linker promotes methyltransferase catalyzed norbornene transfer for rapid bioorthogonal tetrazine ligation. Chem. Sci. 8(12), 7947–7953.
Wilkinson A.A., Jagu E., Ubych K., Coulthard S., Rushton A.E., Kennefick J., Su Q., Neely R.K., Fernandez-Trillo P. (2020) Site-selective and rewritable labeling of DNA through enzymatic, reversible, and click chemistries. ACS Cent. Sci. 6(4), 525–534.
Goyvaerts V., Van Snick S., D’Huys L., Vitale R., Helmer Lauer M., Wang S., Leen V., Dehaen W., Hofkens J. (2020) Fluorescent SAM analogues for methyltransferase based DNA labeling. Chem. Commun. 56(22), 3317–3320.
Liu Q., Deiters A. (2014) Optochemical control of deoxyoligonucleotide function via a nucleobase-caging approach. Acc. Chem. Res. 47(1), 45–55.
Hemphill J., Govan J., Uprety R., Tsang M., Deiters A. (2014) Site-specific promoter caging enables optochemical gene activation in cells and animals. J. Am. Chem. Soc. 136(19), 7152–7158.
Vaníková Z., Janoušková M., Kambová M., Krásný L., Hocek, M. (2019) Switching transcription with bacterial RNA polymerase through photocaging, photorelease and phosphorylation reactions in the major groove of DNA. Chem. Sci. 10(14), 3937–3942.
Klose R.J., Zhang Y. (2007) Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 8(4), 307–318.
Heimes M., Kolmar L., Brieke C. (2018) Efficient cosubstrate enzyme pairs for sequence-specific methyltransferase-directed photolabile caging of DNA. Chem. Commun. 54(90), 12718–12721.
Anhäuser L., Muttach F., Rentmeister A. (2018) Reversible modification of DNA by methyltransferase-catalyzed transfer and light-triggered removal of photo-caging groups. Chem. Commun. 54(5), 449–451.
Anhäuser L., Klöcker N., Muttach F., Mäsing F., Špaček P., Studer A., Rentmeister A. (2020) A benzophenone-based photocaging strategy for the N7 position of guanosine. Angew. Chem. Int. Ed. 59(8), 3161–3165.
Park M., Patel N., Keung A.J., Khalil A.S. (2019) Engineering epigenetic regulation using synthetic read-write modules. Cell. 176(1–2), 227–238.e20.
Xie Q., Wu T.P., Gimple R.C., Li Z., Prager B.C., Wu Q., Yu Y., Wang P., Wang Y., Gorkin D.U., Zhang C., Dowiak A.V., Lin K., Zeng C., Sui Y., Kim L.J.Y., Miller T.E., Jiang L., Lee C.H., Huang Z., Fang X., Zhai K., Mack S.C., Sander M., Bao S., Kerstetter-Fogle A.E., Sloan A.E., Xiao A.Z., Rich J.N. (2018) N6-methyladenine DNA modification in glioblastoma. Cell. 175(5), 1228–1243.e20.
Ovcharenko A., Weissenboeck F.P., Rentmeister A. (2021) Tag-free internal RNA labeling and photocaging based on mRNA methyltransferases. Angew. Chem. Int. Ed. 60(8), 4098–4103.
Michailidou F., Rentmeister A. (2021) Harnessing methylation and AdoMet-utilising enzymes for selective modification in cascade reactions. Org. Biomol. Chem. 19(17), 3756–3762.
Pignot M., Siethoff C., Linscheid M., Weinhold E. (1998) Coupling of a nucleoside with DNA by a methyltransferase. Angew. Chem. Int. Ed. 37(20), 2888–2891.
Pljevaljcic G., Pignot M., Weinhold E. (2003) Design of a new fluorescent cofactor for DNA methyltransferases and sequence-specific labeling of DNA. J. Am. Chem. Soc. 125(12), 3486–3492.
Comstock L.R., Rajski S.R. (2005) Conversion of DNA methyltransferases into azidonucleosidyl transferases via synthetic cofactors. Nucl. Acids Res. 33(5), 1644–1652.
Comstock L.R., Rajski S.R. (2005) Methyltransferase-directed DNA strand scission. J. Am. Chem. Soc. 127(41), 14136–14137.
Townsend A.P., Roth S., Williams H.E.L., Stylianou E., Thomas N.R. (2009) New S-adenosyl-L-methionine analogues: synthesis and reactivity studies. Org. Lett. 11(14), 2976–2979.
Weller R.L., Rajski S.R. (2005) DNA methyltransferase-moderated click chemistry. Org. Lett. 7(11), 2141–2144.
Weller R.L., Rajski S.R. (2006) Design, synthesis, and preliminary biological evaluation of a DNA methyltransferase-directed alkylating agent. ChemBioChem. 7(2), 243–245.
Du Y., Hendrick C.E., Frye K.S., Comstock L.R. (2012) Fluorescent DNA labeling by N-mustard analogues of S-adenosyl-L-methionine. ChemBioChem. 13(15), 2225–2233.
Zhang C., Weller R.L., Thorson J.S., Rajski S.R. (2006) Natural product diversification using a non-natural cofactor analogue of S-adenosyl-L-methionine. J. Am. Chem. Soc. 128(9), 2760–2761.
Hymbaugh Bergman S.J., Comstock L.R. (2015) N-mustard analogs of S-adenosyl-L-methionine as biochemical probes of protein arginine methylation. Bioorg. Med. Chem. 23(15), 5050–5055.
Sirasunthorn N., Jailwala A., Gerber A., Comstock L.R. (2019) Evaluation of N-mustard analogues of S-adenosyl-L-methionine with eukaryotic DNA methyltransferase 1. ChemistrySelect. 4(35), 10525–10531.
Hymbaugh S.J., Pecor L.M., Tracy C.M., Comstock L.R. (2019) Protein arginine methyltransferase 1-dependent labeling and isolation of histone H4 through N‑mustard analogues of S-adenosyl-L-methionine. ChemBioChem. 20(3), 379–384.
Coward J.K., Pegg A.E. (1987) Specific multisubstrate adduct inhibitors of aminopropyltransferases and their effect on polyamine biosynthesis in cultured cells. Adv. Enzyme Regulat. 26(C), 107–113.
Польшаков В.И., Батуев Е.А., Манцызов А.Б. (2019) Методы спектроскопии ЯМР для скрининга и изучения взаимодействия биомишень–лиганд. Успехи химии. 88(1), 59–98.
Qu W., Catcott K.C., Zhang K., Liu S., Guo J.J., Ma J., Pablo M., Glick J., Xiu Y., Kenton N., Ma X., Duclos R.I., Zhou Z.S. (2016) Capturing Unknown substrates via in situ formation of tightly bound bisubstrate adducts: S-adenosyl-vinthionine as a functional probe for AdoMet-dependent methyltransferases. J. Am. Chem. Soc. 138(9), 2877–2880.
Dalhoff C., Hüben M., Lenz T., Poot P., Nordhoff E., Köster H., Weinhold E. (2010) Synthesis of S-adenosyl-L-homocysteine capture compounds for selective photoinduced isolation of methyltransferases. ChemBioChem. 11(2), 256–265.
Lenz T., Poot P., Gräbner O., Glinski M., Weinhold E., Dreger M., Köster H. (2010) Profiling of methyltransferases and other S-adenosyl-L-homocysteine-binding proteins by Capture Compound Mass Spectrometry (CCMS). J. Visualized Exp. 46(46), 2264.
Brown L.J., Baranowski M., Wang Y., Schrey A.K., Lenz T., Taverna S.D., Cole P.A., Sefkow M. (2014) Using S-adenosyl-L-homocysteine capture compounds to characterize S-adenosyl-L-methionine and S-adenosyl-L-homocysteine binding proteins. Anal. Biochem. 467, 14–21.
Hong W., Dowden J. (2011) Facile synthesis of N-6 adenosine modified analogue toward S-adenosyl methionine derived probe for protein arginine methyltransferases. Chinese Chem. Lett. 22(12), 1439–1442.
Iyamu I.D., Huang R. (2020) Development of fluorescence polarization-based competition assay for nicotinamide N-methyltransferase. Anal. Biochem. 604, 113833.
Burgos E.S., Walters R.O., Huffman D.M., Shechter D. (2017) A simplified characterization of: S-adenosyl-L-methionine-consuming enzymes with 1-Step EZ-MTase: a universal and straightforward coupled-assay for in vitro and in vivo setting. Chem. Sci. 8(9), 6601–6612.
Ottink O.M., Nelissen F.H.T., Derks Y., Wijmenga S.S., Heus H.A. (2010) Enzymatic stereospecific preparation of fluorescent S-adenosyl-L-methionine analogs. Anal. Biochem. 396(2), 280–283.
Hickey S.F., Hammond M.C. (2014) Structure-guided design of fluorescent S-adenosyl-L methionine analogs for a high-throughput screen to target SAM-I riboswitch RNAs. Chem. Biol. 21(3), 345–356.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология