

Молекулярная биология, 2022, T. 56, № 2, стр. 275-295
РНК-хроматиновый интерактом. Что? Где? Когда?
Г. К. Рябых a, b, *, Д. Е. Мыларщиков a, С. В. Кузнецов c, А. И. Сигорских a, Т. Ю. Пономарёва a, А. А. Жарикова a, b, d, А. А. Миронов a, b
a Факультет биоинженерии и биоинформатики Московского государственного университета им. М.В. Ломоносова
119234 Москва, Россия
b Институт проблем передачи информации им. А.А. Харкевича
127051 Москва, Россия
c ООО Бостонджин
115114 Москва, Россия
d Научно-исследовательский институт физико-химической биологии им. А.Н. Белозерского Московского государственного университета им. М.В. Ломоносова
119992 Москва, Россия
* E-mail: ryabykhgrigory@gmail.com
Поступила в редакцию 20.08.2021
После доработки 13.10.2021
Принята к публикации 14.10.2021
- EDN: COLJSF
- DOI: 10.31857/S002689842202015X
Аннотация
Еще в начале 60-х годов прошлого века заметили, что с хроматином ассоциировано большое количество различных РНК. Что это за РНК и где на хроматине они локализованы? Когда и в каких процессах эти РНК выполняют свою нормальную или патогенную функцию? В данном обзоре мы описываем современные подходы, позволяющие в какой-то мере ответить на эти вопросы. Мы разбираем экспериментальные методы, позволяющие получить полный РНК-хроматиновый интерактом клетки или полногеномные карты взаимодействий отдельных РНК с хроматином, а также методы обработки экспериментальных данных. Мы акцентируем внимание на некодирующих РНК, которые функционируют в тесном контакте с хроматином и ассоциированными с хроматином белковыми комплексами. Разнообразие биологических примеров, полученных с помощью этих методов, не оставляет сомнений в том, что за счет тонкой настройки архитектуры хроматина, приводящей к изменению уровня экспрессии генов, взаимодействующие с хроматином РНК играют важную роль в работе клеточных систем.
ВВЕДЕНИЕ
Известно, что значительная часть генома эукариот транскрибируется с образованием большого количества разнообразных РНК, включая мРНК и такие некодирующие РНК (нкРНК), как микроРНК, малые ядерные и малые ядрышковые РНК, энхансерные РНК, длинные нкРНК и другие [1]. Кодирующие и некодирующие транскрипты могут выполнять свои функции не только в цитоплазме, но и в ядре клетки, где активно участвуют в процессах регуляции транскрипции, а также в ремоделировании и поддержании пространственной структуры хроматина [2]. Классическими примерами таких РНК могут служить MALAT1, NEAT1, XIST, TERC, HOTAIR и другие [2].
Механизмы взаимодействия нкРНК с хроматином, модификаторами хроматина или с другими белками изучают с помощью многочисленных разработанных за последнее время экспериментальных подходов (см. обзор [3]). Среди них можно выделить два класса методов: первый определяет взаимодействия конкретной РНК со всеми локусами хроматина (далее “один-против-всех”) и позволяет установить сайты связывания одной конкретной РНК во всем геноме – карту контактов (RAP [4], CHART-seq [5], ChIRP-seq [6], dChIRP-seq [7], ChOP-seq [8] и CHIRT-seq [9]), в то время как методы второго класса определяют взаимодействия всех РНК со всеми локусами ДНК (далее “все-против-всех”) и позволяют получить данные обо всех потенциальных РНК-хроматиновых взаимодействиях в клетке (MARGI [10], GRID-seq [11], ChAR-seq [12], iMARGI [13, 14], RADICL-seq [15], Red-C [16]). Обзору работ по РНК-хроматиновым взаимодействиям и посвящена настоящая статья.
ЭКСПЕРИМЕНТЫ “ОДИН-ПРОТИВ-ВСЕХ”
В этой части обзора мы рассмотрим работы, посвященные изучению полногеномных взаимодействий 42 нкРНК с хроматином (см. табл. 1) в парадигме “один-против-всех”.
Таблица 1.
РНК, контакты которых с хроматином установлены с помощью методов группы “один-против-всех”
| РНК | Организм | Метод | Поиск ДНК-мотивов | Поиск триплексов | Год, ссылка |
|---|---|---|---|---|---|
| ANRIL | Homo sapiens | ChIRP | MEME SUITE | Triplex Domain Finder | 2021, [17] |
| B2 | Mus musculus | CHART | – | – | 2016, [18] |
| Bloodlinc | M. musculus | ChIRP | MEME | – | 2017, [19] |
| Charme | M. musculus | ChIRP | – | – | 2018, [20] |
| DACOR1 | H. sapiens | ChIRP | – | – | 2015, [21] |
| Dali | M. musculus | CHART | – | Triplexator | 2014, [22] |
| DINO | H. sapiens | ChIRP | – | – | 2016, [23] |
| EPR | M. musculus | ChIRP | HOMER | Triplex Domain Finder | 2020, [24] |
| Eprn | M. musculus | ChIRP | – | – | 2017, [25] |
| ERV-9 | H. sapiens, M. musculus | ChIRP | – | – | 2017, [26] |
| Evx1as | M. musculus | ChIRP | – | – | 2016, [27] |
| Firre | H. sapiens | RAP | – | – | 2014, [28] |
| HAND2-AS1 | H. sapiens | ChIRP | – | – | 2019, [29] |
| Haunt | M. musculus | ChIRP | – | – | 2015, [30] |
| HOTAIR | H. sapiens | ChIRP | MEME | – | 2011, [6] |
| Hotchon | M. musculus | ChIRP | – | – | 2015, [31] |
| HOTTIP | H. sapiens | ChIRP | HOMER | – | 2019, [32] |
| IAP | M. musculus | ChIRP | – | – | 2021, [33] |
| LED | H. sapiens | ChIRP | – | – | 2015, [34] |
| Linc-ASEN | H. sapiens | ChIRP | MEME | – | 2020, [35] |
| lincDUSP | H. sapiens | ChIRP | – | – | 2018, [36] |
| LINE1 | M. musculus | ChIRP | – | – | 2021, [33] |
| lnc-NR2F1 | H. sapiens, M. musculus | ChIRP | MEME | – | 2019, [37] |
| LncHSC-2 | M. musculus | ChIRP | DREME | – | 2015, [38] |
| MALAT1 | H. sapiens, M. musculus | CHART, RAP, ChIRP | MEME-ChIP | – | 2014, [39, 40], 2020, [41] |
| MEG3 | H. sapiens, M. musculus | ChOP, ChIRP | MEME-ChIP | Triplexator | 2015, [8] 2017, [42] |
| NEAT1 | H. sapiens | CHART | MEME-ChIP | – | 2014, [39] |
| Panct1 | M. musculus | ChIRP | – | – | 2017, [43] |
| PAR-TERRA | M. musculus | CHIRT | – | – | 2017, [44] |
| Paupar | M. musculus | CHART | MEME-ChIP | – | 2014, [45] |
| PCGEM1 | H. sapiens | ChIRP | – | – | 2013, [46] |
| roX1 | Drosophila. melanogaster, D. virilis, D. willistoni, D. busckii | dchIRP, ChIRP | – | – | 2014, [7] 2016, [47] |
| roX2 | D. melanogaster, D. virilis, D. willistoni, D. busckii |
CHART, ChIRP | MEME | – | 2011, [5, 6] 2014, [7] 2016, [47] |
| SLEAR | H. sapiens | ChIRP | MEME | – | 2020, [48] |
| SRA | H. sapiens | ChIRP | – | – | 2015 [49] 2020, [50] |
| TERC | H. sapiens | ChIRP | MEME | – | 2011, [6] |
| TERRA | M. musculus | CHIRT | MEME | – | 2017, [9, 44], 2019, [51] |
| Tug1 | M. musculus | ChIRP | MEME-ChIP | – | 2016, [52] |
| U1 | M. musculus | RAP | – | – | 2014, [40] |
| XIST | M. musculus | RAP, CHART | – | – | 2013, [4, 53] 2016, [55] 2019, [56, 57] 2020, [58] |
| 116HG | M. musculus | ChIRP | – | – | 2013, [59] |
| 7SK | H. sapiens, M. musculus | ChIRP | – | – | 2013, [60] 2016, [61] 2021, [62] |
Объектом анализа часто служат РНК, факт взаимодействия которых с хроматином установлен в более ранних работах: XIST [4, 53–58], NEAT1 [39], U1 [40], HOTAIR [6] и другие. Еще один способ выбора объекта исследования основан на дифференциальном анализе транскриптома. Например, поиск нкРНК, специфичных для клеток рака толстой кишки, с помощью дифференциального анализа транскриптома, позволил отобрать потенциальную хроматин-ассоциированную РНК (хаРНК), функционально связанную с этим типом рака – lincDUSP [36].
Экспериментальные подходы к анализу взаимодействий конкретных РНК с хроматином
Группа методов “один-против-всех” (RAP, CHART-seq, ChIRP-seq, dChIRP-seq, ChOP-seq, CHIRT-seq) во многом однородна. Взаимодействия РНК–ДНК фиксируются комбинацией или единственным сшивающим агентом, например, формальдегидом, дисукцинимидилглутаратом, глутаральдегидом или ультрафиолетовым излучением. Хроматин фрагментируется ДНКазой и/или ультразвуком. Сшитые комплексы (РНК-геномная ДНК) захватываются биотинилированными ДНК-олигонуклеотидами, антисмысловыми по отношению к целевой РНК, и отбираются с помощью стрептавидиновых шариков. Далее геномную ДНК обычно элюируют в присутствии РНКазы H и очищают от белков с помощью протеазы K. В конце секвенируют фрагменты геномной ДНК, ранее сшитые с целевой РНК, и анализируют полученные данные (рис. 1).
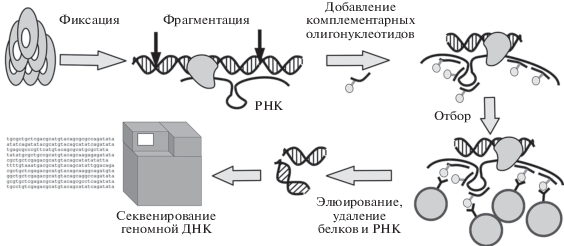
Доступные участки целевой РНК, как правило, заранее неизвестны, поэтому подбор антисмысловых зондов является одной из главных проблем методов “один-против-всех”. Например, для решения этой проблемы в методе CHART-seq предлагается использовать малое количество коротких зондов, которые в комплексе с РНК обладают высокой чувствительностью к РНКазе Н [5]. Этот отбор позволяет выявить наиболее доступные для гибридизации зондов области РНК. Напротив, в методе ChIRP-seq предлагается использовать выстилающие наборы зондов, покрывающие РНК без пересечений (в случае HOTAIR зонды покрыли около 50% ее последовательности) [6]. Исключением стала многокопийная РНК TERRA, содержащая повторяющиеся субтеломерные последовательности, что делало затруднительным использование выстилающих зондов. Метод CHIRT-seq, объединивший и оптимизировавший методы ChIRP-seq и CHART-seq [9], решил данную проблему, использовав для специфического захвата TERRA один олигонуклеотид, комплементарный теломерному повтору. Отметим, что все методы, борясь с неспецифическими взаимодействиями зондов, отбраковывают зонды, которые оказались комплементарными другим РНК или участкам геномной ДНК.
Если локус контактов целевой РНК известен заранее, то поиск ее контактов можно ограничить этим локусом, амплифицировав ДНК только этого локуса (метод ChOP-qPCR), как в случае управляющей импринтингом нкРНК Kcnq1ot1 [63]. Аналогичным образом с помощью CHART-qPCR проанализированы РНК STEEL [64], а с помощью ChIRP-qPCR – FMR1 [65].
Домены РНК, ответственные за взаимодействие с хроматином, можно выявлять с использованием делеционного анализа (в ряде работ из XIST удаляли повторы B [56], E [58] и A [4, 54], а из ANRIL – экзон 8 [17]) или домен-специфичного метода dChIRP-seq, как в случае roX1 [7].
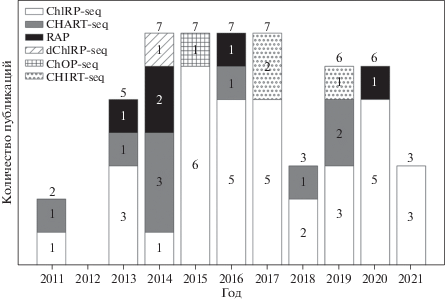
Популярность экспериментальных протоколов менялась со временем. В настоящее время в большинстве работ используют протокол ChIRP-seq (рис. 2).
Основными задачами обработки данных “один-против-всех” являются: во-первых, выделение участков генома, обогащенных контактами данной РНК с хроматином (далее “пики”), во-вторых, фильтрация специфичных пиков от артефактных.
Типичный конвейер обработки данных “один-против-всех” состоит из следующих шагов: удаление ПЦР-дубликатов и картирование прочтений на геном; выделение участков со статистически значимым превышением покрытия над покрытием фона (определение пиков) и/или нормализация на фон; поиск и фильтрация пиков.
В связи с особенностью решаемой биологической задачи в некоторых работах выполняли аллель-специфичное картирование [53, 56]. Чаще всего при не аллель-специфичном выравнивании оставляли уникально картированные на геном прочтения.
Для учета артефактных сигналов используют сравнение с фоновыми данными (“input”), а также фильтрацию с помощью данных, полученных в контрольных экспериментах: геномную ДНК элюируют без добавления РНКазы Н [9] или образец перед выделением обрабатывают РНКазой [37]; используют зонды, не имеющие отношения к целевой РНК, например, комплементарные зондам основного эксперимента [8] или антисмысловые зонды к РНК, которые не транскрибируются в данной клеточной культуре, например, к РНК из другого организма – мРНК гена LacZ [38]. В качестве контроля используют также клеточные линии, в которых изучаемая РНК либо не экспрессировалась [31], либо ее ген был “нокаутирован” [30].
В большинстве работ пики определяли с использованием программы MACS [66], первоначально разработанной для поиска пиков в данных ChIP-seq. Однако использовали также такие программы, как SICER [67], HOMER [68] и SPP [69]. В зависимости от целевой РНК количество обнаруженных пиков варьировало от сотен до десятков тысяч. Любопытно, что при использовании в качестве фона контрольных данных в некоторых работах [5, 22] получены пики, отличные от пиков, полученных при нормировании на “input”, в то время как в [44] показана слабая зависимость между полученными пиками и примененной фоновой моделью.
Для фильтрации случайного шума также применяют биологические реплики с четными и нечетными зондами. Основной конвейер обработки таких данных состоит из генерации консенсусного трека для четных и нечетных зондов и фильтрации пиков по порогам обогащения, покрытия и корреляции между четными и нечетными данными (подробнее этот подход описан в [6]).
Другой подход к использованию контрольных данных для исключения неспецифических пиков состоит в том, чтобы отфильтровать те пики, которые попадают в области, обогащенные пиками из контрольных экспериментов (со смысловыми зондами, зондами против LacZ, с предобработкой РНКзой) [19, 29].
Определение пиков проводят не всегда – в некоторых случаях треки покрытия сглаживают с помощью SPP, после чего нормируют на фон и/или треки, полученные из других экспериментов или с другого аллеля [55, 56].
Отметим, что РНК может взаимодействовать с геномной ДНК несколькими способами. Напрямую – формируя такие структуры, как РНК–ДНК-триплексы (при этом образуются хугстиновские связи между РНК и ДНК) и R-петли (уотсон–криковское спаривание РНК–ДНК), или через белкового посредника [3].
Поиск прямого взаимодействия некодирующих РНК с ДНК можно проводить с использованием нескольких подходов как независимо, так и в комбинации. Простейший подход ищет потенциальные комплементарные взаимодействия с ДНК в местах, ассоциированных с пиками РНК-хроматиновых контактов. Однако применение, например, алгоритма EMBOSS Water [70] не выявило значительной комплементарности между транскриптами Paupar и Dali и их соответствующими контактами [22, 45]. С помощью программы Triplexator [71] показано, что РНК Dali не образует триплексные структуры РНК-ДНК [22]. Таким образом, РНК Paupar и Dali, вероятно, не связываются с ДНК напрямую. С другой стороны, с помощью программы Triplexator обнаружены потенциальные триплексы у MEG3 и HOTAIR [8], а “Triplex Domain Finder” [72] – у EPR [24] и ANRIL [17]. Здесь следует отметить, что программы поиска потенциальных триплексов имеют невысокое качество предсказания [73].
Для обнаружения потенциального белкового партнера проводят de novo анализ ДНК-мотивов по данным связывания изучаемой РНК с хроматином методами MEME, DREME, MEME-ChIP, MEME SUITE [74] и HOMER [68] (см. табл. 1). Далее обогащенные мотивы можно сравнивать с известными мотивами ДНК-связывающих белков, например, с помощью TOMTOM [75]. В нкРНК LncHSC-2 [38] обнаружен мотив связывания изоформы гемопоэтического фактора транскрипции E2A. Показано пересечение пиков связывания в данных ChIP-seq E2A и ChIRP-seq LncHSC-2. Сделанные наблюдения, а также результаты нокдауна гена E2A, показали, что LncHSC-2 отвечает за связывание E2A на некоторых сайтах-мишенях.
Поиск ДНК-мотивов может дать неожиданный результат. Например, в нкРНК MEG3 [8], HOTAIR, roX2 [6], TERRA [9], ANRIL [17], linc-ASEN [35] и Tug1 [52] идентифицированы GA-богатые ДНК-мотивы, которые, как известно, могут образовывать триплексные структуры [76–78]. Это указывает на потенциально важную роль GA-богатых мотивов в полногеномном таргетинге длинных нкРНК. И если взглянуть на MEG3, то выстраивается цельная картина: за нацеливание на хроматин отвечает одна часть РНК, а за привлечение EZH2 (часть комплекса PRC2), с помощью которой происходит модификация хроматина, отвечает совсем другая часть РНК MEG3 [8].
Полученные пики можно связать с регуляторными областями генов и другими геномными областями с помощью программ GREAT [79], CEAS [80] (lincDUSP [36], TERRA [9, 44]). Гены, ассоциированные с контактами, можно охарактеризовать по их принадлежности к тому или иному процессу, выполнив анализ онтологий генов (GO-анализ, программы Panther [81], DAVID [82], QuickGO [83], Enrichr [84]). В дальнейшем анализе данные “один-против-всех” можно сравнивать с другими экспериментальными данными (например, с GRID-seq, ChIP-seq, RNA-seq, Hi-C и др.) и геномными аннотациями (SINE, LINE, CpG-острова, цвета хроматина, энхансеры и т.д.). Например, сравнение с данными RNA-seq после нокдауна или сверхэкспрессии изучаемой РНК (ANRIL [17], lnc-NR2F1 [37], Bloodlinc [19], EPR [24], LED [34]) позволяет определить первичные мишени среди генов, чья экспрессия значительно изменялась при манипулировании изучаемой РНК. Другой вариант последующего анализа: определение колокализации сайтов связывания нкРНК с сайтами связывания белков или белковых комплексов, способствующих активации транскрипции (например, MSL, Mediator, PGC-1α, факторы транскрипции bHLH, INO80 и др.), метками активного хроматина (H3K4me3, H3K4me1, H3K27ac, H3K9ac, РНК-полимераза II и др.), а также ассоциация сайтов связывания нкРНК с репрессирующими гистоновыми метками. Такие стратегии позволили показать, что нкРНК roX [5, 6], Dali [22], Hotchon [31], Evx1as [27], Tug1 [52], lnc-NR2F1 [37], HAND2-AS1 [29], ERV-9 [26], Charme [20] и LED [34] являются активирующими. С другой стороны, обогащение репрессирующими гистоновыми метками, колокализация с сайтами связывания PRC1/PRC2 (или их субъединицами), BAF, P-TEFb и другими белковыми партнерами свидетельствуют о том, что XIST [4, 53–58] HOTAIR [6], MEG3 [8], Linc-ASEN [35], 7SK [61] относятся к репрессирующим РНК. Однако это вовсе не значит, что РНК может быть только активирующей или только репрессирующей. Например, в зависимости от образования комплекса с TrxG или с PRC2 нкРНК SRA будет способствовать установлению либо активирующих (H3K4me3), либо репрессирующих (H3K27me3) гистоновых меток в соответствующем геномном локусе и в зависимости от этого будет выполнять либо активирующую, либо репрессирующую функцию [49]. Paupar [45], ANRIL [17], TERRA [9, 51] также ведут себя двойственно.
Данные “один-против-всех” часто используются для поиска различий между состояниями: между ортологами roX1 и roX2 нескольких видов Drosophila [47], до и после теплового шока (нкРНК B2 мыши) [18], нокдауна белкового партнера РНК HOTAIR, EZH2 [6], между разными временны́ми точками дифференциации клеток (XIST [4], Evx1as [27], PAR-TERRA [44], ERV-9 [26]), до и после дополнительной обработки клеток ингибитором элонгации транскрипции флавопиридолом (MALAT1, U1 [40]) или ингибитором бромодомена JQ1 (7SK [61]).
В отличие от белоккодирующих генов, гены длинных нкРНК часто менее консервативны на уровне последовательности [85, 86], однако другие характеристики нкРНК: синтеничные отношения с соседними генами, сходство коротких фрагментов последовательностей и вторичная структура – часто остаются консервативными (roX1/2 [47]). Важно отметить, что среди разбираемых в данном обзоре нкРНК встречаются как неконсервативные (DACOR1 [21]), так и консервативные РНК (Dali [22], Paupar [45], Firre [28], Bloodlinc [19], DINO [23], Charme [20], HAND2-AS1 [29], lnc-NR2F1 [37], EPR [24], Eprn [25], 7SK [61], MALAT1, NEAT1 [39]).
Биологические примеры
Дозовая компенсация X-хромосомы у Drosophila, roX1/2. Рибонуклеопротеин MSL (male-specific lethal), в состав которого входят нкРНК roX2 и roX1, обеспечивает эквивалентную экспрессию генов, расположенных на Х-хромосоме, между мужскими (XY) и женскими (XX) особями Drosophila за счет увеличения транскрипции с единственной мужской Х-хромосомы примерно в 2 раза [87]. Полученные с помощью методов CHART-seq и ChIRP-seq в 2011 году полногеномные карты контактов roX2 с хроматином подтвердили, что roX2 в основном локализуется на Х-хромосоме и связывается в тех же участках хроматина, как и белок MSL3 (субъединица MSL-комплекса) [5, 6]. Анализ ДНК-мотивов roX2 в данных ChIRP-seq выявил мотив, почти идентичный мотиву MSL [6]. Плотность контактов roX2 увеличивается с 5′- до 3′-конца каждого гена на Х-хромосоме, что соответствует представлению о том, что комплекс roX-MSL усиливает элонгацию, а не инициацию транскрипции [88].
Модификация метода ChIRP-seq (dChIRP-seq – “domain-specific chromatin isolation by RNA purification” [7]) позволила уточнить, какие именно домены РНК roX1 участвуют в MSL- и CLAMP-опосредованном взаимодействии с хроматином. Карты связывания другой нкРНК, roX1 [89], показали схожие сайты связывания с хроматином, что подтверждает ранее известные данные о функциональной избыточности и взаимозаменяемости roX1 и roX2. Сравнение dChIRP-seq roX1 и ChIRP-seq roX2 с данными Hi-C позволили предложить модель конформации Х-хромосомы, в которой локус roX2 и некоторые CES-сайты (специфические сайты на Х-хромосоме, с которыми взаимодействуют обе РНК и MSL-белки) кластеризуются в территорию дозовой компенсации, в то время как roX1-локус находится вне этой территории [7].
Поиск ортологов roX1 и roX2 выявил 19 и 28 новых ортологов соответственно, у 35 видов рода Drosophila, эволюционно разошедшихся около 40 млн лет назад [47]. Сравнительный анализ данных ChIRP-seq по взаимодействию ортологов roX1 и roX2 с хроматином у D. melanogaster, D. willistoni, D. virilis и D. busckii показал, что сайты связывания roX1/2 ассоциированы с Х-хромосомой и в большинстве своем эволюционно динамичны. Обнаружено, что новые сайты могут возникать из интронных полипиримидиновых трактов. Более детальный анализ показал, что для сайтов связывания roX1/2 важна близость, а не точное расположение относительно генов. В случае слияния аутосомы с Х-хромосомой и образования нео-Х-хромосомы (как у D. willistoni) новые Х‑сцепленные гены также могут подвергаться дозовой компенсации.
Дозовая компенсация X-хромосомы у млекопитающих, XIST. Инактивация Х-хромосомы (“X-chromosome inactivation”, или XCI) – это механизм регулирования экспрессии Х-сцепленных генов у женских особей млекопитающих, позволяющий им компенсировать удвоенное количество половых хромосом [90–92]. В процессе XCI неактивная Х-хромосома (Xi) подвергается реконфигурации в уникальную структуру – тельце Барра. Центральный игрок этого процесса – длинная нкРНК XIST, которая распространяется вдоль Xi, привлекая факторы сайленсинга и индуцируя глобальное подавление топологически ассоциированных доменов (ТАД) [56, 93–95]. Она может выполнять функцию каркаса в сборке репрессивных комплексов, таких как Polycomb (PRC1/PRC2), которые моноубиквитинируют гистон H2A по лизину 119 (H2AK119ub) и триметилируют гистон H3 по лизину 27 (H3K27me3) [95–98], или формировать репрессивный компартмент, например, взаимодействуя непосредственно с рецептором ламина B (LBR) и привлекая Xi в ядерную ламину [54].
В 2013 году были опубликованы две работы, в которых методы “один-против-всех” (RAP [4] и CHART-seq [53]) впервые применили для изучения распространения РНК XIST по Х-хромосоме. Экспериментальные данные получены для нескольких временных точек: перед XCI (эмбриональные стволовые клетки мыши, ESC), после завершения XCI (терминально дифференцированные фибробласты) и три и две промежуточные стадии соответственно. Как и ожидалось, чем ближе клеточная линия приближалась к терминально дифференциированной стадии, тем больше становилось XIST-обогащенных сегментов на Х-хромосоме, тем больше была плотность контактов на этих сегментах по сравнению с плотностью на аутосомах. Проанализировав корреляции между профилями распространения XIST вдоль Xi (по стадиям развития) и геномными и хроматиновыми аннотациями, пришли к выводу, что во время de novo XCI XIST сначала распространяется по богатым генами регионам (“ранние” домены). Любопытно, что XIST с большей вероятностью нацелена на гены в областях активного хроматина, а затем распространяется в области, более бедные генами (“поздние” домены), причем XIST и PRC2 совместно мигрируют в новые регионы внутри “ранних” доменов Xi. Показано, что именно конформация хромосомы, а не аффинность “ранних” доменов играет важную роль в определении ранних сайтов локализации XIST на Хi при de novo XCI [4]. Чтобы выяснить, как распространяется XIST по Xi после завершения XCI, проанализировали распространение XIST по Xi в клеточной линии MEF (эмбриональные фибробласты мыши) через 1, 3 и 8 ч после удаления XIST с помощью заблокированных нуклеиновых кислот LNA-C1 и LNA-4978. Оказалось, что во время поддержания Xi в соматических клетках XIST распространяется как на “ранние”, так и на “поздние” домены одновременно [53]. Таким образом, РНК XIST распространяется по Х-хромосоме по двум разным механизмам в зависимости от стадии: de novo XCI или поддержания XCI.
РНК XIST имеет доменную структуру из консервативных повторяющихся мотивов (повторы A–F) [99], роль которых в распространении XIST известна не до конца. Для выяснения роли этих доменов применили делеционный анализ. После удаления повтора E паттерны распространения данной РНК по Xi на 3-й день дифференцировки ESC сильно коррелировали с паттернами XIST дикого типа, однако на 7-й день микроскопическими методами (FISH и 3D-SIM) выявили значительную дисперсию XIST-ΔE внутри ядра по сравнению с диким типом. Это показывает, что повтор E не влияет на начальное распространение XIST по Хi, но необходим на более поздних стадиях XCI [58].
С помощью методов ChIP-seq и CHART-seq показано [56], что удаление повтора B в клеточной линии MEF (процесс XCI завершен), необходимого XIST для взаимодействия с белком HNRNPK и привлечения через него PRC1 и PRC2, вызывает истощение покрытия XIST и почти полную потерю меток H2AK119ub и H3K27me3 на Xi. Причем дефект распространения XIST связан с потерей PRC1 и PRC2, что подтверждают данные независимых двойных нокаутов по компонентам PRC1 и PRC2. Также показано, что повтор A, с которым взаимодействует PRC2 [97], необходим XIST на начальных этапах XCI для подавления экспрессии генов в “ранних” доменах и изменения структуры хромосомы [4]. Так, с повтором A напрямую взаимодействует LBR [95, 100] при de novo XCI, что способствует привлечению покрытой XIST ДНК в ядерную ламину и распространению XIST на другие участки Х-хромосомы, которые физически стали ближе к локусу транскрипции XIST [54].
В дальнейшем была опубликована модель пошагового сворачивания Xi. При de novo XCI XIST сначала распространяется по богатым генами “ранним” доменам (которые сильно коррелируют с A-компартментами укладки хроматина), а далее по “поздним” доменам (B-компартменты). В этот момент A/B-компартменты XIST-зависимо ремоделируются в S1/S2-компартменты (специфичные только для Xi). Далее архитектурный белок SMCHD1 ослабляет (но не удаляет полностью) структуру ТАДов Xi, объединяет S1/S2 и формирует два мегадомена, разделенных Dxz4-локусом. Удаление SMCHD1 в клетках с завершенной XCI вызывает повторное появление S1/S2-компартментов и обогащение XIST в S1 (но не в S2), что приводит к аберрантному обогащению H3K27me3 на Xi, сегментарной эрозии гетерохроматина и неудачному подавлению экспрессии генов. Более того, истощение XIST, HNRNPK или PRC1 препятствует S1/S2-компартментализации хроматина [55, 57].
TERRA. На теломерах активно синтезируется гетерогенная популяция длинных нкРНК, получивших название TERRA. Известно, что TERRA является неотъемлемой частью архитектуры теломер. Однако цитологические исследования показали, что только около половины обнаруживаемых транскриптов TERRA локализованы в теломерах. С помощью CHIRT-seq удалось показать, что TERRA связывает хроматиновые мишени по всему геному: как в цис- (на теломерах), так и в транс-положениях [9, 51].
С помощью CHIRT-seq на женских клетках ESC, MEF и двух промежуточных стадиях обнаружена субпопуляция TERRA, транскрибируемая из псевдоаутосомных регионов (PAR) Х-хромосом – PAR-TERRA [44]. Оказалось, что РНК PAR-TERRA необходима для гомологичного спаривания Х-хромосом (X–X) и собственно для процесса XCI, который управляется Xic (X-inactivation center) через нкРНК XITE, TSIX и XIST. Во время раннего развития счетный механизм определяет количество Х-хромосом и инициирует XCI в клетках с двумя или более Х-хромосомами. XIST инициирует сайленсинг всей случайно выбранной хромосомы Xi, тогда как TSIX противодействует XCI на оставшейся активной Xa. Переход от двухаллельной к моноаллельной экспрессии гена TSIX фиксирует выбор аллеля. Переход TSIX в моноаллельное состояние происходит во время гомологичного спаривания между двумя аллелями Xic. Показано, что PAR-TERRA взаимодействует с Xic в течение всего периода спаривания Xic-Xic, а взаимодействия Xic-Xic, PAR-PAR и Xic-PAR регулируются PAR-TERRA.
Уточнение молекулярно-биологического механизма действия NEAT1 и MALAT1. Известно, что высококонсервативная нкРНК MALAT1 локализуется в ядерных спеклах [101], взаимодействует с серин/аргинин-богатыми белками, участвующими в сплайсинге, и может регулировать экспрессию генов и альтернативный сплайсинг [102]. Для более детального изучения молекулярной функции MALAT1 в 2014 году с помощью методов RAP–RNA и RAP–DNA получены полногеномные карты контактов MALAT1 с другими РНК и с хроматином соответственно [40]. Локализация MALAT1 на хроматине оказалась сильно связанной с активно транскрибируемыми генами, причем наибольшее обогащение MALAT1 наблюдалось примерно на 500 н. ниже аннотированного сигнала полиаденилирования. Эксперимент RAP–DNA при подавленной элонгации транскрипции показал, что обогащение MALAT1 в ранее активных генах значительно снижено, т.е. локализация MALAT1 на хроматине зависит от транскрипции его ДНК-мишеней. Кроме того, локализация MALAT1 на хроматине зависит от экзон-интронной структуры генов: мультиэкзонные и альтернативно сплайсируемые гены наиболее обогащены контактами MALAT1. Одноэкзонные гены негистоновых белков менее обогащены, а гены гистонов редко контактируют с MALAT1.
Длинная нкРНК NEAT1 входит в состав ядерных параспеклей [103] и участвует в регуляции транскрипции [104, 105]. Она, как и MALAT1, взаимодействует с сотнями активных генов, причем со многими эти РНК взаимодействуют совместно [39], однако их паттерны связывания различаются: NEAT1 связывает TSS и TTS (сайты инициации и терминации транскрипции) генов, а MALAT1 – тела генов и TTS. Дополнительные эксперименты CHART-seq с подавлением элонгации транскрипции показали, что NEAT1 более заметно реагирует на изменение уровня транскрипции его ДНК-мишеней, чем MALAT1 – ингибирование элонгации транскрипции ведет к отчетливому смещению локализации NEAT1 в направлении сайтов инициации транскрипции [39].
U1. В 2010 году заметили, что сплайсосомная РНК U1 связана с другим процессом – защитой транскриптома клетки. U1 предотвращает преждевременное расщепление и полиаденилирование (PCPA) зарождающихся транскриптов [106]. В 2014 году с помощью методов RAP–RNA и RAP–DNA получены полногеномные карты контактов U1 с другими РНК и с хроматином соответственно [40]. Оказалось, что локализация U1 на хроматине ассоциирована с транскрипцией ДНК-мишеней, при этом 5′- и 3′-концы активных генов обогащены контактами U1 больше, чем их тела. После подавления элонгации транскрипции локализация U1 изменилась: контакты U1 на 3′‑концах генов в значительной степени исчезли, но на 5′-концах генов обогащение U1 сохранялось. Предполагается, что U1 локализуется на хроматине двумя способами: 1) U1 связывается с зарождающимися РНК, защищая транскриптом от PCPA, что отражается в обогащении 3′-концов генов контактами U1; 2) U1 связывается с 5′-концами генов по механизму, который не зависит от элонгации транскрипции, и поэтому может участвовать в регуляции инициации транскрипции [40].
Ингибирование транскрипции и истощение SNRNP70 (компонента малого ядерного рибонуклеопротеина U1) на 90% приводит к снижению связывания MALAT1 с активными генами (данные ChIRP-seq [38]), что подтверждает гипотезу о том, что связанный с активной РНК-полимеразой II малый ядерный рибонуклеопротеин U1 прямо влияет на локализацию некоторых длинных нкРНК на хроматине [41].
HOTAIR. Длинная межгенная нкРНК HOTAIR транскрибируется из HOXC-локуса, может связывать комплекс PRC2 и влиять на степень его обогащения на генах-мишенях по всему геному [107–109]. Как HOTAIR направляет PRC2 к генам-мишеням? Чтобы понять это, в 2011 году с помощью ChIRP-seq получены карты контактов HOTAIR с хроматином на клетках рака молочной железы [6]. Обнаружены 832 сайта связывания HOTAIR с хроматином, которые располагались в основном в энхансерах и интронах, а также ассоциировались с сайтами связывания субъединиц PRC2 (EZH2 и SUZ12). Далее провели ChIRP-seq HOTAIR на клетках MDA-231, истощенных по EZH2-субъединице, которая непосредственно связывается с HOTAIR. Оказалось, что паттерн локализации HOTAIR на хроматине в значительной степени сохранился, что указывает на способность HOTAIR связываться с хроматином без PRC2. В совокупности эти результаты подтверждают, что HOTAIR может активно привлекать комплексы, модифицирующие хроматин.
lnc-NR2F1. Ген длинной нкРНК lnc-NR2F1 (или NR2F1-AS1) располагается в геноме рядом с белоккодирующим геном NR2F1, который кодирует фактор транскрипции, участвующий в нейрогенезе. Последовательность lnc-NR2F1 удивительно консервативна у мыши и человека, что не характерно для длинных нкРНК. Данные ChIRP-seq для lnc-NR2F1 мыши анализировали по стандартной схеме [37]: GREAT-анализ выявил ассоциации пиков с конкретными генами, а GO-анализ отобранных генов показал обогащение этого набора генами, вовлеченными в нейронные процессы. Сравнение карт контактов lnc-NR2F1 с общедоступными данными ChIP-seq показало, что пики lnc-NR2F1 колокализуются с энхансерными хроматиновыми метками H3K27ac и H3K4me3. Согласно анализу ДНК-мотивов сайтов связывания lnc-NR2F1, подобные мотивы найдены у некоторых факторов транскрипции, связанных с нейрогенезом (Ngn2 и Ascl1).
Так как lnc-NR2F1 человека имеет несколько изоформ, предположили, что разные домены lnc-NR2F1 могут иметь различную локализацию на хроматине. С помощью ChIRP-seq картированы контакты двух изоформ lnc-NR2F1: короткой и длинной [37]. Число сайтов связывания длинной изоформы оказалось примерно в 10 раз больше, чем короткой: 4404 против 415, причем эти множества сайтов слабо перекрывались. Это, а также GO-анализ генов-мишеней, согласуется с гипотезой о том, что длинная изоформа участвует в нейрогенезе [37].
Консервативность lnc-NR2F1, сходство множеств генов человека и мыши, ассоциированных с контактами этой РНК, а также соответствующих мотивов ДНК указывают на возможные консервативные функции lnc-NR2F1 у мыши и человека [37].
Firre. Длинная межгенная нкРНК Firre локализуется на Х-хромосоме и взаимодействует с ядерным матричным фактором hnRNPU через повторяющиеся последовательности. Заметное обогащение локуса Firre сайтами связывания CTCF и меткой H3K4me3, а также сниженное число сайтов связывания XIST, LaminB1 и метки H3K27me3 в этом локусе свидетельствуют о том, что этот ген избегает инактивации Х-хромосомы [28]. Используя метод RAP, определили сайты связывания Firre с хроматином и обнаружили значительно обогащенные пики на некоторых хромосомах, однако преобладал сигнал в области ~5 млн п.н. вокруг сайта транскрипции. Дополнительные FISH-эксперименты подтвердили фокусную локализацию Firre в ее собственном локусе. По-видимому, Firre может служить локальным организующим фактором для топологического сближения транс-сайтов и геномного локуса.
SRA. Колокализация активной гистоновой метки H3K4me3 и репрессирующей метки H3K27me3 определяет бивалентные сайты, которые маркируют гены, готовые к индукции экспрессии, в том числе гены, участвующие в дифференцировке плюрипотентных стволовых клеток. За метилирование H3K4 и H3K27 ответственны белки группы Trithorax (TrxG) и PRC2 соответственно. Длинная нкРНК SRA (Steroid Receptor RNA Activator) может связываться как с TrxG, так и с PRC2. Используя метод ChIRP-seq, в геноме плюрипотентных стволовых клеток человека [49] идентифицировали 7899 сайтов связывания SRA, из которых 1570 несли метку H3K4me3, 735 – H3K27me3 и 894 сайта имели сигнатуру бивалентного домена (8% от всех картированных бивалентных доменов). Отсюда следует, что в зависимости от сайта SRA может доставлять одну или обе эти модификации. Пересекая сайты связывания SRA с сайтами p68 (SRA-ассоциированная РНК-хеликаза), CTCF, NANOG, предположили, что CTCF способствует установлению бивалентных состояний в местах, где также присутствует SRA: NANOG привлекает на хроматин SRA и связанные с ним комплексы TrxG и PRC2, а p68 облегчает SRA-опосредованное метилирование H3K4.
Выполнив ChIRP-seq на клеточной линии K562 эритробластов человека [50], обнаружили 2790 сайтов связывания SRA по всему геному, большинство из которых располагались на расстоянии до 50 т.п.н. от сайта инициации транскрипции (TSS). Анализ транскриптома показывает, что SRA способствует экспрессии эритроид-ассоциированных генов и при этом действует как репрессор генов, ассоциированных с лейкоцитами. Кроме того, подавление SRA с помощью малых шпилечных РНК снижает экспрессию эритроид-специфических маркеров TFRC и GYPA и подавляет экспрессию глобиновых генов. Таким образом показано, что нкРНК SRA способствует транскрипции эритроидных генов.
DACOR1. Для эпигенома опухолевых клеток характерна глобальная потеря метилирования ДНК, которое вносит вклад в геномную нестабильность и аберрантную экспрессию генов. Длинная нкРНК DACOR1 взаимодействует с поддерживающей ДНК-метилтрансферазой DNMT1 в клеточной линии HCT116 (рак толстой кишки), экспрессируется на высоком уровне в нормальных клетках здоровой толстой кишки и на сниженном – в клонах раковых клеток толстой кишки. С помощью ChIRP-seq найдено 338 сайтов связывания DACOR1 с хроматином: 161 сайт располагается около 150 аннотированных генов и 177 в межгенных областях. При сравнении сайтов связывания DACOR1, расположенных рядом с аннотированными генами, с дифференциально метилированными областями (DMR), определенными в когорте опухолей толстой кишки и в соответствующих нормальных тканях, выявили перекрывание 31 сайта с DMR. Эти результаты показывают, что, взаимодействуя как с DNMT1, так и с хроматином, DACOR1 потенциально привлекает и/или собирает DNMT1-комплекс в определенных геномных локусах для регуляции эпигенетических модификаций и, следовательно, экспрессии конкретных генов и путей [21].
p53-связанные РНК: LED, DINO, SLEAR. Белок p53 связывается с энхансерами, чтобы регулировать ключевые гены-мишени. Картирование 6270 р53-регулируемых энхансеров показало, что большинство из них не содержит сайтов связывания р53. Поскольку длинные нкРНК являются важными регуляторами динамики хроматина, предположили, что p53-индуцированные длинные нкРНК вносят вклад в активацию энхансеров с помощью p53 [34]. Из индуцированных р53 нкРНК были выбраны три – RP3-510D11.2, loc643401 и LED, но только подавление LED ослабляло функцию р53. Выполнив ChIRP-seq, получили 1698 сайтов связывания LED с хроматином, которые затем сопоставили с аннотацией состояний хроматина [110]. Сайты связывания LED присутствуют во всех состояниях хроматина, однако значительное обогащение наблюдается в сильных энхансерах. Дальнейшее сопоставление LED-ассоциированных энхансеров с данными GRO-seq, гистоновыми метками, а также с изменением уровней гистоновых меток после нокдауна LED позволило сделать вывод, что LED регулирует энхансеры, влияя на установку активной энхансерной гистоновой метки H3K9ac. Показано также, что длинные нкРНК DINO [23] и SLEAR [48] ассоциированы с p53 и непосредственно участвуют в связывании p53 с некоторыми из его генов-мишеней.
РНК, регулирующие гены HOX: Haunt, HOTTIP. Идентифицировав длинные нкРНК, участвующие в плюрипотентности и дифференцировке эмбриональных стволовых клеток (ESC), на примере РНК Haunt постарались определить функции ESC-специфичных нкРНК [30]. Ген Haunt расположен на ~40 т.п.н. выше кластера HOXA. Помимо нокаута, нокдауна и сверхэкспрессии Haunt с последующим анализом дифференциальной экспрессии генов и других экспериментов выполнен ChIRP-seq для РНК Haunt. В недифференцированных ESC контакты Haunt непрерывно покрывали геномную область размером 1 млн п.н. с сигналами, сосредоточенными между ближайшими к гену Haunt HOXA-кластером и геном Skap2. Сигналы ChIRP-seq резко снижались ниже локуса Haunt в непосредственной близости от гена Hoxa1 и уменьшались дальше от 5'- к 3'-концу кластера HOXA, что сопровождалось появлением длинного участка репрессивных меток H3K27me3, покрывающих всю область HOXA (105 т.п.н.). Не обнаружено никаких значительных пиков Haunt за пределами ее гена и близлежащих локусов, что подтверждает цис-функцию Haunt на хроматине. После добавления ретиноевой кислоты (морфоген, регулирующий активацию генов HOX in vivo и in vitro) РНК Haunt стала чаще контактировать с 5'-стороной области HOXA с пиком в локусах Hoxa5 и Hoxa6, показывая 6–7-кратное увеличение плотности контактов Для сравнения, уровень ассоциации Haunt с хроматином не изменился в дистальном Hoxa13 и увеличился только в 2 раза в локусах Haunt и Skap2. Таким образом, вместо неспецифического связывания хроматина из-за повышенной экспрессии после обработки ESC ретиноевой кислотой, усиленная ассоциация Haunt с локусами HOXA может быть специфически связана с функцией РНК Haunt. Сильное и прямое взаимодействие Haunt с 5'-стороной кластера HOXA указывает на то, что Haunt прямо участвует в модуляции индукции HOXA.
Данные ChIRP-seq позволили показать, что РНК HOTTIP является цис-регулятором поздних генов HOXA (HOXA9–HOXA13). Однако анализ 3767 сайтов связывания показал, что помимо регуляции генов HOXA, HOTTIP регулирует гемопоэтический ландшафт хроматина и транскрипционную программу путем взаимодействия со специфическими для гемопоэза факторами транскрипции и эпигенетическими регуляторами [32].
ЭКСПЕРИМЕНТЫ “ВСЕ-ПРОТИВ-ВСЕХ”
Начиная с 2017 года, опубликовано шесть работ, в которых предложено несколько экспериментальных протоколов и биоинформатических подходов, позволяющих определять полный РНК-хроматиновый интерактом (протоколы “все-против-всех”): MARGI [10], GRID-seq [11], ChAR-seq [12], iMARGI [13, 14], RADICL-seq [15], Red-C [16].
Экспериментальные подходы к анализу взаимодействий всех РНК с хроматином
Все методы идеологически используют один и тот же основной подход – сначала клетки фиксируют, фрагментируют ДНК с помощью эндонуклеаз или ультразвука, а затем проводят лигирование пространственно сближенных молекул РНК и локусов ДНК при участии специфически сконструированного линкера, несущего биотиновую метку, необходимую для последующего выделения целевых конструкций. При этом сначала лигируют линкер к РНК, а потом к хроматиновой ДНК (хрДНК). Далее проводят обратную транскрипцию и получают химеры кДНК–линкер–хрДНК (рис. 3). В первой работе [10] все процедуры проводили в ядерном лизате, что требовало много клеток (порядка 400 млн). В других методах процедуры проводили in situ, поэтому клеток требовалось на два порядка меньше.
Рис. 3.
Общая схема протоколов “все-против-всех”. Верхняя панель: GRID, RED-C, RADICL (в RADICL вместо MmeI использована рестриктаза EcoP15I). Нижняя панель: MARGI, iMARGI (показаны этапы после обратной транскрипции, этапы до обратной транскрипции совпадают с GRID, RED-C, RADICL).
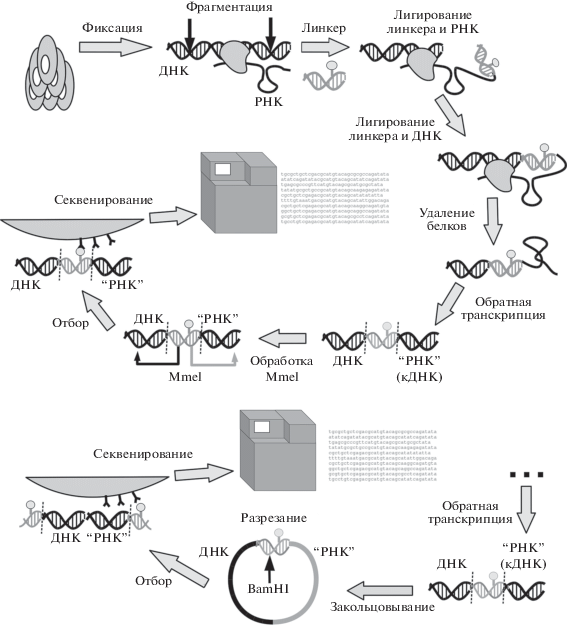
В ряде работ (рис. 3, верхняя панель) линкер содержит сайт узнавания MmeI (GRID [11], Red-C [16]) или EcoP15I (RADICL [15]). Эти рестриктазы замечательны тем, что точка расщепления удалена на 20 или 27 н. от сайта узнавания. В методах GRID и RADICL линкер несет два сайта рестрикции, направленные как в сторону кДНК, так и в сторону хрДНК, в то время как в Red-C линкер содержит только один сайт, направленный в сторону хрДНК (рис. 3, верхняя панель). Фрагменты, полученные после обработки рестриктазами, выделяют на стрептавидине и секвенируют либо с одного конца (GRID и RADICL), либо с двух (Red-C).
В методах MARGI и iMARGI [10, 13, 14] конструкцию кДНК–линкер–хрДНК закольцовывают, после чего кольцо линеаризуют рестриктазой BamHI, сайт которой присутствует в линкере, так, что один конец содержит фрагмент линкера и кДНК, а второй – хрДНК и другой фрагмент линкера (рис. 3, нижняя панель). Далее конструкцию отбирают на стрептавидиновых шариках и секвенируют с двух концов.
В работах использовали разнообразные контроли. В частности, оценивали фоновые взаимодействия, добавляя к клеткам, выбранным для исследования, клетки другого организма (например, дрозофилы), после чего определяли долю обнаруженных межвидовых контактов (MARGI, GRID [10, 11]). Эти контрольные эксперименты показали, что лишь небольшая доля (около 2%) контактов являются межвидовыми. В методах “все-против-всех” корректность протокола проверяют по соответствию полногеномных данных результатам некоторых экспериментов “один-против-всех”.
Отметим, что в используемых методах фиксации предпочтительно сшивают нуклеиновые кислоты с белками, а нуклеиновые кислоты сшиваются между собой менее эффективно. Есть РНК–ДНК-контакты, обусловленные просто процессом транскрипции, которые должны породить большое количество тривиальных контактов (полимеразный след). Поэтому в ряде работ применяют дополнительные процедуры подготовки библиотек. В частности, в методе MARGI [10] используют два варианта фиксации – жесткую с 1%-ным формальдегидом (pxMARGI), и фиксацию смесью формальдегида и дисукцинимидилглутарата (diMARGI), где ожидается меньший уровень полимеразного следа. Для уменьшения шума в работе [15] (RADICL) использовали два дополнительных варианта подготовки биологического материала. В первом для подавления полимеразного следа клетки инкубировали с ингибитором инициации транскрипции актиномицином D. Во втором хроматин после сшивки сразу же расшивали и обрабатывали протеиназой K в денатурирующих условиях, чтобы оставить только прямые РНК–ДНК-взаимодействия. В табл. 2 представлены клеточные линии, использованные в экспериментах “все-против-всех”.
Таблица 2.
Клеточные линии, использованные в экспериментах “все-против-всех”
| Метод | Организм, клеточная линия | Ссылки |
|---|---|---|
| MARGI | H. sapiens: H9 ESC, HEK293T | [10] |
| GRID-seq | H. sapiens: MDA-MB-231, MM.1S M. musculus: ES D. melanogaster: S2 |
[11] |
| iMARGI | H. sapiens: HEK293T и HFF | [13] |
| iMARGI | H. sapiens: клетки эндотелия | [14] |
| RADICL-seq | M. musculus: mESC и прогениторные клетки олигодендроцитов (mOPC) | [15] |
| Red-C | H. sapiens: K562, фибробласты | [16] |
Обработка данных экспериментов “все-против-всех”
В ходе биоинформатического анализа секвенированные фрагменты проходят проверку качества, удаляются ПЦР-дубликаты, затем из последовательностей одноконцевых или парноконцевых чтений удаляют технические последовательности, определяя фрагменты, соответствующие РНК- и ДНК-частям, которые картируют на последовательность соответствующего генома или транскриптома. Таким образом получают координаты фрагмента РНК и локуса ДНК, которые, согласно результатам протокола и секвенирования, определены как контактирующие друг с другом. Все методы анализа подразумевают исключительно уникальное картирование и РНК-, и ДНК-частей, что приводит к исключению из рассмотрения хаРНК, пришедших из повторов, и контактов хаРНК с повторами. Отметим, что использование рестриктазы EcoP15I с более далекой точкой расщепления (27 вместо 20 у MmeI) позволило в эксперименте RADICL-seq получить больше уникально картированных фрагментов [15].
Хроматин имеет разную степень доступности, поэтому можно ожидать, что число контактов с гетерохроматином будет ниже просто из-за того, что эндонуклеазы будут реже расщеплять эти участки, внося тем самым систематическую ошибку в данные. Поэтому в ряде работ применяли нормирование на фон. Так, в [11, 16] в качестве фона (внутреннего контроля) использовали профиль транс-контактов мРНК, поскольку предполагается, что мРНК белоккодирующих генов в основной своей массе не должны специфически контактировать с хроматином. Для выделения наиболее липких участков хроматина применяли также поиск пиков с помощью программы MACS [10]. Во всех работах проводили аннотацию РНК-частей с использованием разметок (ENSEMBL hg38 release 84 [10], ENSEMBL GRCh38.83 [11], NCBIM37 mm9 [11], BDGP5.78 dm3 [11], FlyBase r5.57 dm3 [12], GENCODE release M14 mm10 [15], GENCODE release 19 hg19 [16]). Аннотации генов часто перекрываются, например, в интроне мРНК может быть закодирована малая ядрышковая РНК или микроРНК. Предложена процедура, с помощью которой при проведении аннотации РНК-частей контактов разрешались конфликтные ситуации, возникающие при пересечении генов в разметке: РНК-часть контакта приписывали гену с большей плотностью покрытия [16]. Отмечено также довольно большое число контактов неаннотированных РНК [11, 16]. Предложен способ сборки потенциальных транскриптов из этих фрагментов [16]. С использованием достаточно жестких фильтров обнаружено около 2000 новых хаРНК, которые назвали Х-РНК.
Общие характеристики карт контактов
Установлено, что наибольшее количество контактов образуют мРНК, причем в контактах участвуют как экзоны, так и интроны. Контакты обнаружены также у антисмысловых РНК, малых ядерных и малых ядрышковых РНК, микроРНК, piРНК и других биотипов РНК. Протокол RADICL-seq порождает более длинные фрагменты, чем протоколы GRID-seq и Red-C, поэтому с его использованием удалось уникально картировать порядка 10% РНК-частей. В mESC среди повторов найдено больше всего малых ядерных РНК и SINE (по 1/3), а в mOPC повышена представленность LINE и LTR. Отмечена весьма высокая корреляция между уровнем транскрипции отдельных РНК и числом их контактов [11, 16].
Анализ распределения расстояний между источником РНК и ее контактом на хроматине показал, что средняя плотность контактов убывает с расстоянием между геном РНК и ее ДНК-контактом. Кроме того, большинство РНК чаще контактирует с хроматином той же хромосомы, где расположен ген, чем с другими хромосомами. Однако ряд РНК этими свойствами не обладает. В частности, MALAT1, NEAT1 и малые ядерные РНК имеют контакты на всем геноме, а XIST контактирует только со своей хромосомой. Так, в частности, показано, что чем выше экспрессия РНК (по данным CAGE), тем медленнее убывает плотность контактов с расстоянием [15]. Отмечено также, что РНК, транскрибируемые с SINE, предпочтительно контактируют с ДНК на расстоянии 10 т.п.н.–1 млн п.н. от своего гена, а LINE и LTR – на расстоянии более 100 т.п.н. [15]. Там же отмечено, что подавление транскрипции актиномицином D сохраняет часть цис-контактов.
Контакты известных РНК
В работах “все-против-всех” сравнили карты контактов отдельных РНК и результаты экспериментов “один-против-всех”. Обнаружено достаточно хорошее соответствие данным по MALAT1 [11, 15, 16]. Однако согласно [15], контакты MALAT1 и 7SK, определенные RADICL-seq, лучше соответствуют контактам этих РНК, определенным RAP и ChIRP-seq соответственно, чем контакты, установленные GRID-seq. Здесь трудно сравнивать результаты разных экспериментов, поскольку они проводились на разных клеточных линиях и в разных условиях. Поэтому трудно сказать, обусловлены эти расхождения экспериментальными протоколами или различиями в обработке данных. Также GRID-seq находил меньше специфических далеких цис- и транс-взаимодействий, особенно в случае некодирующих РНК. Показана также клеточная специфичность профилей контактов РНК, особенно РНК-маркеров клеточных линий.
Контакты с промоторами и хроматином в разных состояниях
Значительная часть РНК-ДНК-контактов приходится на активный хроматин. В частности, уровень покрытия ДНК контактами положительно коррелирует с гистоновыми метками транскрипционно активного хроматина (H3K27ac и H3K4me3) [10]. Бо́льшая часть контактов приходится на эухроматин (при сравнении с DHS-seq и ATAC-seq [15]), ДНК-части обогащены в промоторах и межгенных участках [11]. Отметим, что это обогащение может быть артефактом экспериментов, поскольку открытый хроматин более доступен для нуклеаз и поэтому он дает больше контактов.
Рассмотрение контактов различных РНК с хроматином [16] показало, что некоторые РНК предпочитают гетерохроматин, Kcnq1ot1 взаимодействует с хроматином, подавленным Polycomb, и с сайтами связывания CTCF, а энхансерные РНК – преимущественно с промоторами активно транскрибируемых генов и другими энхансерами. Малые ядерные РНК предпочитали активный хроматин, а vlincRNA (очень длинные нкРНК [111]) и Х-РНК – гетерохроматин на расстоянии более 10 млн п.н. от своего гена. Показано также, что некоторые микроРНК чаще контактируют с гетерохроматином и B-компартментами, что указывает на организацию гетерохроматина вокруг ядрышка.
Показано, что РНК с высоким уровнем контактов с хроматином ассоциированы с белками (по данным fRIP для K562), в частности, с белками комплекса Polycomb, HDAC и DNMT1 [16]. Значительная часть хаРНК взаимодействовала также с ADAR.
Сравнение карт контактов с данными Hi-C
Изучение связи РНК-хроматиновых контактов с архитектурой хроматина [11, 15] показало, что РНК–ДНК- и ДНК–ДНК-взаимодействия генов мыши и дрозофилы хорошо коррелировали между собой в радиусах 1 млн п.н. и 200 т.п.н. соответственно [11]. Бо́льшая часть РНК из генов внутри ТАДов контактировала с ДНК преимущественно внутри своих же ТАДов. Отмечено, что границы ТАДов обогащены ДНК-частями во всех линиях и во всех экспериментальных условиях [15]. Там же показано, что ассоциированные с хроматином РНК предпочитают взаимодействовать с тем же типом хроматинового компартмента, из которого они происходят. Отмечено, что до 30% близких взаимодействий можно объяснить пространственной укладкой хроматина.
Протоколы полногеномного изучения архитектуры хроматина и РНК-ДНК-интерактома используют одни и те же подходы: фиксацию клеток и лигирование близко расположенных друг к другу молекул. В результате биоинформатической обработки данных в протоколах Hi-C остается ~75% от исходных чтений, в то время как в случае РНК–ДНК-взаимодействий пригодной для анализа остается только четверть данных, что говорит о несовершенстве экспериментальных подходов при массовом изучении хаРНК.
Другие наблюдения
Согласно [13], 5 из 10 наиболее часто взаимодействующих пар генов определены в iMARGI как гибридные транскрипты в TCGA (The Cancer Genome Atlas). Помимо этого, проанализированы 96 новых опухолевых образцов, в которых нашли 42 гибридных транскрипта, 37 из которых совпадали с РНК–ДНК-контактами в нормальных клетках. Исходя из этих результатов, предложена новая модель образования гибридных транскриптов: два транскрипта сближаются в пространстве (либо из-за сближения их генов, либо из-за того, что РНК оказалась в пространственной близости другого гена), а затем происходит транс-сплайсинг или геномная перестройка.
Применение протокола iMARGI позволило показать, что при искусственно вызванном стрессе, имитирующем сахарный диабет, дисфункция клеток эндотелия обусловлена нарушением экспрессии генов, опосредованным хаРНК [14]. Данные iMARGI указывают на индуцированное стрессом возникновение ряда контактов РНК–хроматин, в том числе, с участием ингибитора активатора плазминогена SERPINE1 и нкРНК LINC00607. Эти гены известны как коэкспрессируемые в дисфункциональных клетках эндотелия, включая клетки, полученные от больных диабетом. Нокдаун LINC00607 привел к подавлению SERPINE1 и других генов, вносящих вклад в эндотелиальную дисфункцию, подтверждая тем самым предположение о том, что взаимодействия РНК с хроматином вносят вклад в регуляцию транскрипции во время дисфункции эндотелия.
ВЫВОДЫ
Данные экспериментов “один-против-всех” (как и “все-против-всех”) отражают физическую близость РНК и конкретных хроматиновых локусов, что делает их центральными для понимания механизма действия нкРНК. Однако, чтобы определить функциональную роль РНК в соответствующем ДНК-локусе, необходимы дополнительные полногеномные данные, например, о структуре хроматина, экспрессии генов или о локализации ДНК-связывающих и хроматин-модифицирующих белков. В частности, более детально партнеров изучаемой РНК выявляют, используя модификации основных методов “один-против-всех”, такие как RAP-RNA [40], CHART-MS [39], CHART-RNA [112], ChIRP-RNA [113] и другие. Помимо методов “один-против-всех”, “всех-против-всех” и их производных существует множество других подходов, позволяющих исследовать РНК-ДНК-взаимодействия не только прямо (поиск R-петель [114], триплексных структур [115]), но и косвенно (определение предположительно хаРНК с помощью комбинации методов изучения РНК-белковых взаимодействий с СhIP-seq) [116].
К преимуществам методов “все-против-всех” перед методами “один-против-всех” относится то, что они не требуют первичного знания о последовательности изучаемых РНК, что позволяет получать данные о контактах с хроматином всех РНК в клетке. Однако у всех описанных выше методов “все-против-всех” есть характерные проблемы, обусловленные общей схемой методов. Во-первых, лишь небольшая доля секвенированных фрагментов (10–30%) РНК–ДНК-контактов сохраняется при картировании из-за требования уникальности. Во-вторых, в итоговых данных присутствует много фоновых неспецифических контактов (транс-контактов мРНК) и много проксимальных контактов (когда РНК контактирует со своим геном), что значительно снижает количество осмысленных контактов и разрешение методов в целом. В-третьих, из-за относительно низкой глубины секвенирования в результирующих данных “все-против-всех” мы видим в основном высокоэкспрессирующиеся РНК. При слиянии контактов из нескольких реплик можно получить набор из 100 млн контактов для млекопитающих, однако этого все равно недостаточно для получения достаточно высокого разрешения. Несмотря на это, паттерны контактов единичных РНК в методах “все-против-всех” хорошо согласуются с паттернами контактов этих же РНК в методах “один-против-всех”, а также с паттернами специфических белок-хроматиновых взаимодействий, что указывает на биологическую достоверность результатов. Таким образом, можно сказать, что “КПД” этих методов невелик, однако они показали свою релевантность и нашли значительное количество нкРНК, которые активно взаимодействуют с хроматином.
Важный этап анализа данных изучения взаимодействий РНК–хроматин – определение истинных взаимодействий между РНК и хроматином. В данном обзоре мы показали многообразие подходов, однако две работы [25, 62] подчеркивают важность использования в качестве отрицательного контроля клеточную линию с нокаутом изучаемой РНК. В этих работах не обнаружено значительного обогащения контактов РНК 7SK и Eprn в клетках дикого типа по сравнению с клетками с нокаутом. Это позволяет сделать вывод о том, что эти РНК не взаимодействуют с хроматином, хотя имеют значительное обогащение контактов относительно “input”.
В подходах “все-против-всех” для определения релевантности данных используют контроли, однако они не генерируют данные для определения фоновых взаимодействий (так называемый “input”), а фон определяют лишь из контактов мРНК, которые считают неспецифическими. Основным источником фоновых неспецифических контактов, по-видимому, является фиксация случайных взаимодействий отрицательно заряженных РНК с положительно заряженными белками хроматина. Уровень неспецифичности, в работах класса “все-против-всех”, по-видимому, наблюдается также и в экспериментах класса “один-против-всех”. Однако существенно большее количество контактов конкретной РНК, которое можно получить в эксперименте “один-против-всех”, спасает ситуацию. Необходимым представляется дальнейшее развитие методов обработки данных с тем, чтобы более четко отделить неспецифические контакты от специфических. Еще одна важная и пока нерешенная проблема – множественное картирование прочтений. Здесь тоже необходимы новые методы анализа данных.
Для лучшего понимания РНК-хроматиновых взаимодействий необходим комплексный подход к анализу данных. Уже предпринята попытка сравнительного анализа всех доступных данных “один-против-всех” [117], однако, в том числе из-за описанных выше проблем, не удается интегрировать данные, полученные методами “один-против-всех” и “все-против-всех”.
Помимо дополнительных экспериментов, направленных на сравнение карт контактов нкРНК в клетках дикого типа и, например, в культуре опухолевых клеток или в клетках, обработанных специальными агентами, вызывающими определенный клеточный ответ, несомненный интерес представляет сравнение карт контактов у разных видов. Эволюционный анализ контактов ортологичных РНК может помочь понять причины связывания тех или иных целевых локусов и их изменения, как в случае нкРНК roX1/2 [47]. Еще одно интересное направление изучения РНК-хроматиновых взаимодействий – получение карт контактов до и после индукции стволовых клеток к дифференцировке, а также сравнение карт контактов на разных этапах клеточного цикла. Эти эксперименты интересны как в технологиях “один-против-всех”, так и “все-против-всех”.
Интерпретацию полученных результатов затрудняют альтернативные изоформы нкРНК. Карта контактов конкретной нкРНК отражает суммарную картину, поэтому определение вклада той или иной изоформы считается перспективной задачей биоинформатики.
Одно из важных направлений – получение методами “один-против-всех” полногеномных карт контактов белоккодирующих РНК, так как в данных “все-против-всех” РНК этого класса имеют большое количество контактов в геноме, даже при добавлении ингибитора транскрипции – актиномицина D. Интерес вызывают также некодирующие функции мРНК или подтверждение правильности использования контактов мРНК в качестве неспецифических при анализе данных “все-против-всех”.
Сейчас видны несколько путей развития методов “все-против-всех”. Можно ставить новые задачи, например, сравнивать карты контактов до и после индукции дифференцировки стволовых клеток, а также на разных стадиях клеточного цикла. Можно применять другие методы фиксации, например, ультрафиолетовое излучение. Методы фиксации показывают преимущественно связи нуклеиновых кислот с белками, поэтому в этих экспериментах видны в основном контакты, опосредованные белками – РНК–белок–ДНК. Поэтому после образования химер РНК–ДНК можно с помощью антител выделить специфические тройные комплексы РНК–белок–ДНК, содержащие интересующий белок. Это позволит существенно повысить мощность метода. При этом наибольший интерес вызывают белки, определяющие архитектуру хроматина (CTCF) и модификаторы хроматина (метилтрансферазы).
Основные выводы, к которым нам хотелось привести читателя, состоят в следующем. Во-первых, взаимодействия РНК–хроматин играют важную роль в работе клеточных систем. Во-вторых, существует ряд методов, позволяющих находить эти контакты. В-третьих, мы находимся в начале пути. Еще предстоит разработать методы анализа РНК-хроматиновых взаимодействий в одной клетке, а также методы, основанные на in situ секвенировании.
Работа поддержана грантом Российского фонда фундаментальных исследований (№ 20-04-00459 А).
Настоящая статья не содержит каких-либо исследований с участием людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Djebali S., Davis C.A., Merkel A., Dobin A., Lassmann T., Mortazavi A., Tanzer A., Lagarde J., Lin W., Schlesinger F., Xue C., Marinov G.K., Khatun J., Williams B.A., Zaleski C., Rozowsky J., Röder M., Kokocinski F., Abdelhamid R.F., Alioto T., Antoshechkin I., Baer M.T., Bar N.S., Batut P., Bell K., Bell I., Chakrabortty S., Chen X., Chrast J., Curado J., Derrien T., Drenkow J., Dumais E., Dumais J., Duttagupta R., Falconnet E., Fastuca M., Fejes-Toth K., Ferreira P., Foissac S., Fullwood M.J., Gao H., Gonzalez D., Gordon A., Gunawardena H., Howald C., Jha S., Johnson R., Kapranov P., King B., Kingswood C., Luo O.J., Park E., Persaud K., Preall J.B., Ribeca P., Risk B., Robyr D., Sammeth M., Schaffer L., See L.-H., Shahab A., Skancke J., Suzuki A.M., Takahashi H., Tilgner H., Trout D., Walters N., Wang H., Wrobel J., Yu Y., Ruan X., Hayashizaki Y., Harrow J., Gerstein M., Hubbard T., Reymond A., Antona-rakis S.E., Hannon G., Giddings M.C., Ruan Y., Wold B., Carninci P., Guigó R., Gingeras T.R. (2012) Landscape of transcription in human cells. Nature. 489(7414), 101–108.
Engreitz J.M., Ollikainen N., Guttman M. (2016) Long non-coding RNAs: spatial amplifiers that control nuclear structure and gene expression. Nat. Rev. Mol. Cell. Biol. 17, 756–770.
Mishra K., Kanduri C. (2019) Understanding long noncoding RNA and chromatin interactions: what we know so far. Non-Coding RNA. 5(4), 1–28.
Engreitz J.M., Pandya-Jones A., McDonel P., Shishkin A., Sirokman K., Surka C., Kadri S., Xing J., Goren A., Lander E.S., Plath K., Guttman M. (2013) The Xist lncRNA exploits three-dimensional genome architecture to spread across the X chromosome. Science. 341(6147), 1–8.
Simon M.D., Wang C.I., Kharchenko P.V., West J.A., Chapman B.A., Alekseyenko A.A., Borowsky M.L., Kuroda M.I., Kingston R.E. (2011) The genomic binding sites of a noncoding RNA. Proc. Natl. Acad. Sci. USA. 108(51), 20497–20502.
Chu C., Qu K., Zhong F.L., Artandi S.E., Chang H.Y. (2011) Genomic maps of long noncoding RNA occupancy reveal principles of RNA-chromatin interactions. Mol. Cell. 44, 667–678.
Quinn J.J., Ilik I.A., Qu K., Georgiev P., Chu C., Akhtar A., Chang H.Y. (2014) Revealing long noncoding RNA architecture and functions using domain-specific chromatin isolation by RNA purification. Nat. Biotechnol. 32, 933–940.
Mondal T., Subhash S., Vaid R., Enroth S., Uday S., Reinius B., Mitra S., Mohammed A., James A.R., Hoberg E., Moustakas A., Gyllensten U., Jones S.J.M., Gustafsson C.M., Sims A.H., Westerlund F., Gorab E., Kanduri C. (2015) MEG3 long noncoding RNA regulates the TGF-β pathway genes through formation of RNA-DNA triplex structures. Nat. Commun. 6(7743), 1–17.
Chu H.P., Cifuentes-Rojas C., Kesner B., Aeby E., Lee H., Wei C., Oh H.J., Boukhali M., Haas W., Lee J.T. (2017) TERRA RNA antagonizes ATRX and protects telomeres. Cell. 170, 86–101.
Sridhar B., Rivas-Astroza M., Nguyen T.C., Chen W., Yan Z., Cao X., Hebert L., Zhong S. (2017) Systematic mapping of RNA-chromatin interactions in vivo. Curr. Biol. 27, 602–609.
Li X., Zhou B., Chen L., Gou L.T., Li H., Fu X.D. (2017) GRID-seq reveals the global RNA-chromatin interactome. Nat. Biotechnol. 35, 940–950.
Bell J.C., Jukam D., Teran N.A., Risca V.I., Smith O.K., Johnson W.L., Skotheim J.M., Greenleaf W.J., Stra-ight A.F. (2018) Chromatin-associated RNA sequencing (ChAR-seq) maps genome-wide RNA-to-DNA contacts. Elife. 7, 1–28.
Yan Z., Huang N., Wu W., Chen W., Jiang Y., Chen J., Huang X., Wen X., Xu J., Jin Q., Zhang K., Chen Z., Chien S., Zhong S. (2019) Genome-wide colocalization of RNA–DNA interactions and fusion RNA pairs. Proc. Natl. Acad. Sci. USA. 116, 3328–3337.
Calandrelli R., Xu L., Luo Y., Wu W., Fan X., Nguyen T., Chen C.-J., Sriram K., Tang X., Burns A.B., Natarajan R., Chen Z.B., Zhong S. (2020) Stress-induced RNA–chromatin interactions promote endothelial dysfunction. Nat. Commun. 11, 1–13.
Bonetti A., Agostini F., Suzuki A.M., Hashimoto K., Pascarella G., Gimenez J., Roos L., Nash A.J., Ghilotti M., Cameron C.J.F., Valentine M., Medvedeva Y.A., Noguchi S., Agirre E., Kashi K., Samudyata, Luginbühl J., Cazzoli R., Agrawal S., Luscombe N.M., Blanchette M., Kasukawa T., Hoon M., Arner E., Lenhard B., Plessy C., Castelo-Branco G., Orlando V., Carninci P. (2020) RADICL-seq identifies general and cell type–specific principles of genome-wide RNA-chromatin interactions. Nat. Commun. 11, 1–14.
Gavrilov A.A., Zharikova A.A., Galitsyna A.A., Luzhin A.V., Rubanova N.M., Golov A.K., Petrova N.V., Logacheva M.D., Kantidze O.L., Ulianov S.V., Magnitov M.D., Mironov A.A., Razin S.V. (2020) Studying RNA-DNA interactome by Red-C identifies noncoding RNAs associated with various chromatin types and reveals transcription dynamics. Nucl. Acids Res. 48(12), 6699–6714.
Alfeghaly C., Sanchez A., Rouget R., Thuillier Q., Igel-Bourguignon V., Marchand V., Branlant C., Motorin Y., Behm-Ansmant I., Maenner S. (2021) Implication of repeat insertion domains in the trans-activity of the long non-coding RNA ANRIL. Nucl. Acids Res. 49(9), 4954–4970.
Zovoilis A., Cifuentes-Rojas C., Chu H.P., Hernandez A.J., Lee J.T. (2016) Destabilization of B2 RNA by EZH2 activates the stress response. Cell. 167(7), 1788–1802.
Alvarez-Dominguez J.R., Knoll M., Gromatzky A.A., Lodish H.F. (2017) The super-enhancer-derived alncRNA-EC7/bloodlinc potentiates red blood cell development in trans. Cell. Rep. 19(12), 2503–2514.
Ballarino M., Cipriano A., Tita R., Santini T., Desideri F., Morlando M., Colantoni A., Carrieri C., Nicoletti C., Musarò A., O’Carroll D., Bozzoni I. (2018) Deficiency in the nuclear long noncoding RNA Charme causes myogenic defects and heart remodeling in mice. EMBO J. 37, 1–16.
Merry C.R., Forrest M.E., Sabers J.N., Beard L., Gao X.-H., Hatzoglou M., Jackson M.W., Wang Z., Markowitz S.D., Khalil A.M. (2015) DNMT1-associated long non-coding RNAs regulate global gene expression and DNA methylation in colon cancer. Hum. Mol Genet. 24(21), 6240–6253.
Chalei V., Sansom S.N., Kong L., Lee S., Montiel J.F., Vance K.W., Ponting C.P. (2014) The long non-coding RNA Dali is an epigenetic regulator of neural differentiation. Elife. 3, 1–24.
Schmitt A.M., Garcia J.T., Hung T., Flynn R.A., Shen Y., Qu K., Payumo A.Y., Peres-da-Silva A., Broz D.K., Baum R., Guo S., Chen J.K., Attardi L.D., Chang H.Y. (2016) An inducible long noncoding RNA amplifies DNA damage signaling. Nat. Genet. 48(11), 1370–1376.
Zapparoli E., Briata P., Rossi M., Brondolo L., Bucci G., Gherzi R. (2020) Comprehensive multi-omics analysis uncovers a group of TGF-β-regulated genes among lncRNA EPR direct transcriptional targets. Nucl. Acids Res. 48(16), 9053–9066.
Li M.A., Amaral P.P., Cheung P., Bergmann J.H., Kinoshita M., Kalkan T., Ralser M., Robson S., Meyenn F., Paramor M., Yang F., Chen C., Nichols J., Spector D.L., Kouzarides T., He L., Smith A. (2017) A lncRNA fine tunes the dynamics of a cell state transition involving lin28, let-7 and de novo DNA methylation. Elife. 6, 1–24.
Hu T., Pi W., Zhu X., Yu M., Ha H., Shi H., Choi J.-H., Tuan D. (2017) Long non-coding RNAs transcribed by ERV-9 LTR retrotransposon act in cis to modulate long-range LTR enhancer function. Nucl. Acids Res. 45(8), 4479–4492.
Luo S., Lu J.Y., Liu L., Yin Y., Chen C., Han X., Wu B., Xu R., Liu W., Yan P., Shao W., Lu Z., Li H., Na J., Tang F., Wang J., Zhang Y.E., Shen X. (2016) Divergent lncRNAs regulate gene expression and lineage differentiation in pluripotent cells. Cell Stem Cell. 18(5), 637–652.
Hacisuleyman E., Goff L.A., Trapnell C., Williams A., Henao-Mejia J., Sun L., McClanahan P., Hendrickson D.G., Sauvageau M., Kelley D.R., Morse M., Engreitz J., Lander E.S., Guttman M., Lodish H.F., Flavell R., Raj A., Rinn J.L. (2019) Topological organization of multichromosomal regions by the long intergenic noncoding RNA Firre. Nat. Struct. Mol. Biol. 21(2), 198–206.
Wang Y., Zhu P., Luo J., Wang J., Liu Z., Wu W., Du Y., Ye B., Wang D., He L., Ren W., Wang J., Sun X., Chen R., Tian Y., Fan Z. (2019) LncRNA HAND2-AS1 promotes liver cancer stem cell self-renewal via BMP signaling. EMBO J. 38, 1–17.
Yin Y., Yan P., Lu J., Song G., Zhu Y., Li Z., Zhao Y., Shen B., Huang X., Zhu H., Orkin S.H., Shen X. (2015) Opposing roles for the lncRNA haunt and its genomic locus in regulating HOXA gene activation during embryonic stem cell differentiation. Cell Stem Cell. 16, 504–516.
Carlson H.L., Quinn J.J., Yang Y.W., Thornburg C.K., Chang H.Y., Stadler H.S. (2015) LncRNA-HIT functions as an epigenetic regulator of chondrogenesis through its recruitment of p100/CBP complexes. PLoS Genet. 11(12), 1–30.
Luo H., Zhu G., Xu J., Lai Q., Yan B., Guo Y., Fung T.K., Zeisig B.B., Cui Y., Zha J., Cogle C., Wang F., Xu B., Yang F.-C., Li W., So C.W.E., Qiu Y., Xu M., Huang S. (2019) HOTTIP lncRNA promotes hematopoietic stem cell self-renewal leading to AML-like disease in mice. Cancer Cell. 36, 645–659.
Liu J., Gao M., He J., Wu K., Lin S., Jin L., Chen Y., Liu H., Shi J., Wang X., Chang L., Lin Y., Zhao Y.-L., Zhang X., Zhang M., Luo G.-Z., Wu G., Pei D., Wang J., Bao X., Chen J. (2021) The RNA m6A reader YTHDC1 silences retrotransposons and guards ES cell identity. Nature. 591(7849), 322–326.
Leveille N., Melo C.A., Rooijers K., Dıaz-Lagares A., Melo S.A., Korkmaz G., Lopes R., Moqadam F.A., Maia A.R., Wijchers P.J., Geeven G., den Boer M.L., Kalluri R., de Laat W., Esteller M., Agami R. (2015) Genome-wide profiling of p53-regulated enhancer RNAs uncovers a subset of enhancers controlled by a lncRNA. Nat. Commun. 6(6520), 1–12.
Lee H.C., Kang D., Han N., Lee Y., Hwang H.J., Lee S.-B., You J.S., Min B.S., Park H.J., Ko Y.-G., Gorospe M., Lee J.-S. (2020) A novel long noncoding RNA Linc-ASEN represses cellular senescence through multileveled reduction of p21 expression. Cell Death Differentiation. 27(6), 1844–1861.
Forrest M.E., Saiakhova A., Beard L., Buchner D.A., Scacheri P.C., LaFramboise T., Markowitz S., Kha-lil A.M. (2018) Colon cancer-upregulated long non-coding RNA lincDUSP regulates cell cycle genes and potentiates resistance to apoptosis. Sci. Rep. 8(7324), 1–12.
Ang C.E., Ma Q., Wapinski O.L., Fan S., Flynn R.A., Lee Q.Y., Coe B., Onoguchi M., Olmos V.H., Do B.T., Dukes-Rimsky L., Xu J., Tanabe K., Wang L., Elling U., Penninger J.M., Zhao Y., Qu K., Eichler E.E., Srivastava A., Wernig M., Chang H.Y. (2019) The novel lncRNA lnc-NR2F1 is pro-neurogenic and mutated in human neurodevelopmental disorders. Elife. 8, 1–29.
Luo M., Jeong M., Sun D., Park H.J., Rodriguez B.A.T., Xia Z., Yang L., Zhang X., Sheng K., Darlington G.J., Li W., Goodell M.A. (2015) Long non-coding RNAs control hematopoietic stem cell function. Cell Stem Cell. 16, 426–438.
West J.A., Davis C.P., Sunwoo H., Simon M.D., Sadreyev R.I., Wang P.I., Tolstorukov M.Y., Kingston R.E. (2014) The long noncoding RNAs NEAT1 and MALAT1 bind active chromatin sites. Mol. Cell. 55, 791–802.
Engreitz J.M., Sirokman K., McDonel P., Shishkin A.A., Surka C., Russell P., Grossman S.R., Chow A.Y., Guttman M., Lander E.S. (2014) RNA-RNA interactions enable specific targeting of noncoding RNAs to nascent pre-mRNAs and chromatin sites. Cell. 159, 188–199.
Yin Y., Lu J.Y., Zhang X., Shao W., Xu Y., Li P., Hong Y., Cui L., Shan G., Tian B., Zhang Q.C., Shen X. (2020) U1 snRNP regulates chromatin retention of noncoding RNAs. Nature. 580(7801), 147–150.
Iyer S., Modali S.D., Agarwal S.K. (2017) Long noncoding RNA MEG3 is an epigenetic determinant of oncogenic signaling in functional pancreatic neuroendocrine tumor cells. Mol. Cell. Biol. 37(22), 1–17.
Chakraborty D., Paszkowski-Rogacz M., Berger N., Ding L., Mircetic J., Fu J., Iesmantavicius V., Choudhary C., Anastassiadis K., Stewart A.F., Buchholz F. (2017) lncRNA Panct1 maintains mouse embryonic stem cell identity by regulating TOBF1 recruitment to Oct-Sox sequences in early G1. Cell Rep. 21, 3012–3021.
Chu H.-P., Froberg J.E., Kesner B., Oh H.J., Ji F., Sadreyev R., Pinter S.F., Lee J.T. (2017) PAR-TERRA directs homologous sex chromosome pairing. Nat. Struct. Mol. Biol. 24(8), 620–631.
Vance K.W., Sansom S.N., Lee S., Chalei V., Kong L., Cooper S.E., Oliver P.L., Ponting C.P. (2014) The long non-coding RNA Paupar regulates the expression of both local and distal genes. EMBO J. 33(4), 296–311.
Yang L., Lin C., Jin C., Yang J.C., Tanasa B., Li W., Merkurjev D., Ohgi K.A., Meng D., Zhang J., Evans C.P., Rosenfeld M.G. (2013) LncRNA-dependent mechanisms of androgen-receptor-regulated gene activation programs. Nature. 500(7464), 598–602.
Quinn J.J., Zhang Q.C., Georgiev P., Ilik I.A., Akhtar A., Chang H.Y. (2016) Rapid evolutionary turnover underlies conserved lncRNA–genome interactions. Genes Develop. 30, 191–207.
Fan Z., Chen X., Liu L., Zhu C., Xu J., Yin X., Sheng Y., Zhu Z., Wen L., Zuo X., Zheng X., Zhang Y., Xu J., Huang H., Zhou F., Sun L., Luo J., Zhang D., Chen X., Cui Y., Hao Y., Cui Y., Zhang X., Chen R. (2020) Association of the polymorphism rs13259960 in SLEAR with predisposition to systemic lupus erythematosus. Arthritis Rheumatol. 72(6), 985–996.
Wongtrakoongate P., Riddick G., Fucharoen S., Felsenfeld G. (2015) Association of the long non-coding RNA steroid receptor RNA activator (SRA) with TrxG and PRC2 complexes. PLoS Genet. 11(10), 1–20.
Sawaengdee W., Cui K., Zhao K., Hongeng S., Fucharoen S., Wongtrakoongate P. (2020) Genome-wide transcriptional regulation of the long non-coding RNA steroid receptor RNA activator in human erythroblasts. Front. Genet. 11(850), 1–15.
Marion R.M., Montero J.J., de Silanes I.L., Grana-Castro O., Martınez P., Schoeftner S., Palacios-Fabrega J.A., Blasco M.A. (2019) TERRA regulate the transcriptional landscape of pluripotent cells through TRF1-dependent recruitment of PRC2. Elife. 8, 1–32.
Long J., Badal S.S., Ye Z., Wang Y., Ayanga B.A., Galvan D.L., Green N.H., Chang B.H., Overbeek P.A., Danesh F.R. (2016) Long noncoding RNA Tug1 regulates mitochondrial bioenergetics in diabetic nephropathy. J. Clin. Invest. 126(11), 4205–4218.
Simon M.D., Pinter S.F., Fang R., Sarma K., Rutenberg-Schoenberg M., Bowman S.K., Kesner B.A., Maier V.K., Kingston R.E., Lee J.T. (2013) High-resolution Xist binding maps reveal two-step spreading during X-chromosome inactivation. Nature. 504(7480), 465–469.
Chen C.-K., Blanco M., Jackson C., Aznauryan E., Ollikainen N., Surka C., Chow A., Cerase A., McDonel P., Guttman M. (2016) Xist recruits the X chromosome to the nuclear lamina to enable chromosome-wide silencing. Science. 354(6311), 468–472.
Wang C.Y., Jégu T., Chu H.P., Oh H.J., Lee L.T. (2018) SMCHD1 merges chromosome compartments and assists formation of super-structures on the inactive X. Cell. 174, 406–421.
Colognori D., Sunwoo H., Kriz A.J., Wang C.Y., Lee J.T. (2019) Xist deletional analysis reveals an interdependency between Xist RNA and polycomb complexes for spreading along the inactive X. Mol. Cell. 74, 101–117.
Wang C.Y., Colognori D., Sunwoo H., Wang D., Lee J.T. (2019) PRC1 collaborates with SMCHD1 to fold the X-chromosome and spread Xist RNA between chromosome compartments. Nat. Commun. 10(2950), 1–18.
Pandya-Jones A., Markaki Y., Serizay J., Chitiashvili T., Leon W.R.M., Damianov A., Chronis C., Papp B., Chen C.-K., McKee R., Wang X.-J., Chau A., Sabri S., Leonhardt H., Zheng S., Guttman M., Black D.L., Plath K. (2020) A protein assembly mediates Xist localization and gene silencing. Nature. 587(7832), 145–151.
Powell W.T., Coulson R.L., Crary F.K., Wong S.S., Ach R.A., Tsang P., Yamada N.A., Yasui D.H., LaSalle J.M. (2013) A Prader-Willi locus lncRNA cloud modulates diurnal genes and energy expenditure. Hum. Mol. Genetics. 22(21), 4318–4328.
Liu W., Ma Q., Wong K., Li W., Ohgi K., Zhang J., Aggarwal A.K., Rosenfeld M.G. (2013) Brd4 and JMJD6-associated anti-pause enhancers in regulation of transcriptional pause release. Cell. 155, 1581–1595.
Flynn R.A., Do B.T., Rubin A.J., Calo E., Lee B., Kuchelmeister H., Rale M., Chu C., Kool E.T., Wysocka J., Khavari P.A., Chang H.Y. (2016) 7SK-BAF axis controls pervasive transcription at enhancers. Nat. Struct. Mol. Biol. 23(3), 231–238.
Studniarek C., Tellier M., Martin P.G.P., Murphy S., Kiss T., Egloff S. (2021) The 7SK/P-TEFb snRNP controls ultraviolet radiation-induced transcriptional reprogramming. Cell Rep. 35(2), 1–14.
Pandey R.R., Mondal T., Mohammad F., Enroth S., Redrup L., Komorowski J., Nagano T., Mancini-DiNardo D., Kanduri C. (2008) Kcnq1ot1 antisense noncoding RNA mediates lineage-specific transcriptional silencing through chromatin-level regulation. Mol. Cell. 32, 232–246.
Man H.S.J., Sukumar A.N., Lam G.C., Turgeon P.J., Yan M.S., Ku K.H., Dubinsky M.K., Ho J.J.D., Wang J.J., Das S., Mitchell N., Oettgen P., Sefton M.V., Marsden P.A. (2018) Angiogenic patterning by STEEL, an endothelial-enriched long noncoding RNA. Proc. Natl. Acad. Sci. USA. 115(10), 2401–2406.
Colak D., Zaninovic N., Cohen M.S., Rosenwaks Z., Yang W.-Y., Gerhardt J., Disney M.D., Jaffrey S.R. (2014) Promoter-bound trinucleotide repeat mRNA drives epigenetic silencing in fragile X syndrome. Science. 343(6174), 1002–1005.
Zhang Y., Liu T., Meyer C.A., Eeckhoute J., Johnson D.S., Bernstein B.E., Nusbaum C., Myers R.M., Brown M., Li W., Liu X.S. (2008) Model-based analysis of ChIP-Seq (MACS). Genome Biol. 9(9), 1–9.
Xu S., Grullon S., Ge K., Peng W. (2014) Spatial clustering for identification of ChIP-enriched regions (SICER) to map regions of histone methylation patterns in embryonic stem cells. Methods in Molecular Biology (Methods and Protocols). 1150, 97–111.
Heinz S., Benner C., Spann N., Bertolino E., Lin Y.C., Laslo P., Cheng J.X., Murre C., Singh H., Glass C.K. (2010) Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell. 38, 576–589.
Kharchenko P.V., Tolstorukov M.Y., Park P.J. (2008) Design and analysis of ChIP-seq experiments for DNA-binding proteins. Nat. Biotechnol. 26(12), 1351–1359.
Rice P., Longden L., Bleasby A. (2000) EMBOSS: ehe European molecular biology open software suite. Trends Genet. 16(6), 276–277.
Buske F.A., Bauer D.C., Mattick J.S., Bailey T.L. (2012) Triplexator: detecting nucleic acid triple helices in genomic and transcriptomic data. Genome Res. 22, 1372–1381.
Kuo C.-C., Hanzelmann S., Cetin N.S., Frank S., Zajzon B., Derks J.-P., Akhade V.S., Ahuja G., Kanduri C., Grummt I., Kurian L., Costa I.G. (2019) Detection of RNA–DNA binding sites in long noncoding RNAs. Nucl. Acids Res. 47(6), 1–12.
Matveishina E., Antonov I., Medvedeva Y.A. (2020) Practical guidance in genome-wide RNA:DNA triple helix prediction. Int. J. Mol. Sci. 21(830), 1–12.
Bailey T.L., Johnson J., Grant C.E., Noble W.S. (2015) The MEME suite. Nucl. Acids Res. 43, 39–49.
Gupta S., Stamatoyannopoulos J.A., Bailey T.L., Noble W.S. (2007) Quantifying similarity between motifs. Genome Biol. 8, 1–9.
Martianov I., Ramadass A., Barros A.S., Chow N., Akoulitchev A. (2007) Repression of the human dihydrofolate reductase gene by a non-coding interfering transcript. Nature. 445(7128), 666–670.
Khomyakova E.B., Gousset H., Liquier J., Huynh-Dinh T., Gouyette C., Takahashi M., Florentiev V.L., Taillandier E. (2000) Parallel intramolecular DNA triple helix with G and T bases in the third strand stabilized by Zn2+ ions. Nucl. Acids Res. 28(18), 3511–3516.
Besch R., Giovannangeli C., Kammerbauer C., Degitz K. (2002) Specific inhibition of ICAM-1 expression mediated by gene targeting with triplex-forming oligonucleotides. J. Biol. Chem. 277(36), 32473–32479.
McLean C.Y., Bristor D., Hiller M., Clarke S.L., Schaar B.T., Lowe C.B., Wenger A.M., Bejerano G. (2010) GREAT improves functional interpretation of cis-regulatory regions. Nat. Biotechnol. 28(5), 495–501.
Shin H., Liu T., Manrai A.K., Liu S.X. (2009) CEAS: cis-regulatory element annotation system. Bioinformatics. 25(19), 2605–2606.
Mi H., Muruganujan A., Ebert D., Huang X., Tho-mas P.D. (2019) PANTHER version 14: more genomes, a new PANTHER GO-slim and improvements in enrichment analysis tools. Nucl. Acids Res. 47, 419–426.
Huang D.W., Sherman B.T., Lempicki R.A. (2009) Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protocols. 4(1), 44–57.
Binns D., Dimmer E., Huntley R., Barrell D., O’Donovan C., Apweiler R. (2009) QuickGO: a web-based tool for Gene Ontology searching. Proteomics. 25(22), 3045–3046.
Kuleshov M.V., Jones M.R., Rouillard A.D., Fernandez N.F., Duan Q., Wang Z., Koplev S., Jenkins S.L., Jagodnik K.M., Lachmann A., McDermott M.G., Monteiro C.D., Gundersen G.W., Ma’ayan A. (2016) Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucl. Acids Res. 44, 90–97.
Chodroff R.A., Goodstadt L., Sirey T.M., Oliver P.L., Davies K.E., Green E.D., Molnár Z., Ponting C.P. (2010) Long noncoding RNA genes: caaonservation of sequence and brain expression among diverse amniotes. Genome Biol. 11(7), 1–16.
Ulitsky I., Shkumatava A., Jan C.H., Sive H., Bartel D.P. (2011) Conserved function of lincRNAs in vertebrate embryonic development despite rapid sequence evolution. Cell. 147(7), 1537–1550.
Gelbart M.E., Kuroda M.I. (2009) Drosophila dosage compensation: A complex voyage to the X chromosome. Development, 136(9), 1399–1410.
Larschan E., Bishop E.P., Kharchenko P.V., Core L.J., Lis J.T., Park P.J., Kuroda M.I. (2011) X chromosome dosage compensation via enhanced transcriptional elongation in Drosophila. Nature. 471(7336), 115–118.
Meller V.H., Rattner B.P. (2002) The roX genes encode redundant male-specific lethal transcripts required for targeting of the MSL complex. EMBO J. 21(5), 1084–1091.
Disteche C.M. (2012) Dosage compensation of the sex chromosomes. Annu. Rev. Genet. 46, 537–560.
Lee J.T. (2012) Epigenetic regulation by long noncoding RNAs. Science. 338(6113), 1435–1439.
Wutz A. (2011) Gene silencing in X-chromosome inactivation: advances in understanding facultative heterochromatin formation. Nat. Rev. Genetics. 12(8), 542–553.
Giorgetti L., Lajoie B.R., Carter A.C., Attia M., Zhan Y., Xu J., Chen C.J., Kaplan N., Chang H.Y., Heard E., Dekker J. (2016) Structural organization of the inactive X chromosome in the mouse. Nature. 535(7613), 575–579.
Splinter E., de Wit E., Nora E.P., Klous P., van de Werken H.J.G., Zhu Y., Kaaij L.J.T., van IJcken W., Gribnau J., Heard E., de Laat W. (2011) The inactive X chromosome adopts a unique three-dimensional conformation that is dependent on Xist RNA. Genes Development. 25(13), 1371–1383.
Minajigi A., Froberg J.E., Wei C., Sunwoo H., Kesner B., Colognori D., Lessing D., Payer B., Boukhali M., Haas W., Lee J.T. (2015) A comprehensive Xist interactome reveals cohesin repulsion and an RNA-directed chromosome conformation. Science. 349(6245), 1–10.
Schoeftner S., Sengupta A.K., Kubicek S., Mechtler K., Spahn L., Koseki H., Jenuwein T., Wutz A. (2006) Recruitment of PRC1 function at the initiation of X inactivation independent of PRC2 and silencing. EMBO J. 25(13), 3110–3122.
Zhao J., Sun B.K., Erwin J.A., Song J.J., Lee J.T. (2008) Polycomb proteins targeted by a short repeat RNA to the mouse X chromosome. Science. 322(5902), 750–756.
Chu C., Zhang Q.C., da Rocha S.T., Flynn R.A., Bharadwaj M., Calabrese J.M., Magnuson T., Heard E., Chang H.Y. (2015) Systematic discovery of Xist RNA binding proteins. Cell. 161(2), 404–416.
Brockdorff N. (2018) Local tandem repeat expansion in Xist RNA as a model for the functionalisation of ncRNA. Non-Coding RNA. 4(4), 1–11.
McHugh C.A., Chen C.-K., Chow A., Surka C.F., Tran C., McDonel P., Pandya-Jones A., Blanco M., Burghard C., Moradian A., Sweredoski M.J., Shishkin A.A., Su J., Lander E.S., Hess S., Plath K., Guttman M. (2015) The Xist lncRNA interacts directly with SHARP to silence transcription through HDAC3. Nature. 521(7551), 232–236.
Hutchinson J.H., Ensminger A.W., Clemson C.M., Lynch C.R., Lawrence J.B., Chess A. (2007) A screen for nuclear transcripts identifies two linked noncoding RNAs associated with SC35 splicing domains. BMC Genomics. 8(39), 1–16.
Zhang X., Hamblin M.H., Yin K.J. (2017) The long noncoding RNA Malat1: its physiological and pathophysiological functions. RNA Biol. 14(12), 1705–1714.
Clemson C.M., Hutchinson J.N., Sara S.A., Ensminger A.W., Fox A.H., Chess A., Lawrence J.B. (2009) An architectural role for a nuclear noncoding RNA: NEAT1 RNA is essential for the structure of paraspeckles. Mol. Cell. 33(6), 717–726.
Imamura K., Imamachi N., Akizuki G., Kumakura M., Kawaguchi A., Nagata K., Kato A., Kawaguchi Y., Sato H., Yoneda M., Kai C., Yada T., Suzuki Y., Yamada T., Ozawa T., Kaneki K., Inoue T., Kobayashi M., Kodama T., Wada Y., Sekimizu K., Akimitsu N. (2014) Long noncoding RNA NEAT1-dependent SFPQ relocation from promoter region to paraspeckle mediates IL8 expression upon immune stimuli. Mol. Cell. 53(3), 393–406.
Hirose T., Virnicchi G., Tanigawa A., Naganuma T., Li R., Kimura H., Yokoi T., Nakagawa S., Bénard M., Fox A.H., Pierron G. (2014) NEAT1 long noncoding RNA regulates transcription via protein sequestration within subnuclear bodies. Mol. Biol. Cell. 25, 169–183.
Kaida D., Berg M.G., Younis I., Kasim M., Singh L.N., Wan L., Dreyfuss G. (2010) U1 snRNP protects pre-mRNAs from premature cleavage and polyadenylation. Nature. 468(7324), 664–668.
Gupta R.A., Shah N., Wang K.C., Kim J., Horlings H.M., Wong D.J., Tsai M.-C., Hung T., Argani P., Rinn J.L., Wang Y., Brzoska P., Kong B., Li R., West R.B., van de Vijver M.J., Sukumar S., Chang H.Y. (2010) Long non-coding RNA HOTAIR reprograms chromatin state to promote cancer metastasis. Nature. 464(7291), 1071–1076.
Rinn J.L., Kertesz M., Wang J.K., Squazzo S.L., Xu X., Brugmann S.A., Goodnough H., Helms J.A., Farnham P.J., Segal E., Chang H.Y. (2007) Functional demarcation of active and silent chromatin domains in human HOX loci by noncoding RNAs. Cell. 129(7), 1311–1323.
Tsai M.-C., Manor O., Wan Y., Mosammaparast N., Wang J.K., Lan F., Shi Y., Segal E., Chang H.Y. (2010) Long noncoding RNA as modular scaffold of histone modification complexes. Science. 329(5992), 689–693.
Ernst J., Kheradpour P., Mikkelsen T.S., Shoresh N., Ward L.D., Epstein C.B., Zhang X., Wang L., Issner R., Coyne M., Ku M., Durham T., Kellis M., Bernstein B.E. (2011) Mapping and analysis of chromatin state dynamics in nine human cell types. Nature. 473(7345), 43–49.
Laurent G.S., Shtokalo D., Dong B., Tackett M.R., Fan X., Lazorthes S., Nicolas E., Sang N., Triche T.J., McCaffrey T.A., Xiao W., Kapranov P. (2013) VlincRNAs controlled by retroviral elements are a hallmark of pluripotency and cancer. Genome Biol. 14, 1–20.
Wang Y., Hu S.-B., Wang M.-R., Yao R.-W., Wu D., Yang L., Chen L.-L. (2018) Genome-wide screening of NEAT1 regulators reveals cross-regulation between paraspeckles and mitochondria. Nat. Cell Biol. 20(10), 1145–1158.
Binder S., Hösler N., Riedel D., Zipfel I., Buschmann T., Kämpf C., Reiche K., Burger R., Gramatzki M., Hackermüller J., Stadler P.F., Horn F. (2017) STAT3-induced long noncoding RNAs in multiple myeloma cells display different properties in cancer. Sci. Rep. 7(7976), 1–13.
Guh C.Y., Hsieh Y.H., Chu H.P. (2020) Functions and properties of nuclear lncRNAs – from systematically mapping the interactomes of lncRNAs. J. Biomed. Sci. 27(44), 1–14.
Cetin N.S., Kuo C.C., Ribarska T., Li R., Costa I.G., Grummt I. (2019) Isolation and genome-wide characterization of cellular DNA:RNA triplex structures. Nucl. Acids Res. 47(5), 2306–2321
Rom A., Melamed L., Gil N., Goldrich M.J., Kadir R., Golan M., Biton I., Perry R.B.-T., Ulitsky I. (2019) Regulation of CHD2 expression by the Chaserr long noncoding RNA gene is essential for viability. Nat. Commun. 10(5092), 1–15.
Zhang G., Lan Y., Xie A., Shi J., Zhao H., Xu L., Zhu S., Luo T., Zhao T., Xiao Y., Li X. (2019) Comprehensive analysis of long noncoding RNA (lncRNA) – chromatin interactions reveals lncRNA functions dependent on binding diverse regulatory elements. J. Biol. Chem. 294(43), 15613–15622.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология