

Молекулярная биология, 2022, T. 56, № 5, стр. 783-794
Белок НВх потенцирует реактивацию вируса гепатита В
С. А. Брезгин a, b, А. П. Костюшева a, *, Н. И. Пономарева a, b, В. И. Гегечкори c, Н. П. Кирдяшкина d, С. Р. Айвазян e, Л. Н. Дмитриева e, Л. Н. Кокорева e, В. П. Чуланов a, b, e, f, Д. С. Костюшев a, b
a Институт медицинской паразитологии, тропических и трансмиссивных заболеваний им. Е.И. Марциновского, Первый Московский государственный медицинский университет им. И.М. Сеченова
(Сеченовский Университет)
119048 Москва, Россия
b Научно-технологический университет “Сириус”
354340 Сочи, Россия
c Кафедра фармацевтической и токсикологической химии, Первый Московский государственный медицинский университет им. И.М. Сеченова (Сеченовский Университет)
119991 Москва, Россия
d Научно-исследовательский институт медицины труда им. академика Н.Ф. Измерова
105275 Москва, Россия
e Кафедра инфекционных болезней, Первый Московский государственный медицинский университет
им. И.М. Сеченова (Сеченовский Университет)
119048 Москва, Россия
f Национальный медицинский исследовательский центр фтизиопульмонологии и инфекционных заболеваний
127994 Москва, Россия
* E-mail: dkostushev@gmail.com
Поступила в редакцию 31.03.2022
После доработки 31.03.2022
Принята к публикации 12.04.2022
- EDN: QAKLNM
- DOI: 10.31857/S0026898422050044
Аннотация
Вирус гепатита В (ВГВ) вызывает одно из распространенных инфекционных заболеваний – хронический гепатит В. Во всем мире в контакте с ВГВ находятся около 2 млрд. человек, при этом до 250 млн хронически инфицированы. У пациентов с хронической инфекцией и сопутствующими заболеваниями, прием лекарственных препаратов может приводить к резкому усилению репликации вируса – реактивации инфекции, часто с развитием декомпенсации печени и летальным исходом. Механизмы реактивации инфекции в основном связаны с подавлением иммунного надзора и активацией провирусных сигнальных каскадов. Определение механизмов реактивации ВГВ-инфекции необходимо для рационального использования лекарственных средств и снижения смертности пациентов с хронической формой инфекции. Нами впервые изучена роль вирусного белка НВх в реактивации ВГВ. На модели метилированной рекомбинантной формы генома ВГВ выявлена возможность реактивации вируса из транскрипционно-неактивного состояния, а также изучена возможность реактивации инфекции под действием генотоксических соединений (доксорубицин, пероксид водорода) и препаратов таргетной терапии (сунитиниб, бортезомиб). Показано, что НВх дикого типа и, в большей степени, мутированная форма этого белка без сигнала ядерного экспорта потенцирует репликацию вируса и способствует его реактивации. Впервые показана возможность реактивации ВГВ из транскрипционно-неактивного состояния. Доксорубицин и пероксид водорода вызывают реактивацию ВГВ как на модели транскрипционно-активной, так и неактивной формы генома ВГВ. Сунитиниб вызывает слабую реактивацию ВГВ, тогда как бортезомиб не вызывает реактивации вируса на моделях in vitro.
ВВЕДЕНИЕ
Хронический гепатит В (ХГВ) – одно из распространенных инфекционных заболеваний, смертность от исходов которого превышает 1 млн человек в год [1] – вызывается вирусом гепатита В (ВГВ). Полное излечение ХГВ при использовании современных противовирусных препаратов невозможно. В редких случаях при длительном приеме противовирусных препаратов может происходить сероконверсия по антигенам ВГВ, сопровождаемая подавлением вирусной репликации и снижением вирусной нагрузки с помощью иммуноопосредованных механизмов [2]. Снижение вирусной репликации ассоциировано со значительным уменьшением рисков развития гепатоцеллюлярной карциномы и цирроза печени [1, 3].
При лечении пациентов с ХГВ либо со скрытой формой ХГВ с сопутствующими заболеваниями прием лекарственных препаратов часто приводит к реактивации инфекции – резкому увеличению уровней репликации вируса [4–7]. Последствия такой реактивации могут варьировать от бессимптомных до фатальной декомпенсации функции печени. В этой связи для снижения/предотвращения нежелательных реакций и снижения смертности пациентов с ХГВ и сопутствующими заболеваниями необходим мониторинг маркеров ВГВ-инфекции и рациональное назначение лекарственных средств. Понимание вирусологических основ реактивации ВГВ-инфекции при приеме различных препаратов позволит подбирать нужные препараты и определять оптимальные стратегии лечения подобных пациентов, тем самым сводя к минимуму риски неблагоприятных исходов.
Вирусный белок HBx является основным фактором, ответственным за регуляцию репликации ВГВ в зараженных клетках. Белок HBx участвует в инициации и поддержании транскрипции всех вирусных РНК с матриц кольцевой ковалентно замкнутой ДНК (ккзДНК) ВГВ [8]. В зараженных клетках HBx локализуется как в ядре, так и в цитоплазме. Преимущественная локализация белка определяется уровнем экспрессии HBx и, соответственно, уровнями вирусной репликации. Низкий уровень HBx в клетке способствует ядерной локализации белка, при среднем уровне белок распределен равномерно по клетке, в то время как при высоких значениях вирусной репликации HBx локализуется преимущественно в цитоплазме [9, 10]. В активации вирусного цикла принимают участие как цитоплазматическая, так и внутриядерная формы HBx, причем обе формы вносят практически одинаковый вклад в активацию транскрипции с ккзДНК [9].
НВх осуществляет многофакторную регуляцию транскрипции ккзДНК, главным образом, за счет разрушения комплекса SMC5/6 (structural maintenance of chromosomes 5/6 ) [11] и ремоделирования хроматина, ассоциированного с ккзДНК [12]. HBx связывает Е3-содержащую убиквитин-лигазу DDB1, тем самым способствуя убиквитинированию SMC5/6, что, в свою очередь, приводит к деградации комплекса. В отсутствие HBx либо при нарушении взаимодействия HBx с DDB1 происходит восстановление комплексов SMC5/6, которые связываются с ккзДНК и блокируют транскрипцию вирусного генома. Другой механизм активации вирусного цикла белком HBx в ядре – эпигенетическое ремоделирование хроматина ккзДНК за счет привлечения факторов, вызывающих образование эухроматина (гистоновых деацетилаз HDAC1 и Sirt1, ацетилтрансфераз p300, CBP, PCAF, а также ряда факторов транскрипции, включая E2F1) [13]. Усиление транскрипции ВГВ цитоплазматическим HBx связано с активацией ряда сигнальных путей. За счет C-концевого региона HBx проявляет тропность к мембране митохондрий (значительная часть цитоплазматического HBx связывается с митохондриями) [14]. В результате HBx вызывает образование значительного количества активных форм кислорода, которые вносят одноцепочечные и двухцепочечные разрывы в ДНК инфицированной клетки и активируют сигнальные пути, участвующие в репарации ДНК, включая сигнальный путь АТМ/ATR [15]. Известно, что повреждение генома и кислородный стресс влияют на репликацию многочисленных вирусов [16–18].
Белки ATM (ataxia telangiectasia mutated) и ATR (ataxia telangiectasia and Rad3-related) принадлежат к семейству фосфоинозитид-3-киназа-зависимых киназ и являются ключевыми регуляторами ответа на повреждение ДНК. Мишенями АТМ и ATR служат в том числе факторы прохождения клеточного цикла – киназы контрольных точек 1 и 2 (Chk2, Chk1, checkpoint kinase 2 и 1) соответственно. Впервые роль ATR в репликации ВГВ показали Zhao и соавт. [19, 20]. В частности, показано, что усиление репликации ВГВ происходит за счет ATR-зависимого фосфорилирования Chk1, p53 и Н2АХ. Кроме того, Kim и соавт. [15] показали, что цитоплазматический НВх индуцирует образование активных форм кислорода в клетке, активируя АТМ и вызывая фосфорилирование Chk2. Недавно нашей группой показано, что химиотерапевтический препарат доксорубицин, а также пероксид водорода усиливают транскрипцию АТМ и ATR и вызывают многократное усиление репликации ВГВ [21]. Помимо этого, Luo и соавт. впервые обнаружили участие пути ATR-Chk1 в ключевом этапе поддержания персистенции вируса – образовании ккзДНК ВГВ из предшественника. Подавление образования ATR и Chk1 снижало формирование ккзДНК. В последней работе, посвященной данной проблеме, Lubyova и соавт. [22] показали, что химиотерапевтические лекарственные препараты активируют путь АТМ-Chk2, вызывая фосфорилирование корового белка ВГВ (HBcAg), увеличение инкапсидирования прегеномной РНК ВГВ и образование промежуточных форм генома.
В целом, возможность реактивации ВГВ-инфекции при действии различных препаратов остается малоизученной. Известна возможность реактивации вируса при воздействии иммуносупрессоров, антрациклинов (доксорубицин, эпирубицин) и распространенных химиопрепаратов, а также ритуксимаба и других препаратов, разрушающих В-клетки, ингибиторов фактора некроза опухолей-α, ингибиторов тирозинкиназ, ингибиторов кальцинейрина (циклоспорин, такролимус), использовании антиметаболитов (азатиоприн, 6-меркаптопурин, метотрексат), ингибиторов контрольных точек иммунного ответа PD1/PD1-L/CTLA4 (ниволумаб, пембролизумаб, ипимумаб). Зарегистрированы также единичные случаи реактивации при применении ряда препаратов прямого противовирусного действия для лечения инфекции, вызванной вирусом гепатита С. Описаны различные механизмы, посредством которых в реактивации вируса участвуют разные группы препаратов. В целом, можно выделить два основных механизма: подавление иммунного ответа и повреждение клеточного генома человека с активацией провирусных сигнальных путей. Частота реактивации, вызываемой разными группами препаратов, может варьировать от нескольких процентов до 50–70% [23]. Тем не менее, возможность реактивации ВГВ под действием большинства традиционных химиотерапевтических препаратов до сих пор не анализировали.
К серологическим факторам риска реактивации инфекции относятся: наличие HBsAg, HBeAg в сыворотке крови, высокая вирусная нагрузка (уровни ДНК ВГВ в сыворотке крови >10 000 МЕ/мл) и уровни анти-HBc-антител (≥6.41 МЕ/мл) [23]. Известно, что высокая вирусная нагрузка является одним из факторов риска реактивации ВГВ-инфекции, однако роль НВх, основного регулятора вирусной транскрипции, а также цитоплазматической и ядерной форм белка НВх в реактивации ВГВ-инфекции, фактически не изучена.
В нашей работе изучено влияние белка HBx дикого типа и белка HBxNESM с ядерной локализацией (мутация в сигнале ядерного экспорта, NES (nuclear exportation signal) нарушает выход белка в цитоплазму) [24] на реактивацию ВГВ-инфекции при действии ДНК-повреждающих агентов (доксорубицин, пероксид водорода) и препаратов таргетной терапии (ингибитор тирозинкиназ сунитиниб, ингибитор протеасом бортезомиб).
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Получение рекомбинантной ккзДНК и метилированной рекомбинантной ккзДНК. Рекомбинантная ккзДНК (рккзДНК) ВГВ получена с использованием технологии “minicircle” в системе бактериальной рекомбинации ZYCY10P3S2T E. сoli, как показано ранее [25], и выделена с помощью коммерческого набора MaxiPrep Plasmid Kit (“QIAGEN”, ФРГ). рккзДНК метилировали с помощью M.SssI CpG-метилтрансферазы (“СибЭнзим”, Россия) в соответствии с инструкцией производителя. рккзДНК (1 мг) инкубировали с M.SssI CpG-метилтрансферазой при 37°C в течение 30 мин с последующей очисткой набором Qiagen PCR purification kit (“QIAGEN”, ФРГ). Метилирование ккзДНК подтверждали путем рестрикционного анализа с использованием чувствительной к метилированию рестриктазы Hpa II, как показано ранее [26].
Культивирование клеток человека. Линию клеток HepG2 культивировали в среде DMEM с высоким содержанием глюкозы (4.5 г/л) с добавлением 10% фетальной сыворотки крупного рогатого скота (FBS), 2 мкМ L-глутамина и 1% пенициллина/стрептомицина. Клетки рассевали в культуральные планшеты до достижения ~60% конфлуентности ко дню трансфекции. Клетки трансфицировали с помощью реагента Lipofectamine3000 (“Thermo Fisher Scientific”, США), как описано ранее [21], плазмидой, кодирующей одну из форм белка HBx (HBx, addgene #65463; HBxNESM, addgene #24932) либо контрольной плазмидой, не кодирующей HBx, и рккзДНК в соотношении 1 : 1.
Обработка клеток. Через 48 ч после трансфекции, клетки обрабатывали препаратами либо пероксидом водорода в течение 1 ч в концентрациях, указанных в табл. 1. После инкубации в течение 1 ч клетки дважды промывали фосфатным буфером и добавляли полную культуральную среду.
Таблица 1.
Концентрация веществ, использованных в работе
| Вещество | Концентрация |
|---|---|
| Доксорубицин | 0.2 мкM |
| Н2О2 | 0.4 мМ |
| Сунитиниб | 1.25 мкM |
| Бортезомиб | 50 нМ |
| ДМСО | 1.25 мкM |
Выделение нуклеиновых кислот. Нуклеиновые кислоты выделяли с помощью набора AmpliSens Riboprep (“AmpliSens”, Россия) в соответствии с инструкциями производителя на 4 сут после трансфекции. Для анализа ккзДНК нуклеиновые кислоты обрабатывали T5 экзонуклеазой (“New England Biolabs”, США) в течение 60 мин при 37°C с инактивацией фермента при 70°C в течение 20 мин.
ПЦР-анализ. Количественную полимеразную цепную реакцию (кПЦР) в режиме реального времени проводили с использованием флуоресцентных зондов TaqMan. В образце нуклеиновых кислот анализировали ДНК ВГВ. В образце, обработанном T5 экзонуклеазой, определяли ккзДНК ВГВ. Праймеры для амплификации ДНК и ккзДНК ВГВ использовали согласно [27, 28]. Уровни ДНК ВГВ и ккзДНК нормировали на уровни ДНК β-глобина человека. Относительные уровни рассчитывали с помощью метода ΔΔCt.
Иммунохимический анализ. Клетки HepG2 рассаживали в культуральные планшеты с покровными стеклами. В конечной точке эксперимента клетки фиксировали 4%-ным раствором параформальдегида в течение 10 мин, после чего отмывали 3 раза Трис-гидрохлоридом (50 мМ, рН 8.0) с последующей инкубацией в течение 30 мин в блокирующем буфере (0.02% Тритон Х-100, 10% лошадиная сыворотка, 150 мМ NaCl в растворе Трис-гидрохлорида (50 мМ, рН 8.0). Далее стекла инкубировали с первичными поликлональными кроличьими анти-НВх-антителами (Abcam ab39716, Великобритания) в течение 1 ч при комнатной температуре, затем стекла отмывали 3 раза в течение 10 мин отмывочным буфером (0.02% Тритон Х-100, 200 мМ NaCl в растворе Трис-гидрохлорида (50 мМ, рН 8.0) и инкубировали со вторичными анти-кроличьими антителами козы, конъюгированными с меткой Alexa Fluor 594 (Abcam ab150080) и реактивом для окрашивания ядер Hoechst33342 (Abcam ab228551) при комнатной температуре в течение 1 ч. Стекла заключали в среду Fluoroshield (Abcam ab104135). Визуализацию проводили на флуоресцентном микроскопе Leica DMI6000 с иммерсионными объективами ×100. Работу проводили с использованием оборудования ЦКП ИБР им. Н.К. Кольцова РАН.
Статистический анализ. Статистическую обработку данных проводили в программе GraphPad Prism 7 с помощью t-критерия Стьюдента. Различия считали значимыми при р < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Сверхэкспрессия HBx и HBxNESM усиливает цикл вируса гепатита В
Первым этапом изучения влияния HBx на реактивацию ВГВ-инфекции при воздействии различных препаратов стала оценка влияния НВх и мутированной формы НВх на репликацию ВГВ per se.
Для оценки отличий в клеточной локализации НВх клетки HepG2 трансфицировали плазмидами, кодирующими один из белков HBx (HBx дикого типа либо HBxNESM), а также рккзДНК. Через 48 ч после трансфекции проводили иммуноцитохимический анализ распределения белка НВх в клетках HepG2 (рис. 1а). Как и ожидалось, белок дикого типа распределен по ядру и цитоплазме клеток, в то время как белок с мутантным сигналом ядерного экспорта NES (HBxNESM) локализовался исключительно в ядре и не детектировался в цитоплазме (рис. 1а).
Рис. 1.
Влияние HBx и HBxNESM на репликацию ВГВ. а – Иммуноцитохимическое выявление белка HBx (красное свечение). Ядра окрашены красителем Hoescht33324 (синее свечение). б – Относительные уровни внутриклеточной ДНК ВГВ (черные столбцы) и ккзДНК ВГВ (серые столбцы) при действии HBx либо HBxNESM на модели с рккзДНК по данным ПЦР в реальном времени. Нормирование проведено на уровни ДНК β-глобина человека. Планки погрешностей соответствуют стандартным отклонениям. Уровни значимости: +p < 0.05; Δp < 0.01; οp < 0.005; *p < 0.001.
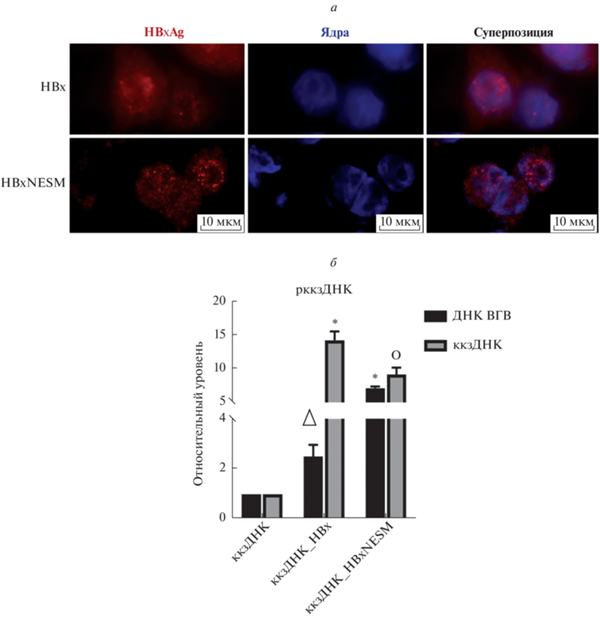
Далее с помощью ПЦР-анализа уровней суммарной внутриклеточной ДНК ВГВ и ккзДНК на модели рккзДНК изучены эффекты HBx и HBxNESM на репликацию ВГВ (рис. 1б). Показано, что оба вида белка НВх вызывают значительное увеличение уровней ДНК ВГВ (НВх – в ~2.5 раза и HBxNESM – в ~7 раз) и ккзДНК ВГВ (НВх – в ~14 раз и НВхNESM – в ~9 раз) (рис. 1б). Белок HBxNESM вызывал более значимое увеличение уровней ДНК ВГВ, чем НВх дикого типа, но менее существенно повышал уровни ккзДНК (рис. 1б). Следовательно, НВх дикого типа и НВхNESM усиливают репликацию ВГВ, но с неодинаковой выраженностью.
Белок НВх усиливает реактивацию ВГВ-инфекции при действии генотоксических агентов
Далее было изучено влияние HBx дикого типа и HBxNESM на реактивацию ВГВ при действии генотоксических агентов (доксорубицина –докс, и пероксида водорода – Н2О2) и препаратов таргетной терапии (сунитиниба – Сун, и бортезомиба – Борт). Таргетные препараты широко используются в терапии онкологических заболеваний, однако механизм их действия основан не на прямом повреждении генома, как в случае генотоксических агентов, а на блокаде отдельных сигнальных путей.
С этой целью клетки, трансфицированные плазмидой НВх, НВхNESM либо некодирующей плазмидой, а также рккзДНК, обрабатывали соединениями в выбранных концентрациях. Контрольный образец обрабатывали раствором диметилсульфоксида (ДМСО).
Обработка клеток доксорубицином, пероксидом водорода либо сунитинибом приводила к увеличению уровней ДНК и ккзДНК ВГВ в ~2–7 раз (рис. 2а–в). Напротив, бортезомиб снижал параметры ВГВ (рис. 2г). В условиях сверхэкспрессии НВх либо HBxNESM уровни реактивации ВГВ были значительно выше при использовании генотоксических агентов (рис. 2а, б). Уровни ккзДНК/ДНК ВГВ увеличивались в ~40–200 раз при обработке доксорубицином и пероксидом водорода в клетках с гиперпродукцией НВх/HBxNESM (рис. 2а, б), в то время как сунитиниб приводил к ~4–5-кратному увеличению уровней ДНК ВГВ (рис. 2в). В контрольной группе с бортезомибом отмечено снижение вирусной репликации, однако в условиях сверхэкспрессии НВх/HBxNESM бортезомиб индуцировал ~2–5-кратное увеличение уровней ДНК и ккзДНК ВГВ (рис. 2г). Повышение уровней вирусной репликации при использовании препаратов таргетной терапии совместно с НВх было сопоставимым с действием НВх с ДМСО (рис. 1, рис. 2в, г).
Рис. 2.
Влияние белков HBx на реактивацию ВГВ-инфекции под действием генотоксических агентов и препаратов для таргетной терапии. Уровни репликации ВГВ при действии доксорубицина (а), пероксида водорода (б), сунитиниба (в) и бортезомиба (г). ДНК ВГВ и ккзДНК анализировали с помощью ПЦР в реальном времени. Контроль – клетки, трансфицированные рккзДНК; Докс, Н2О2, Сун, Борт – клетки, трансфицированные рккзДНК и обработанные соответствующим агентом. Планки погрешностей соответствуют стандартным отклонениям. *p < 0.05; **p < 0.01; ***p < < 0.005; ****p < 0.001.
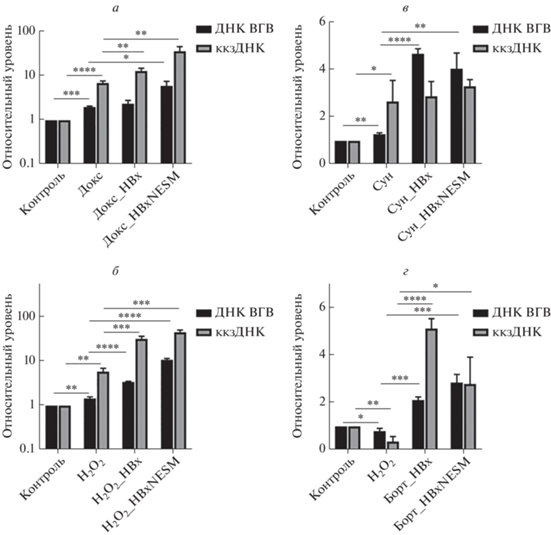
Таким образом, сверхэкспрессия различных форм HBx потенцирует реактивацию ВГВ-инфекции при действии генотоксических агентов. Сунитиниб слабо влияет на реактивацию ВГВ, бортезомиб не вызывает реактивации ВГВ in vitro.
Реактивация транскрипционно-неактивной метилированной ккзДНК ВГВ
Основная форма генома ВГВ, ккзДНК, в ходе патогенеза ХГВ и при приеме различных препаратов [29] подвергается различным эпигенетическим модификациям, таким как модификация гистонов и метилирование ДНК [30]. Метилирование ккзДНК снижает транскрипцию и подавляет репликацию ВГВ. Действительно, гиперметилирование ккзДНК приводит к снижению образования прегеномной РНК ВГВ на >90% [26]. Криптическая транскрипционно-неактивная ккзДНК может служить своего рода депо для восстановления вирусной репликации и реактивации ВГВ-инфекции. Особенно это касается пациентов с низкими либо недетектируемыми уровнями вирусных маркеров (ДНК ВГВ), и с анти-HBc-антителами (маркером контакта организма с ВГВ). Вместе с этим, возможность реактивации транскрипционно-неактивной формы генома ВГВ ранее не была исследована. В данной работе впервые изучена возможность реактивации репликации гиперметилированного генома вируса при действии различных форм НВх-белка, а также и при действии генотоксических агентов и препаратов для таргетной терапии (б).
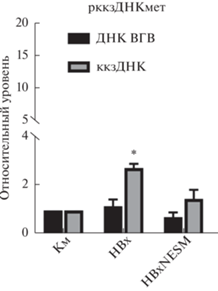
Нами показано, что увеличение репликации гиперметилированной рккзДНК ВГВ (~трехкратное повышение уровней ккзДНК) происходит только при использовании НВх дикого типа (рис. 3). HBxNESM не вызывает статистически значимого увеличения уровней вирусных интермедиатов.
Рис. 3.
Влияние белков НВх на репликацию транскрипционно-неактивной рккзДНК. Планки погрешностей соответствуют стандартным отклонениям. *p < 0.001. Км – контрольный образец с метилированной рккзДНК ВГВ.
В рамках данной работы впервые выявлена возможность реактивации гиперметилированной рккзДНК ВГВ под действием генотоксических агентов (рис. 4а, б). Доксорубицин не влиял на уровни ДНК ВГВ, однако вызывал увеличение уровней ккзДНК в ~5.5 раз. Обработка пероксидом водорода повышала уровни как ДНК ВГВ, так и ккзДНК ВГВ в ~4 раза (рис. 4б).
Рис. 4.
Реактивация транскрипционно-неактивной метилированной ккзДНК ВГВ. Уровни ДНК ВГВ и ккзДНК ВГВ при обработке клеток с метилированной рккзДНК доксорубицином (а), пероксидом водорода (б), сунитинибом (в) и бортезомибом (г). Планки погрешностей соответствуют стандартным отклонениям. *p < 0.05; **p < 0.01; ***p < 0.005; ****p < 0.001.
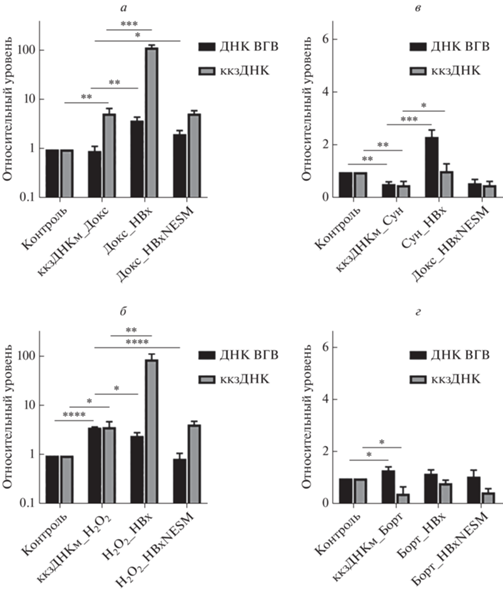
С другой стороны, среди препаратов таргетной терапии только бортезомиб незначительно увеличивал уровни ДНК ВГВ (рис. 4г). Реактивация вирусной репликации происходила только при продукции белка НВх дикого типа и действии генотоксических соединений. Совместное действие сунитиниба либо бортезомиба с НВх не усиливало репликацию ВГВ в сравнении с действием индивидуальных факторов (рис. 3, рис. 4в, г). При экспрессии HBxNESM уровни вирусных маркеров не отличались от уровней в образцах, обработанных химическим агентом. Наиболее значительная реактивация (вплоть до 100-кратного увеличения ккзДНК) происходила при одновременной продукции НВх и обработке генотоксическими соединениями (рис. 4а, б).
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Реактивацию ВГВ можно купировать с помощью современных препаратов прямого противовирусного действия [31]. Тем не менее, у пациентов с хронической формой инфекции нередко возможны тяжелые либо даже смертельные последствия реактивации ВГВ. По различным данным смертность при реактивации ВГВ может варьировать от 0–20 до >50% [31]. Нами изучена роль ключевого фактора транскрипции ВГВ – белка НВх – в реактивации ВГВ генотоксическими препаратами и препаратами таргетной терапии. Белок НВх необходим для инициации и поддержания репликации ВГВ. Взаимодействуя с ккзДНК в ядре, НВх активирует транскрипцию вирусных РНК. В цитоплазме НВх активирует пути трансдукции сигналов, способствующие вирусной репликации, включая пути выживания клетки, метаболизма, пролиферации и транскрипции. HBx взаимодействует более чем со 100 различными белками в клетке [32]. Поскольку цитоплазматический НВх индуцирует образование активных форм кислорода и повреждение генома клетки, этот белок может также стимулировать репликацию ВГВ за счет активации путей репарации повреждений ДНК (например, АТМ и ATR) [21]. Вместе с этим, роль НВх в реактивации ВГВ до сих пор оставалась неизученной. В данной работе впервые показано, что белок НВх может значительно потенцировать реактивацию ВГВ-инфекции, причем потенцирование происходит при использовании как НВх дикого типа, так и НВхNESM (рис. 2а, б). Интересно, что усиление реактивации наблюдается только при действии НВх с генотоксическими агентами, в то время как потенцирования действия препаратов таргетной терапии не происходит (рис. 2в, г). Последнее указывает не только на различия в механизмах реактивации препаратами разных групп, но и позволяет предположить существование особенностей влияния НВх на жизненный цикл ВГВ в измененных условиях функционирования клеток.
Как отмечено выше, существуют два принципиальных механизма реактивации ВГВ: за счет подавления иммунного ответа (кортикостероиды, ритуксимаб, ингибиторы фактора некроза опухолей-α и др.) и индукции вирусной репликации в инфицированных клетках, главным образом за счет активации путей репарации повреждений ДНК (доксорубицин, винкристин, цисплатин, бусульфан, циклофосфамид и др.) [33]. Тем не менее, возможность реактивации ВГВ за счет непосредственного воздействия большинства лекарственных препаратов с установленным иммуносупрессорным действием на инфицированные клетки не изучали.
Ряд препаратов таргетной терапии, включая ингибиторы протеасом и ингибиторы тирозинкиназ, имеют средний (от 1 до 10%) риск реактивации ВГВ у хронически инфицированных пациентов [19]. В отличие от генотоксических соединений (доксорубицин и пероксид водорода), которые индуцируют выраженную реактивацию ВГВ-инфекции (рис. 2), препарат таргетной терапии бортезомиб не вызывает реактивацию ВГВ, только сунитиниб слабо увеличивает репликацию вируса (рис. 2в, г). Реактивация ВГВ под действием генотоксических соединений связана с активацией сигнальных каскадов АТМ и ATR. В то же время механизмы действия препаратов таргетной терапии до сих пор не изучены.
Сунитиниб – мультитаргетный ингибитор тирозинкиназ. Мишенью сунитиниба служат рецепторы фактора роста эндотелия сосудов, FMS-подобной тирозинкиназы-3 и колониестимулирующего фактора [34]. Помимо этого, сунитиниб оказывает миелосупрессорное действие за счет подавления активности c-kit [35]. Предполагается, что механизмом реактивации ВГВ при действии сунитиниба как раз и является миелосупрессорная активность. Вместе с этим, на модели клеток in vitro нами впервые показана слабая реактивирующая активность сунитиниба (рис. 2в). Следовательно, сунитиниб может вызывать реактивацию ВГВ-инфекции у пациентов с хроническим гепатитом В как за счет миелосупрессорных эффектов, так и за счет прямого воздействия на ВГВ-инфицированные клетки. Ранее показали, что сунитиниб вызывает повреждение ДНК и увеличивает образование фокусов γ-H2AX (маркеров разрывов ДНК) в клетках, а также незначительно усиливает экспрессию ATM и ATR [21, 22], что может стимулировать жизненный цикл вируса. Кроме того, сунитиниб вызывает остановку клеточного цикла в фазе G1/S за счет увеличения экспрессии фактора транскрипции DEC1 [23]. Известно, что прекращение деления клеток, инфицированных ВГВ, способствует накоплению ккзДНК ВГВ в клетках и увеличению вирусной репликации [36]. Определение механизмов провирусного действия сунитиниба требует дальнейших исследований.
Бортезомиб – ингибитор протеасом и антинеопластический агент, используемый при множественной миеломе и некоторых лимфомах. Бортезомиб также негативно влияет на функции В-клеток и плазматических клеток, включая ВГВ-специфичные [19]. Обработка бортезомибом культивируемых клеток in vitro не вызывала реактивации ВГВ. Напротив, нами отмечено снижение параметров вирусного цикла, особенно уровней ккзДНК ВГВ (рис. 2г). С использованием в качестве модели трансгенных мышей Bandi и соавт. показали, что бортезомиб значительно снижает (около 90%) параметры вирусного цикла [24], что согласуется с нашими данными. Возможными механизмами снижения вирусного цикла могут быть увеличение времени жизни вирусной полимеразы за счет блокады протеасомной деградации и ускорение инкапсидирования прегеномной РНК ВГВ. С другой стороны, влияние бортезомиба на вирусный цикл в работе Bandi и соавт. было дозозависимым: при этом средняя доза (1 мг/кг) подавляла вирусный цикл, а высокая (5 мг/кг) оказывала провирусное действие [24]. Описано также увеличение уровней HBx в клетке, вызванное действием других ингибиторов протеасом [37–39], за счет блокирования протеасомного разрушения НВх, что может приводить к увеличению вирусного цикла, объясняя провирусное действие высоких доз бортезомиба.
Один из наименее изученных аспектов вирусного гепатита В – эпигенетическая гетерогенность внутриядерного пула ккзДНК, основной формы генома ВГВ, и возможность реактивации/восстановления репликации вируса из транскрипционно-инактивированного состояния [40]. Эти аспекты приобретают особую актуальность в связи с разработкой методов эпигенетического подавления активности ккзДНК и других перспективных подходов к функциональному излечению ХГВ [41, 42]. Молекулы ккзДНК содержат три–четыре канонических островка CpG, которые служат мишенями для ДНК-метилтрансфераз. В зависимости от стадии заболевания и вирусологических особенностей при ХГВ выявляют молекулы ккзДНК с разной степенью метилирования. Метилирование ккзДНК ассоциировано со снижением вирусной репликации, частой конверсией ВГВ по HBeAg, в целом, оно ассоциировано с более низкой вирусной нагрузкой [30]. В нашей работе впервые установлена возможность восстановления вирусной репликации белком НВх из гиперметилированного состояния генома ВГВ. Экспрессия белка НВх дикого типа более чем в 3 раза увеличивала уровни ккзДНК (рис. 3). Более 10 лет назад было показано, что внутриядерный НВх связывается с минихромосомами ккзДНК и эпигенетически активирует транскрипцию вируса [12]. Однако возможность активации транскрипции гиперметилированной (транскрипционно-неактивной) ккзДНК белком НВх не изучали. Мы впервые показали, что внутриядерный белок HBxNESM не вызывает активацию вирусной репликации, в то время как НВх дикого типа способствует восстановлению цикла ВГВ (рис. 3). Эти данные указывают на то, что восстановление репликации из гиперметилированного состояния ккзДНК может быть связано не с прямым взаимодействием НВх с минихромосомами, а с активацией цитоплазматических провирусных каскадов. Кроме того, впервые показана возможность реактивации ВГВ из гиперметилированного состояния при воздействии генотоксических агентов (рис. 4а, б). Подобная реактивация потенцируется только белком НВх дикого типа (рис. 4а, б).
Известно, что после разрешения острой ВГВ-инфекции ккзДНК может сохраняться в гепатоцитах человека в течение всей жизни. Отсутствие активной вирусной инфекции в таких случаях объясняют развитием иммунологического контроля и подавлением активности ккзДНК [43]. Действительно, у лиц с ослабленным иммунитетом и историей острой ВГВ-инфекции в анамнезе возможна реактивация инфекции, обусловленная активацией транскрипции остаточной ккзДНК [44]. Наши результаты впервые указывают на возможность реактивации транскрипционно-неактивной ккзДНК под действием генотоксических агентов, что может иметь существенное значение для лечения лиц с историей инфекции ВГВ (определяется по наличию анти-HBc-антител).
В заключение следует отметить, что реактивация ВГВ-инфекции остается актуальной проблемой современного здравоохранения. Определение спектра лекарственных препаратов и механизмов реактивации инфекции необходимо для снижения частоты нежелательных побочных явлений и смертности при лечении лиц с историей ВГВ-инфекции.
Работа выполнена при поддержке Российского фонда фундаментальных исследований (№ 20-515-12010 и № 20-015-00442).
В данной работе не использовали людей и животных в качестве объекта исследования.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Kostyusheva A., Kostyushev D., Brezgin S., Volchkova E., Chulanov V. (2018) Clinical implications of hepatitis B virus RNA and covalently closed circular DNA in monitoring patients with chronic hepatitis B today with a gaze into the future: the field is unprepared for a sterilizing cure. Genes (Basel). 9, 483.
Perceau G. Diris N., Estines O., Derancourt C., Levy S., Bernard P. (2006) Late lethal hepatitis B virus reactivation after rituximab treatment of low-grade cutaneous B-cell lymphoma. Br. J. Dermatol. 155, 1053–1056. https://doi.org/10.1111/j.1365-2133.2006.07451.x
Чуланов В.П., Зуева А.П., Костюшев Д.С., Брезгин С.А., Волчкова Е.В., Малеев В.В. (2017) Гепатит С стал излечим. Гепатит В – следующий? Терапевтический архив (архив до 2018 г.) 89, 4–13.
Xu L., Tu Z., Xu G., Hu J.-L., Cai X.-F., Zhan X.-X., Wang Y.-W., Huang Y., Chen J., Huang A.-L. (2015) S‑Phase arrest after vincristine treatment may promote hepatitis B virus replication. World. J. Gastroenterol. 21, 1498–1509. https://doi.org/10.3748/wjg.v21.i5.1498
Chen Y.-F., Chong C.-L., Wu Y.-C., Wang Y.-L., Tsai K.-N., Kuo T.-M., Hong M.-H., Hu C.-P., Chen M.-L., Chou Y.-C., Chang C. (2015) Doxorubicin activates hepatitis B virus replication by elevation of p21 (Waf1/Cip1) and C/EBPα expression. PLoS One. 10, e0131743–e0131743. https://doi.org/10.1371/journal.pone.0131743
Li X., Pan E., Zhu J., Xu L., Chen X., Li J., Liang L., Hu Y., Xia J., Chen J., Chen W., Hu J., Wang K., Tang N., Huang A. (2018) Cisplatin enhances hepatitis B virus replication and PGC-1α expression through endoplasmic reticulum stress. Sci. Rep. 8, 3496. https://doi.org/10.1038/s41598-018-21847-3
Pisaturo M., Macera M., Alessio L., Calo F., Coppola N. (2019) Hepatitis B virus (HBV) reactivation following pharmacological eradication of hepatitis C virus (HCV). Viruses. 11(9), 850. https://doi.org/10.3390/v11090850
Lucifora J., Arzberger S., Durantel D., Belloni L., Strubin M., Levrero M., Zoulim F., Hantz O., Protzer U. (2011) Hepatitis B virus X protein is essential to initiate and maintain virus replication after infection. J. Hepatol. 55, 996–1003.
Cha M.-Y., Ryu D.-K., Jung H.-S., Chang H.-E., Ryu W.-S. (2009) Stimulation of hepatitis B virus genome replication by HBx is linked to both nuclear and cytoplasmic HBx expression. J. Gen. Virol. 90, 978–986.
Henkler F., Hoare J., Waseem N., Goldin R.D., McGarvey M.J., Koshy R., King I.A. (2001) Intracellular localization of the hepatitis B virus HBx protein. J. Gen. Virol. 82, 871–882.
Murphy C.M., Xu Y., Li F., Nio K., Reszka-Blanco N., Li X., Wu Y., Yu Y., Xiong Y., Su L. (2016) Hepatitis B virus X protein promotes degradation of SMC5/6 to enhance HBV replication. Cell. Rep. 16, 2846–2854.
Belloni L., Pollicino T., Nicola F.De, Guerrieri F., Raffa G., Fanciulli M. (2009) Nuclear HBx binds the HBV minichromosome and modifies the epigenetic regulation of cccDNA function. Proc. Natl. Acad. Sci. USA. 106(47), 19975–19979. https://doi.org/10.1073/pnas.0908365106
Rivière L., Gerossier L., Ducroux A., Dion S., Deng Q., Michel M.L., Buendia M.A., Hantz O., Neuveut C. (2015) HBx relieves chromatin-mediated transcriptional repression of hepatitis B viral cccDNA involving SETDB1 histone methyltransferase. J. Hepatol. 63(5), 1093‒1002. https://doi.org/10.1016/j.jhep.2015.06.023
Clippinger A.J., Bouchard M.J. (2008) Hepatitis B virus HBx protein localizes to mitochondria in primary rat hepatocytes and modulates mitochondrial membrane potential. J. Virol. 82, 6798–6811.
Kim S., Lee H.-S., Ji J.-H., Cho M.-Y., Yoo Y.-S., Park Y.-Y., Cha H.-J., Lee Y., Kim Y., Cho H (2015) Hepatitis B virus X protein activates the ATM–Chk2 pathway and delays cell cycle progression. J. Gen. Virol. 96, 2242–2251.
Ivanov A.V., Valuev-Elliston V.T., Tyurina D.A., Ivanova O.N., Kochetkov S.N., Bartosch B., Isaguliants M.G. (2017) Oxidative stress, a trigger of hepatitis C and B virus-induced liver carcinogenesis. Oncotarget. 8, 3895.
Ivanov A.V., Smirnova O.A., Ivanova O.N., Masalova O.V., Kochetkov S.N., Isaguliants M.G. (2011) Hepatitis C virus proteins activate NRF2/ARE pathway by distinct ROS-dependent and independent mechanisms in HUH7 cells. PLoS One. 6, e24957.
Ivanov A.V., Bartosch B., Smirnova O.A., Isaguliants M.G., Kochetkov S.N. (2013) HCV and oxidative stress in the liver. Viruses. 5. 439–469.
Zhao F., Hou N.-B., Song T., He X., Zheng Z.-R., Ma Q.-J., Li L., Zhang Y.-H., Zhong H. (2008) Cellular DNA repair cofactors affecting hepatitis B virus infection and replication. World J. Gastroenterol. 14, 5059.
Zhao F., Hou N.-B., Yang X.-L., He X., Liu Y., Zhang Y.-H., Wei C.-W., Song T., Li L., Ma Q.-J. (2008) Ataxia telangiectasia-mutated-Rad3-related DNA damage checkpoint signaling pathway triggered by hepatitis B virus infection. World J. Gastroenterol. 14, 6163.
Kostyusheva A., Brezgin S., Bayurova E., Gordeychuk I., Isaguliants M., Goptar I., Urusov F., Nikiforova A., Volchkova E., Kostyushev D. (2019) ATM and ATR expression potentiates HBV replication and contributes to reactivation of HBV infection upon DNA damage. Viruses. 11, 997.
Lubyova B., Tikalova E., Krulova K., Hodek J., Zabransky A., Hirsch I., Weber J. (2021) ATM-dependent phosphorylation of hepatitis B core protein in response to genotoxic stress. Viruses. 13, 2438.
Chang Y., Jeong S.W., Jang J.Y. (2021) Hepatitis B virus reactivation associated with therapeutic interventions. Front. Med. 8, 770124.
Forgues M., Marrogi A.J., Spillare E.A., Wu C.-G., Yang Q., Yoshida M., Wang X.W. (2001) Interaction of the hepatitis B virus X protein with the Crm1-dependent nuclear export pathway. J. Biol. Chem. 276, 22797–22803.
Guo X., Chen P., Hou X., Xu W., Wang D., Wang T., Zhang L., Zheng G., Gao Z., He C.-Y. (2016) The recombined cccDNA produced using minicircle technology mimicked HBV genome in structure and function closely. Sci. Rep. 6, 1–10.
Kostyushev D., Brezgin S., Kostyusheva A., Zarifyan D., Goptar I., Chulanov V. (2019) Orthologous CRISPR/Cas9 systems for specific and efficient degradation of covalently closed circular DNA of hepatitis B virus. Cell. Mol. Life. Sci. 76, 1779–1794. https://doi.org/10.1007/s00018-019-03021-8
Kostyushev D., Kostyusheva A., Brezgin S., Zarifyan D., Utkina A., Goptar I., Chulanov V. (2019) Suppressing the NHEJ pathway by DNA-PKcs inhibitor NU7026 prevents degradation of HBV cccDNA cleaved by CRISPR/Cas9. Sci. Rep. 9, 1–11. https://doi.org/10.1038/s41598-019-38526-6
Костюшева А.П., Костюшев Д.С., Брезгин С.А., Зарифьян Д.Н., Волчкова Е.В., Чуланов В.П. (2019) Низкомолекулярные ингибиторы путей репарации двухцепочечных разрывов ДНК усиливают противовирусное действие системы CRISPR/Cas9 на моделях вируса гепатита В. Молекуляр. биология. 53(2), 311‒323.
Tropberger P., Mercier A., Robinson M., Zhong W., Ganem D.E., Holdorf M. (2015) Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc. Natl. Acad. Sci. USA. 112, E5715–E5724. https://doi.org/10.1073/pnas.1518090112
Костюшев Д.С., Зуева А.П., Брезгин С.А., Липатников А.Д., Волчкова Е.В., Малеев В.В., Чуланов В.П. (2018) Роль ДНК-метилтрансфераз в жизненном цикле вируса гепатита B и патогенезе хронического гепатита B. Вопросы вирусологии. 63, 19–29.
Garg H., Sarin S.K., Kumar M., Garg V., Sharma B.C., Kumar A. (2011) Tenofovir improves the outcome in patients with spontaneous reactivation of hepatitis B presenting as acute-on-chronic liver failure. Hepatology. 53, 774–780.
Wu Z.-J., Zhu Y., Huang D.-R., Wang Z.-Q. (2010) Constructing the HBV-human protein interaction network to understand the relationship between HBV and hepatocellular carcinoma. J. Exp. Clin. Cancer Res. 29, 1–10.
Loomba R., Liang T.J. (2017) Hepatitis B reactivation associated with immune suppressive and biological modifier therapies: current concepts, management strategies, and future directions. Gastroenterology. 152, 1297–1309.
Roskoski Jr.R. (2007) Sunitinib: a VEGF and PDGF receptor protein kinase and angiogenesis inhibitor. Biochem. Biophys. Res. Commun. 356, 323–328.
DiNitto J.P., Deshmukh G.D., Zhang Y., Jacques S.L., Coli R., Worrall J.W., Diehl W., English J.M., Wu J.C. (2010) Function of activation loop tyrosine phosphorylation in the mechanism of c-Kit auto-activation and its implication in sunitinib resistance. J. Biochem. 147, 601–609.
Chong C.-L., Chen M.-L., Wu Y.-C., Tsai K.-N., Huang C.-C., Hu C., Jeng K.-S., Chou Y.-C., Chang C. (2011) Dynamics of HBV cccDNA expression and transcription in different cell growth phase. J. Biomed. Sci. 18, 1–16.
Zhang Z., Protzer U., Hu Z., Jacob J., Liang T.J. (2004) Inhibition of cellular proteasome activities enhances hepadnavirus replication in an HBX-dependent manner. J. Virol. 78, 4566–4572.
Kim J.-H., Kang S., Kim J., Ahn B.-Y. (2003) Hepatitis B virus core protein stimulates the proteasome-mediated degradation of viral X protein. J. Virol. 77, 7166–7173.
Hu Z., Zhang Z., Doo E., Coux O., Goldberg A.L., Liang T.J. (1999) Hepatitis B virus X protein is both a substrate and a potential inhibitor of the proteasome complex. J. Virol. 73, 7231–7240.
Tropberger P., Mercier A., Robinson M., Zhong W., Ganem D.E., Holdorf M. (2015) Mapping of histone modifications in episomal HBV cccDNA uncovers an unusual chromatin organization amenable to epigenetic manipulation. Proc. Natl. Acad. Sci. USA. 112, E5715–E5724.
Kostyushev D., Kostyusheva A., Ponomareva N., Brezgin S., Chulanov V. (2022) CRISPR/Cas and hepatitis B therapy: technological advances and practical barriers. Nucl. Acid. Ther. 32, 14–28.
Koumbi L., Karayiannis P. (2016) The epigenetic control of hepatitis B virus modulates the outcome of infection. Front. Microbiol. 6, 1–9. https://doi.org/10.3389/fmicb.2015.01491
Rehermann B., Ferrari C., Pasquinelli C., Chisari F.V. (1996) The hepatitis B virus persists for decades after patients’ recovery from acute viral hepatitis despite active maintenance of a cytotoxic T-lymphocyte response. Nat. Med. 2, 1104–1108.
Yang S., Zeng W., Zhang J., Lu F., Chang J., Guo J.-T. (2021) Restoration of a functional antiviral immune response to chronic HBV infection by reducing viral antigen load: if not sufficient, is it necessary? Emerg. Microbes Infect. 10, 1545–1554. https://doi.org/10.1080/22221751.2021.1952851
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология