

Молекулярная биология, 2022, T. 56, № 5, стр. 832-847
Транскрипционный фактор Sp1 в регуляции экспрессии генов, кодирующих компоненты сигнальных путей MAPK, JAK/STAT и PI3K/Akt
К. А. Иваненко a, В. С. Прасолов a, Э. Р. Хабушева a, *
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
* E-mail: vr.elmira@gmail.com
Поступила в редакцию 18.03.2022
После доработки 15.04.2022
Принята к публикации 17.04.2022
- EDN: VYBGXK
- DOI: 10.31857/S0026898422050081
Аннотация
Транскрипционный фактор Sp1, входящий в семейство белков Sp/KLF, связывает GC-богатые участки регуляторных областей генов и участвует в регуляции пролиферации, апоптоза и дифференцировки клеток, а также ангиогенеза. Высокий уровень экспрессии гена SP1 и нарушение транскрипционной активности кодируемого им белка, обусловленные посттрансляционными модификациями, обнаруживают при раке молочной, поджелудочной и щитовидной железы, раке легкого и желудка, в глиомах, а также при врожденном пороке сердца и нейродегенеративных заболеваниях, включая болезни Гентингтона и Паркинсона. Связываясь с GC-богатыми участками регуляторных областей генов, кодирующих компоненты сигнальных путей MAPK, p38, JAK/STAT, PI3K/Akt, Sp1 участвует в контроле пролиферации, дифференцировки и гибели клеток. Кроме того, киназы, входящие в состав этих сигнальных путей, способны влиять на транскрипционную активность Sp1 путем фосфорилирования определенных аминокислотных остатков, что приводит к изменению эффективности его связывания с кофакторами и регуляторными областями генов. В обзоре представлены данные о взаимосвязи фактора Sp1 с активностью сигнальных путей MAPK, p38, JAK/STAT и PI3K/Akt в норме и при различных патологиях, включая злокачественные заболевания.
ВВЕДЕНИЕ
Белок Sp1 входит в семейство Sp-подобных белков/KLF (Krüppel-like factor) – факторов транскрипции с доменами типа “цинковых пальцев” Сys2Нis2, которые связывают GC-богатые участки в регуляторных областях генов [1]. Белки Sp1, Sp3 и Sp4 содержат два трансактивационных домена, обогащенных остатками глутамина, что отличает их от белка Sp2, у которого такой домен всего один. Sp1 и Sp3 обнаружены во всех тканях человека, а Sp4 находится преимущественно в нейральных клетках. Sp1 человека состоит из 785 аминокислотных остатков и имеет молекулярную массу 81 кДа. Гомологи белка Sp1 и других белков этого семейства найдены в клетках большого числа организмов, включая нематод, мышей, крыс, полосатого данио (Danio rerio), кроликов.
Фактор Sp1 необходим для нормального эмбрионального [2] и постнатального развития; он регулирует процессы пролиферации и апоптоза [4, 5], ангиогенеза [5] и дифференцировки [7–9]. Кроме того, Sp1 вовлечен в развитие противовирусного клеточного ответа, в том числе посредством связывания с вирусными промоторами [2, 10–12]. Обнаружено, что аномальная сверхэкспрессия гена SP1, а также нарушение транскрипционной активности кодируемого им белка связаны с развитием нейродегенеративных заболеваний, таких как болезни Гентингтона [11] и Паркинсона [12]; онкологических заболеваний, включая рак легкого [13], поджелудочной железы [14], молочной железы [15], желудка [16], щитовидной железы [17] и глиому [18]. На данный момент подавление транскрипционной активности Sp1 низкомолекулярными соединениями рассматривают как один из многообещающих подходов к терапии злокачественных заболеваний человека [19].
В обзоре представлены данные об участии Sp1 в регуляции экспрессии генов, кодирующих компоненты сигнальных путей MAPK, p38, JAK/STAT и PI3K/Akt, а также о том, как активность этих сигнальных каскадов связана с транскрипционной активностью белка Sp1 в клетках человека и животных в норме и при ряде патологий, включая злокачественные заболевания человека.
Sр1 И MAP-КИНАЗНЫЙ СИГНАЛЬНЫЙ ПУТЬ ERK
Сигнальные пути, включающие митоген-активируемые протеинкиназы (MAP-киназы, MAPK), регулируют такие клеточные процессы, как пролиферация, дифференцировка, апоптоз, ответ на стресс и выживание клеток [20]. Охарактеризованы четыре основных MAPK-пути: ERK, JNK/SAPK, ERK/BMK1 и p38 [21]. Активация сигнального пути ERK происходит в несколько этапов: сначала лиганд взаимодействует с рецепторной тирозинкиназой, которая активирует малый G-белок (RAS). Затем происходит последовательное фосфорилирование протеинкиназ RAF (серин/треониновая протеинкиназа), MEK (киназа МАРК), ERK1/2 (внеклеточные сигнал-регулируемые киназы 1 и 2) [22]. Киназы ERK1 и ERK2 активируют киназу рибосомного белка S6 (RSK), протеинкиназу, активируемую митогенами и стрессом (MSK), и взаимодействующую с MAPK киназу (MNK) [23]. В злокачественных клетках 30% пациентов с такими заболеваниями, как аденокарцинома поджелудочной железы, рак толстой кишки, легкого, щитовидной железы и миелоидный лейкоз, обнаруживают активирующие мутации в гене RAS [26, 27] и генах RAF, MEK, ERK [26]. Поэтому компоненты MAP-киназного пути рассматривают в качестве перспективных мишеней противоопухолевых препаратов [20]. Компоненты сигнального пути MAPK направляют ДНК-связывающую и трансактивирующую активность Sp1 посредством фосфорилирования, гликозилирования и ацетилирования этого белка. Например, фосфорилирование Sp1 киназой ERK2 приводит к увеличению его ДНК-связывающей способности. Кроме того, фактор Sp1 участвует в регуляции экспрессии генов, кодирующих компоненты сигнального пути MAPK/ERK, влияя тем самым на его активность.
Сверхэкспрессия онкогена Ha-ras в фибробластах NIH-3T3 мыши приводит к снижению уровня экспрессии гена Reck, который кодирует одноименный цистеин-богатый белок с Kazal-мотивами, ингибирующий метастазирование опухоли за счет подавления активности металлопротеазы 9 (MMP9) [29, 30] (рис. 1). Показано, что в ходе RAS-зависимого фосфорилирования ретинобластома-связывающий белок 7 (RbAp46) формирует комплекс с гистоновой деацетилазой HDAC1 и белком Sp1 и подавляет экспрессию гена Reck. Одновременно в таких клетках повышается активность MMP9, что приводит к усилению метастазирования злокачественных клеток in vivo. Показано, что Sp1 и RAS с онкогенным потенциалом (Ha-RasLeu61) увеличивают экспрессию гена белка группы высокой подвижности А1 (HMGA1) в клетках рака толстой кишки человека HCT116, но прямого взаимодействия Sp1 с RAS не обнаружено [29] (рис. 1).
Рис. 1.
Влияние MAP-киназного сигнального пути на транскрипционную активность Sp1. РТК – рецепторные тирозинкиназы; RAS – малый G-белок, RAF – серин/треониновая протеинкиназа; MEK – киназа митоген-активируемой протеинкиназы; ERK – внеклеточная сигнал-регулируемая киназа; RKIP – белок-ингибитор киназы RAF; Elk-1 (ETS-подобный белок 1) и SAP-1 (SRF-вспомогательный белок 1) – транскрипционные факторы; HMGA2 – ген белка группы высокой подвижности А1, EGFR – ген рецептора эпидермального фактора роста; IGF-1R – ген рецептора инсулиноподобного фактора роста 1; ITGA2 – ген интегрина 2; Reck – ген цистеин-богатого белка, содержащего Kazal-мотивы и индуцирующего обратные мутации; PEBP1 – ген белка RKIP; CDKN1A – ген ингибитора циклинзависимой киназы 1; PLAU – ген урокиназы; BIRC5 – ген сурвивина; BMP-2 – ген морфогенетического белка кости 2; Hspb1 – ген белка теплового шока B1. Пунктирными линиями показан процесс выхода мРНК из ядра для синтеза белков.
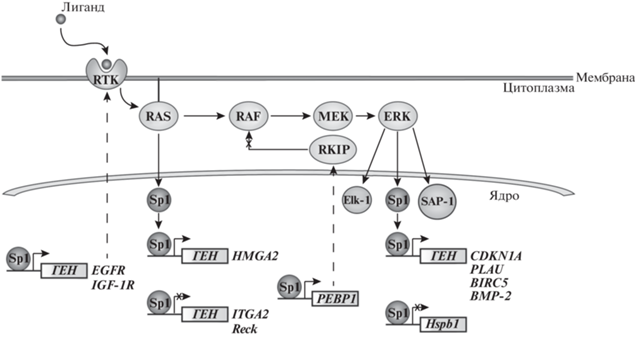
В эпителиальных клетках молочной железы человека эффективность связывания Sp1 с промоторным участком гена интегрина 2 (ITGA2) снижается в ответ на сверхэкспрессию онкогена v-RAS и гена рецептора эпидермального фактора роста типа 2 (ERBB2), что приводит к снижению уровня мРНК гена ITGA2 [30]. Показано, что нокдаун генов SP1, SP2 и SP3 в перевиваемых клетках Panc28 и L3.6pL рака поджелудочной железы человека приводит к снижению в них доли активной киназы RAS за счет снижения уровня экспрессии генов рецепторных тирозинкиназ EGFR и IGF-1R, экспрессия которых регулируется белками Sp1–3 [31] (рис. 1).
Экспрессия гена, кодирующего белок-ингибитор киназы RAF (PEBP1), снижена в клетках злокачественных опухолей человека, таких как меланома [32], гепатоцеллюлярная карцинома [33] и рак молочной железы [34]. В этом процессе участвует фактор транскрипции Sp1, так как нокдаун SP1 приводит к снижению активности промотора PEBP1 [35] (рис. 1).
Известно, что главными мишенями киназы ERK являются транскрипционные факторы Elk-1 (ETS-подобный белок 1) и SAP-1 (SRF-вспомогательный белок 1) [36]. Киназа ERK2 фосфорилирует Sp1, что приводит к увеличению эффективности связывания этого транскрипционного фактора с целевыми участками ДНК [37]. Показано, что в опухолевых клетках с мутациями в генах киназы RAF (B-RAFV600E) и опухолевого супрессора p53 (TP53) Sp1 может запускать экспрессию гена ингибитора циклинзависимой киназы 1 p21CIP1 (CDKN1A). Снижение количества морталина (шаперона семейства белков теплового шока Hsp70), вызывает гиперактивацию MEK/ERK в опухолевых клетках с этими мутациями, что приводит к увеличению экспрессии CDKN1A за счет белка p53. Активатором транскрипции CDKN1A в клетках с мутацией TP53 является Sp1 [38]. Компоненты MAP-киназного пути могут фосфорилировать белок Sp1, что приводит к Sp1-зависимой активации транскрипции: в клетках HeLa (рак шейки матки) и LNCaP (аденокарцинома предстательной железы) активированный Sp1 участвует в транскрипции гена урокиназы человека (PLAU) [39]. В клетках тератокарциномы ATDC5, обработанных интерлейкином 1 (IL-1), Sp1 вовлечен в транскрипцию гена морфогенетического белка кости 2 (BMP-2) [40]; в клетках острого миелоидного лейкоза Kg1 Sp1 регулирует экспрессию гена сурвивина (BIRC5), связанного с устойчивостью раковых клеток к противоопухолевым препаратам [41]. Также MAP-киназный путь ингибирует транскрипцию гена белка теплового шока B1 (Hspb1) в астроцитах мыши вследствие фосфорилирования белка Sp1 [42] (рис. 1).
Sр1 И MAP-СИГНАЛЬНЫЙ ПУТЬ P38
К MAP-киназным путям относится сигнальный каскад, включающий митоген-активируемые протеинкиназы p38. Активность этих киназ изменяется в условиях теплового и осмотического шока, взаимодействия клетки с провоспалительными цитокинами (фактор некроза опухоли-β (TNF) и IL-1), факторами роста (макрофагальный колониестимулирующий фактор (CSF-1)) и при воздействии ультрафиолета. Киназы p38 участвуют в регуляции клеточного цикла, дифференцировки клеток, апоптоза, аутофагии, супрессии опухолей и гипертрофии кардиомиоцитов [21]. Активировать киназы MAPK (MKK) могут различные белки: киназа 1, регулирующая сигналы апоптоза (ASK1) [43], белок DLK1 [44], активируемая трансформирующим фактором роста (TGF) киназа 1 (TAK1) [45]. Три MKK фосфорилируют p38: MKK6 фосфорилирует все четыре изоформы p38, MKK3– все изоформы, кроме p38β и MKK4 – p38α [46]. Нарушение активности сигнального пути p38 связывают с развитием нейродегенеративных заболеваний (болезни Альцгеймера [47], Гентингтона [48] и Паркинсона [49], боковой амиотрофический склероз [50], рассеянный склероз [51]), аутоиммунных заболеваний (ревматоидный артрит [52], болезнь Крона [53], псориаз [54]), бронхиальной астмы [55], сахарного диабета типа 2 [56]) и онкозаболеваний. Ингибиторы p38, такие как BIRB796 и ARRY-614, проходят клинические испытания в качестве средств для лечения болезни Крона, хронической обструктивной болезни легких, миелодиспластического синдрома, сердечно-сосудистых и других заболеваний [57].
Протеинкиназа p38 фосфорилирует белок Sp1, активируя его в клетках различного происхождения, таких как клетки моноцитарного лейкоза человека THP-1 [58], нейробластомы человека SH-SY5Y и кортикальные нейроны крысы [59], эпителиальные клетки бронхов человека [60], эндотелиальные клетки пуповины человека [61], клетки аденокарциномы протоков поджелудочной железы человека [62] (рис. 2а).
Рис. 2.
p38-зависимое фосфорилирование белка Sp1 и транскрипция контролируемых Sp1 генов в клетках человека. а – Активация белка Sp1 и транскрипции Sp1-контролируемых генов в нормальных клетках человека. б – Активация белка Sp1 в злокачественных клетках человека. Экспрессию указанных генов (FLNA – ген филамина А, ALOX5 – ген 5-липоксигеназы, CAV-1 – ген кавеолина-1) регулирует фосфорилированный белком p38 транскрипционный фактор Sp1.
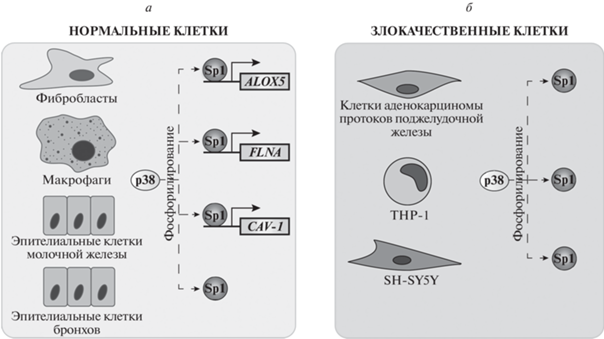
Фосфорилирование фактора Sp1 киназой p38 приводит к повышению его ДНК-связывающей и транскрипционной активности и, как следствие, к увеличению экспрессии Sp1-зависимых генов: в фибробластах активируется транскрипция гена филамина А (FLNA) [63], в макрофагах – гена 5‑липоксигеназы (ALOX5) [64], в клетках эпителия молочной железы человека – гена кавеолина-1 (CAV-1) [65] (рис. 2а). Фосфорилирование киназы MAPK-киназы (MKKK) в результате связывания нейротропного фактора мозга (BDNF) с его рецептором TrkB в клетках хондросаркомы приводит к увеличению эффективности связывания Sp1 с промотором гена MMP-1, вовлеченного в регуляцию инвазии клеток [66].
Sр1 И СИГНАЛЬНЫЙ ПУТЬ JAK/STAT
Интерлейкины, интерфероны, фактор роста макрофагов CSF1, адапторные сигнальные молекулы STAM1 и STAM2, белок StIP, взаимодействующий со STAT, и семейство белков SH2B/Lnk/APS вовлечены в активацию сигнального пути JAK/STAT [69, 70]. Известны негативные регуляторы активности сигнального пути JAK/STAT – супрессоры цитокиновых сигналов (SOCS), белки-ингибиторы активированных STAT (PIAS) и протеин-тирозин-фосфатазы (PTP) [67]. Сигнальный путь JAK/STAT играет важную роль в гемопоэзе, образовании стволовых клеток, развитии иммунной системы, а также в пролиферации, дифференцировке, миграции клеток и регуляции апоптоза [71, 72].
Главными компонентами этого сигнального пути являются два семейства белков: JAK и STAT. В семейство JAK входят тирозинкиназы JAK1, JAK2, JAK3 и TYK2; к семейству STAT относятся факторы транскрипции STAT1, STAT2, STAT3, STAT4, STAT5 (имеет две изоформы a и b) и STAT6 [67]. В настоящее время ингибиторы киназ JAK/STAT-пути применяют при таких заболеваниях, как миелодиспластический синдром [71], ревматоидный артрит [72]; они проходят клинические испытания в качестве средств против рака молочной железы [73], острого лимфобластного лейкоза [74] и немелкоклеточного рака легкого [77, 78].
Взаимодействие лигандов с рецепторами, такими как FLT3 (Feline McDonough Sarcoma-Like Tyrosine kinase 3), рецепторами факторов роста и интерлейкинов, с рецептором эритропоэтина (EpoR), а также конститутивная активность химерного белка BCR-ABL приводят к активации сигнального пути JAK/STAT. Компоненты этого пути могут, в свою очередь, взаимодействовать с компонентами MAP-киназного каскада и сигнального пути PI3K/Akt в клетках острого и хронического миелоидного лейкоза и лимфобластных лейкозов, что опосредует перемещение транскрипционных факторов STAT, c-Myc и Sp1 в ядро, где они связывают регуляторные области промотора гена теломеразной обратной транскриптазы (TERT) [77]. Активация киназы JAK2 в результате стимуляции эндотелиальных клеток человека (линии ECV304 и EAhy926) лизофосфатидилхолином (LPC) приводит к увеличению эффективности связывания Sp1 с промотором гена eNOS, кодирующего эндотелиальную NO-синтазу [78] (рис. 3а).
Рис. 3.
Влияние факторов семейства STAT на транскрипционную активность белка Sp1 и уровень экспрессии его гена в клетках человека. а – Совместная активация экспрессии генов факторами семейства STAT и Sp1. б – Активация транскрипции гена SP1 белками семейства STAT в различных клетках. STAT – переносчики сигнала и активаторы транскрипции, TERT – ген обратной транскриптазы теломеразы, NOS3 – ген эндотелиальной NO-синтазы, ICAM-1 – ген молекулы клеточной адгезии 1, VEGFA – ген фактора роста эндотелия сосудов, CEBPE – ген CCAAT/связывающего белка-энхансера , CCND2 – ген циклина D2, CDKN1A – ген ингибитора циклинзависимой киназы 1.
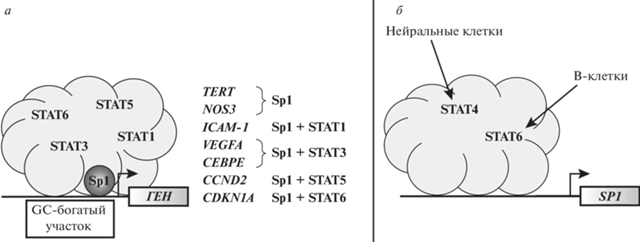
Показано, что одновременное связывание Sp1 и STAT с регуляторными областями необходимо для активации экспрессии ряда генов [79]. Например, связывание белков STAT1 и Sp1 необходимо для активации гена молекулы клеточной адгезии 1 (ICAM-1) в клетках бронхотрахеального эпителия [82, 83]. STAT3 и Sp1 инициируют транскрипцию генов фактора роста эндотелия сосудов (VEGFA) в астроцитах in vivo и в клетках глиобластомы U87MG in vitro [82]. Комплекс STAT3/Sp1 увеличивает экспрессию гена CCAAT/связывающего белка-энхансера (CEBPE) в клетках гепатоцеллюлярной карциномы в ответ на связывание IL-6 с рецепторами [83] (рис. 3а). Прямое связывание Sp1 и STAT6 стимулирует транскрипцию CDKN1A в клетках рака молочной железы [86, 87]. STAT4 может инициировать транскрипцию SP1 в нейральных клетках [86]. Рецептор IL-2 регулирует экспрессию циклина D2 (CCND2) в цитотоксических T-клетках через энхансерный комплекс STAT5 и Sp1 [87]. STAT6 регулирует транскрипцию SP1 в лимфоцитах и B-клетках для остановки пролиферации, индуцированной интерфероном IFN первого типа (рис. 3б). В этом случае важную роль играет STAT2, активирующий STAT6 [88].
Sр1 И СИГНАЛЬНЫЙ ПУТЬ PI3K/AKT
Главными компонентами сигнального пути PI3K/Akt являются фосфоинозитид-3-киназа (PI3K) и протеинкиназа B (Akt). Сигнальный путь PI3K/Akt участвует в регуляции многих клеточных процессов, включая синтез белков, выживание клеток, регуляцию апоптоза и клеточный цикл. На первом этапе этого пути происходит активация рецепторов, сопряженных с G-белками, интегринов, факторов роста, цитокинов и B-клеточных рецепторов. В число основных белков-мишеней Akt входят транскрипционный фактор FOX1 [89], киназа гликогенсинтазы 3 (GSK-3) [90], киназа mTOR [91] и фосфатаза PTEN с двойной субстратной специфичностью – ингибитор превращения фосфатидилинозит-(4,5)-бифосфата (PIP2) в фосфатидилинозит-(3,4,5)-трифосфат (PIP3) [92].
Нарушение активности сигнального пути PI3K/Akt может быть причиной развития аутоиммунных и нейродегенеративных заболеваний, таких как ревматоидный артрит, болезни Альцгеймера, Паркинсона и Гентингтона, а также может приводить к образованию и прогрессии злокачественных заболеваний, таких как глиома, плоскоклеточный рак головы и шеи, колоректальный рак, рак молочной железы, яичников, поджелудочной и предстательной железы, а также меланома [93–95].
Взаимосвязь Sp1 с сигнальным путем PI3K/Akt выявлена в фибробластах кролика in vitro и in vivo: микроРНК miR-29b связывается с 3'-нетранслируемой областью мРНК Sp1 и снижает таким образом количество этого белка в клетках. При этом белок Sp1 необходим для экспрессии гена Col1A1, кодирующего коллаген первого типа [96, 97] (рис. 4а). Таким образом, введение miR-29b в фибробласты приводит к подавлению экспрессии гена Col1A1 и, как следствие, к снижению скорости роста этих клеток. Следует отметить, что TGF1 способен ингибировать miR-29b с последующей активацией киназ PI3K и Akt, которые фосфорилируют белок Sp1, увеличивая тем самым эффективность его связывания с промотором гена Col1A1.
Рис. 4.
Участие транскрипционного фактора Sp1 в экспрессии генов в клетках кролика, мыши и человека, сопряженное с активностью сигнального пути PI3K/Akt. РТК – рецепторные тирозинкиназы, PI3K – фосфоинозитид-3-киназа, Akt – протеинкиназа B, mTOR – серин-треониновая протеинкиназа, TGF1 –трансформирующий фактор роста β1, c-Jun – транскрипционный фактор, Col1A1 – ген коллагена первого типа, Birc5 – ген сурвивина, Bcl2 – ген антиапоптотического белка Bcl2, B4GALT5 – ген 1.4-галактозилтрансферазы V, MMP14 – ген матриксной металлопротеазы 14, MMP2 – ген матриксной металлопротеазы 2, HIF1A – ген фактора 1, индуцируемого гипоксией, DDIT4 – ген, регулируемый при развитии и повреждении ДНК, miR – микроРНК.
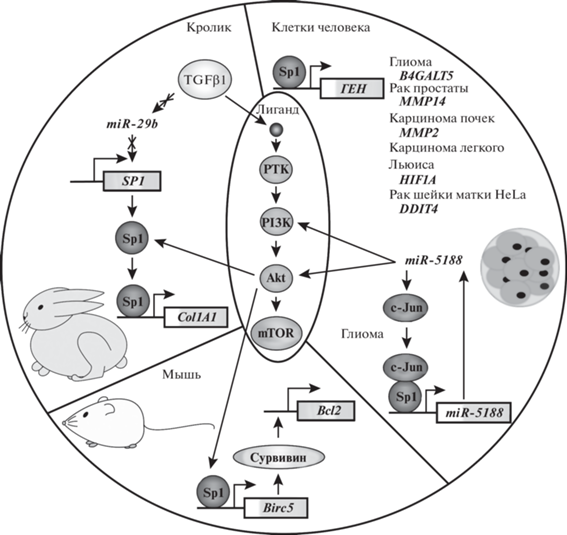
Обработка культуры клеток миелоидных предшественников новорожденных крыс противоопухолевым антибиотиком доксорубицином вызывает значительное снижение уровня фосфорилирования Akt, что приводит к одновременному снижению количества белка Sp1 и повышению p53. Sp1 – это регулятор экспрессии гена сурвивина (Birc5), который активирует экспрессию гена антиапоптотического белка Bcl2. Обработка клеток доксорубицином приводит к снижению уровня экспрессии генов Bcl2 и Birc5 с последующей гибелью клеток и развитием кардиомиопатии у модельных мышей. Добавление к кардиомиоцитам внеклеточных везикул мезенхимальных стволовых клеток C3H/10T1/2 мыши, содержащих микроРНК miR-199a-3p, приводит к восстановлению уровня белков Bcl2 и сурвивина за счет увеличения доли фосфорилированного Akt по сравнению с клетками, обработанными доксорубицином, предотвращая тем самым гибель клеток [98] (рис. 4б). Сходным образом miR-199a-3p защищает кардиомиоциты мышей от гибели in vivo.
На перевиваемых клетках линий U251 и U87 и первичных клетках глиомы человека показано, что сверхэкспрессия микроРНК miR-5188 повышает уровень фосфорилирования PI3K, Akt, а также содержания транскрипционного фактора c-Jun [99]. c-Jun в свою очередь связывает Sp1 и активирует экспрессию miR-5188. При этом повышенный уровень микроРНК miR-5188 в клетках глиомы связан с неблагоприятным прогнозом заболевания. Обработка первичных нейронов мыши ингибитором mTOR темсиролимусом приводит к снижению уровня экспрессии генов, кодирующих компоненты сигнального пути биосинтеза холестерина, таких как Ldlr, Sqle и Dhcr7 [100]. Аналогичный эффект в клетках коры мозга мыши вызывает ингибитор mTOR рапамицин. В промоторах большинства генов, экспрессия которых зависит от mTOR, обнаружены сайты связывания Sp1 и еще двух транскрипционных факторов – SREBP и NF-Y, что указывает на их вовлеченность в mTOR-зависимый контроль экспрессии генов биосинтеза холестерина у животных.
В клетках сердца мышей показана совместная регуляция экспрессии генов-мишеней mTOR белками Sp1 и сиртуином-6. Обнаружено одновременное снижение количества ядерной гистон-ацетилазы SIRT6, повышение активности синтеза белков и фосфорилирования mTOR в клетках сердца при сердечной гипертрофии, вызванной неселективным бета-адреномиметиком – изопреналином [101]. При этом установлено, что нокаут SIRT6 приводит к увеличению связывания Sp1 с промоторными областями генов, кодирующих mTOR и GTPазу Rheb. mTOR и Rheb, в свою очередь, фосфорилируют p70S6K и 4EBP1, которые направляют синтез большого числа белков [106, 107]. Таким образом, у мышей с нокаутом гена SIRT6 спонтанно развивается сердечная гипертрофия, обусловленная сверхактивацией mTOR при участии Sp1, при этом ингибирование mTOR приводит к частичному восстановлению функции сердца. Sp1 ингибирует фосфорилирование киназы p70S6K, которая служит мишенью mTOR.
Показано, что нокаут и нокдаун SP1 приводят к увеличению количества центросом, снижению нуклеации микротрубочек, образованию мультиполярных митотических веретен и микроядер, а также к повышению частоты анеуплоидии в эмбриональных фибробластах мыши и перевиваемых злокачественных клетках человека (U2OS, HeLa) [104].
Направленное подавление экспрессии генов транскрипционных факторов Sp1, Sp2 и Sp3 приводит к снижению уровня экспрессии генов рецепторных тирозинкиназ EGFR и IGF-1R в клетках рака поджелудочной железы человека Panc28 и L3.6pL, вследствие чего в этих клетках уменьшается фосфорилирование киназ mTOR и Ras [31]. Сходный эффект вызывает воздействие метформина, обладающего химиотерапевтической активностью.
Показана вовлеченность транскрипционного фактора Sp1 и сигнального пути PI3K/Akt в злокачественную трансформацию клеток и прогрессию гепатоцеллюлярной карциномы, рака молочной и предстательной железы, рака почки и шейки матки, а также глиомы [105–113]. Добавление к клеткам рака молочной железы линии MCF7 рекомбинантного белка IGF-1 приводит к повышению уровня мРНК гена, кодирующего белок, связывающий инсулиноподобный фактор роста 2 (IGFBP-2), маркер агрессивного течения заболевания [106]. Блокирование активности PI3K ингибитором LY294002 или mTOR рапамицином вызывает снижение уровня мРНК гена IGFBP-2, а повышение активности киназы PI3K рекомбинантным белком IGF-1 усиливает экспрессию IGFBP-2. Кроме того, изменение уровня мРНК этого гена коррелирует с внутриядерным количеством Sp1 и долей белка с фосфорилированными остатками серина. При этом воздействие на клетки белком IGF-1 увеличивает долю ядерного Sp1, а ингибитором PIK3 – снижает. Таким образом, эти данные свидетельствуют о том, что Sp1 вместе с киназами PI3K и mTOR участвует в регуляции экспрессии гена IGFBP-2 в клетках рака молочной железы.
Установлено, что Sp1 и сигнальный путь PIK3/Akt вовлечены в регуляцию гена CTH, кодирующего цистатионин-лиазу, необходимую для усиленной пролиферации клеток гепатоцеллюлярной карциномы человека под действием H2S [105]. Показано, что Sp1 связывается с промотором гена CTH. Sp1 также направляет экспрессию гена рецептора фактора роста сосудистого эндотелия VEGR, участвующего в неоангиогенезе опухоли, в клетках различной природы [111, 112]. Транскрипционный фактор Sp1 индуцирует экспрессию ряда генов, кодирующих регуляторы пролиферации, апоптоза, инвазии и метастазирования, такие как матриксные металлопротеазы (MMP14 – в клетках инвазивного рака предстательной железы человека [109] и MMP2 – в клетках рака почки [110]); 1.4-галактозилтрансфераза V (B4GALT5) в клетках глиомы [111]; фактор, индуцируемый гипоксией 1, HIF1A, в клетках рака легкого [112] и DDIT4 в клетках рака шейки матки HeLa [113] (рис. 4в).
Таким образом, белок Sp1 – это один из ключевых регуляторов экспрессии генов, кодирующих компоненты сигнального пути PI3K/Akt в клетках человека, крысы и кролика. Установлено, что транскрипционная активность Sp1 регулируется киназами этого сигнального пути. Нарушение активности Sp1 и сигнального пути PI3K/Akt приводит к потере целостности и стабильности генома, повышению способности клеток к инвазии, стимуляции прорастания сосудов в опухоль, блокированию апоптоза и активации пролиферации злокачественных клеток, что в совокупности способствует злокачественному перерождению клеток и прогрессии ряда злокачественных заболеваний.
ЗАКЛЮЧЕНИЕ
Sp1 – фактор транскрипции, который связывает GC-богатые участки в промоторах генов. Sp1 регулирует процессы дифференцировки, пролиферации, апоптоза в нормальных клетках, он вовлечен в регуляцию клеточного цикла, репарации ДНК, миграции и инвазии злокачественных клеток. Sp1 взаимодействует с широким спектром белков, формируя транскрипционные комплексы, для регуляции экспрессии генов, в том числе кодирующих компоненты сигнальных путей MAPK/ERK, MAPK/p38, JAK/STAT и PI3K/Akt. Киназы, входящие в эти сигнальные каскады, участвуют в посттрансляционной модификации белка Sp1, регулируя таким образом его транскрипционную активность (табл. 1).
Таблица 1.
Связь белка Sp1 с активностью сигнальных путей ERK, p38, PI3K/Akt при различных патологиях
| Сигнальный путь | Киназа | Наблюдаемый эффект | Механизм | Клетки | Патология |
|---|---|---|---|---|---|
| ERK | RAS | Снижение экспрессии гена Reck | Ha-Ras фосфорилирует RbAp46, который формирует комплекс с Sp1 и Reck | NIH-3T3 | Онкозаболевания |
| Повышение экспрессии гена HGMA1 | Ha-RasLeu61 и Sp1 увеличивают экспрессию гена HMGA1 | HCT116 | |||
| Снижение активности киназы RAS | Нокдаун гена SP1 приводит к снижению экспрессии EGFR и IGF1-R, которые регулируют активность RAS | Panc28, L3.6pL | |||
| Снижение экспрессии гена ITGA2 | v-RAS и ERBB2 фосфорилируют белок Sp1 и уменьшают эффективность его связывания с промотором гена ITGA2 | Клетки эпителия молочной железы | |||
| RAF | Увеличение активности RAF-киназы | Нокдаун гена SP1 приводит к уменьшению экспрессии гена PEBP1, кодирующего ингибитор RAF | Клетки меланомы, гепатоцеллюлярной карциномы, рака молочной железы | ||
| Запуск экспрессии гена CDKN1A | Уменьшение количества морталина в клетках с мутантными генами RAF и p53 вызывает гиперактивацию MEK/ERK, что приводит к повышению экспрессии гена CDKN1A фактором транскрипции Sp1 | SK-MEL28 | |||
| MEK/ERK | Повышение экспрессии гена PLAU | Фосфорилирование Sp1 MAP-киназами приводит к увеличению эффективности его связывания с промотором гена PLAU | HeLa, LNCaP | ||
| Повышение экспрессии гена BMP-2 | Фосфорилирование Sp1 MAP-киназами приводит к увеличению эффективности его связывания с промотором гена BMP-2 после обработки клеток интерлейкином-1 | ATDC5 | Остеоартрит | ||
| MSK/ERK | Повышение экспрессии гена BIRC5 | Фосфорилирование Sp1 MAP-киназами приводит к увеличению эффективности его связывания с промотором гена BIRC5 | Kg1 | Онкозаболевания | |
| ERK1/2 | Снижение экспрессии гена Hspb1 | Фосфорилирование Sp1 киназами ERK1/2 приводит к ингибированию транскрипции Hspb1 | Астроциты мышей | Эпилепсия, ишемическая болезнь, болезнь Альцгеймера | |
| p38 | p38 | Активация Sp1 | p38 фосфорилирует Sp1, что приводит к его активации | THP-1, SH-SY5Y, клетки аденокарциномы протоков поджелудочной железы человека, кортикальные нейроны крысы, клетки эпителия бронхов человека, эндотелиальные клетки пуповины человека | Онкозаболевания |
| Повышение экспрессии гена FLNA | Активация p38 в результате механического воздействия приводит к фосфорилированию Sp1, который запускает экспрессию FLNA | Фибробласты человека | – | ||
| Повышение экспрессии гена ALOX5 | Промотор ALOX5 имеет сайт связывания Sp1, активность которого уменьшается при обработке клеток ингибитором p38 | Макрофаги мышей | Атеросклероз | ||
| Повышение экспрессии гена CAV-1 | Фосфорилирование Sp1 протеинкиназой p38 приводит к увеличению эффективности его связывания с промотором гена CAV-1 | Клетки эпителия молочной железы человека | Онкозаболевания | ||
| MKKK | Повышение экспрессии гена MMP-1 | Связывание нейротропного фактора мозга BDNF с его рецептором TrkB приводит к фосфорилированию киназ MKKK, что приводит к увеличению эффективности связывания Sp1 с промотором гена MMP-1 | Клетки хондросаркомы | ||
| JAK/STAT | Компоненты сигнального пути JAK/STAT | Повышение экспрессии гена TERT | Взаимодействие с компонентами других сигнальных путей (MAPK и PI3K/Akt) опосредует перемещение фактора Sp1 в ядро, где он связывает регуляторные области промотора TERT | Клетки острого и хронического миелоидных лейкозов, лимфобластных лейкозов | |
| JAK2 | Повышение экспрессии гена NOS3 | Стимуляция LPC приводит к активации JAK2, что увеличивает эффективность связывания с промотором гена NOS3 | ECV304, EAhy926 | – | |
| PI3K/Akt | PI3K, Akt | Повышение экспрессии гена Col1A1 | Последовательная активация киназ PI3K и Akt приводит к фосфорилированию Sp1, что увеличивает эффективность его связывания с промотором гена Col1A1 | Фибробласты кролика | Глаукома |
| Akt | Снижение экспрессии генов Birc5 и Bcl2 | Снижение уровня фосфорилирования Akt уменьшает уровень фосфорилирования Sp1, транскрипционным регулятором экспрессии гена Birc5. Продукт гена Birc5 (сурвивин) является активатором экспрессии гена Bcl2 | Кардиомиоциты мыши | Кардиомиопатия | |
| PI3K, Akt | Связывание Sp1 с c-Jun для активации экспрессии miR-5188 | miR-5188 повышает уровень фосфорилирования PI3K, Akt, и уровень c-Jun, который связывается с Sp1 и инициирует транскрипцию miR-5188 | U251, U87, первичные клетки глиомы человека | Онкозаболевания | |
| mTOR | Снижение экспрессии генов Ldlr, Sqle, Dhcr7, cнижение продукции холестерина | Ингибиторы mTOR (темсиролимус и рапамицин) приводят к снижению уровня экспрессии mTOR-зависимых генов биосинтеза холестерина, в промоторах которых есть сайты связывания Sp1 | Первичные нейроны мыши, клетки коры мозга мыши | Неврологические расстройства | |
| Активация mTOR | Нокаут гена SIRT6 приводит к увеличению связывания Sp1 с промотором гена mTOR | Клетки сердечной ткани мышей | Сердечная гипертрофия | ||
| Уменьшение фосфорилирования mTOR | Подавление экспрессии гена SP1 снижает уровень экспрессии генов рецепторных тирозинкиназ EGFR и IGF-1R, которые регулируют активность mTOR | Panc28, L3.6pL | Онкозаболевания | ||
| Akt | Повышение экспрессии гена CTH | В результате фосфорилирования Akt Sp1 напрямую связывается с промотором гена CTH | Клетки гепатоцеллюлярной карциномы человека | ||
| PI3K, mTOR | Повышение экспрессии гена MMP2 | Ингибиторы PI3K и Akt вызывают уменьшение эффективности связывания Sp1 с промотором MMP2 | Клетки рака почки | ||
| PI3K/Akt | Повышение экспрессии гена B4GALT5 | Продукт гена B4GALT5, транскрипцию которого контролирует Sp1, регулирует сигнальный путь PI3K/Akt | Клетки глиомы | ||
| Повышение экспрессии гена DDIT4 | Ингибиторы PI3K и Akt уменьшают экспрессию гена DDIT4, которую контролирует Sp1 | HeLa |
Поскольку транскрипционный фактор Sp1 участвует в регуляции процессов, связанных со злокачественной трансформацией клеток, а повышенный уровень экспрессии гена SP1 характерен для злокачественных клеток различного происхождения, подавление активности Sp1 рассматривают в качестве перспективного подхода к противоопухолевой терапии.
Исследование выполнено за счет гранта Российского научного фонда (проект № 22-14-00353).
Настоящая статья не содержит каких-либо исследований с использованием животных в качестве объектов.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Suske G. (2017) NF-Y and SP transcription factors – New insights in a long-standing liaison. Biochim. Biophys. Acta – Gene Regul. Mech. 1860, 590–597.
Marin M., Karis A., Visser P., Grosveld F., Philipsen S. (1997) Transcription factor Sp1 is essential for early embryonic development but dispensable for cell growth and differentiation. Cell. 89, 619–628.
Vellingiri B., Iyer M., Subramaniam M.D., Jayaramayya K., Siama Z., Giridharan B., Narayanasamy A., Dayem A.A., Cho S.G. (2020) Understanding the role of the transcription factor sp1 in ovarian cancer: from theory to practice. Int. J. Mol. Sci. 21, 1153.
Torabi B., Flashner S., Beishline K., Sowash A., Donovan K., Bassett G., Azizkhan-Clifford J. (2018) Caspase cleavage of transcription factor Sp1 enhances apoptosis. Apoptosis. 23, 65–78.
Ding A., Bian Y.Y., Zhang Z.H. (2020) SP1/TGF–β1/SMAD2 pathway is involved in angiogenesis during osteogenesis. Mol. Med. Rep. 21, 1581–1589.
Soleimani T., Falsafi N., Fallahi H. (2017) Dissection of regulatory elements during direct conversion of somatic cells into neurons. J. Cell. Biochem. 118, 3158–3170.
Gong L., Ji W.-K., Hu X.-H., Hu W.-F., Tang X.-C., Huang Z.-X., Li L., Liu M., Xiang S.-H., Wu E., Woodward Z., Liu Y.Z., Nguyen Q. D., Li D.W.C. (2014) Sumoylation differentially regulates Sp1 to control cell differentiation. Proc. Natl. Acad. Sci. USA. 111, 5574–5579.
Hotter D., Bosso M., Jønsson K.L., Krapp C., Stürzel C.M., Das A., Littwitz-Salomon E., Berkhout B., Russ A., Wittmann S., Dittmer U., Sauter D., Kirchhoff F. (2019) IFI16 targets the transcription factor Sp1 to suppress HIV-1 transcription and latency reactivation. Cell Host Microbe. 25, 858–872.e13.
Dynan W.S., Tjian R. (1983) The promoter-specific transcription factor Sp1 binds to upstream sequences in the SV40 early promoter. Cell. 35, 79–87.
Iwahori S., Shirata N., Kawaguchi Y., Weller S.K., Sato Y., Kudoh A., Nakayama S., Isomura H., Tsu-rumi T. (2007) Enhanced phosphorylation of transcription factor Sp1 in response to herpes simplex virus type 1 infection is dependent on the ataxia telangiectasia-mutated protein. J. Virol. 81, 9653–9664.
Chen-Plotkin A.S., Sadri-Vakili G., Yohrling G.J., Braveman M.W., Benn C.L., Glajch K.E., DiRocco D.P., Farrell L.A., Krainc D., Gines S., MacDonald M.E., Cha J.J. (2006) Decreased association of the transcription factor Sp1 with genes downregulated in Huntington’s disease. Neurobiol. Dis. 22, 233–241.
Yao L., Dai X., Sun Y., Wang Y., Yang Q., Chen X., Liu Y., Zhang L., Xie W., Liu J. (2018) Inhibition of transcription factor SP1 produces neuroprotective effects through decreasing MAO B activity in MPTP/ MPP+ Parkinson’s disease models. J. Neurosci. Res. 96, 1663–1676.
Hsu T.I., Wang M.C., Chen S.Y., Yeh Y.M., Su W.C., Chang W.C., Hung J.J. (2012) Sp1 expression regulates lung tumor progression. Oncogene. 31, 3973–3988.
Jiang N.Y., Woda B.A., Banner B.F., Whalen G.F., Dresser K.A., Lu D. (2008) Sp1, a new biomarker that identifies a subset of aggressive pancreatic ductal adenocarcinoma. Cancer Epidemiol. Biomarkers Prev. 17, 1648–1652.
Wang X.B., Peng W.Q., Yi Z.J., Zhu S.L., Gan Q.H. (2007) Expression and prognostic value of transcriptional factor sp1 in breast cancer. Chinese J. Cancer. 26, 996–1000.
Zhang J., Zhu Z.G., Ji J., Yuan F., Yu Y.Y., Liu B.Y., Lin Y.Z. (2005) Transcription factor Sp1 expression in gastric cancer and its relationship to long-term prognosis. World J. Gastroenterol. 11, 2213–2217.
Chiefari E., Brunetti A., Arturi F., Bidart J.-M., Russo D., Schlumberger M., Filetti S. (2002) Increased expression of AP2 and Sp1 transcription factors in human thyroid tumors: a role in NIS expression regulation? BMC Cancer. 2, 35.
Guan H., Cai J., Zhang N., Wu J., Yuan J., Li J., Li M. (2012) Sp1 is upregulated in human glioma, promotes MMP-2-mediated cell invasion and predicts poor clinical outcome. Int. J. Cancer. 130, 593–601.
Вагапова Э.Р., Лебедев Т.Д., Тихонова А.Д., Гойхман Б.В., Иваненко К.А., Спирин П.В., Прасолов. В.С. (2020) Высокая экспрессия SP1, CSF1R и PAK1 коррелирует с повышенной чувствительностью лейкозных клеток к антибиотику митрамицину. Молекуляр. биология. 54, 522–528.
Guo Y., Pan W., Liu S., Shen Z., Xu Y., Hu L. (2020) ERK/MAPK signalling pathway and tumorigenesis (review). Exp. Ther. Med. 19, 1997–2007.
Zarubin T., Han J. (2005) Activation and signaling of the p38 MAP kinase pathway. Cell Res. 15, 11–18.
McCain J. (2013) The MAPK (ERK) pathway: investigational combinations for the treatment of BRAF-mutated metastatic melanoma. Pharmacy Therapeutics (P&T). 38(2), 96–108.
Lavoie H., Gagnon J., Therrien M. (2020) ERK signalling: a master regulator of cell behaviour, life and fate. Nat. Rev. Mol. Cell Biol. 21, 607–632.
Khan A.Q., Kuttikrishnan S., Siveen K.S., Prabhu K.S., Shanmugakonar M., Al-Naemi H.A., Haris M., Dermime S., Uddin S. (2019) RAS-mediated oncogenic signaling pathways in human malignancies. Semin. Cancer Biol. 54, 1–13.
Bos J.L. (1989) Ras oncogenes in human cancer: a review. Cancer Res. 49, 4682–4689.
Sanchez-Vega F., Mina M., Armenia J., Chatila W.K., Luna A., La K.C., Dimitriadoy S., Liu D.L., Kantheti H.S., Saghafinia S., Chakravarty D., Daian F., Gao Q., Bailey M.H., Liang W.W., Foltz S.M., Shmulevich I., Ding L., Heins Z., Ochoa A., Gross B., Gao J., Zhang H., Kundra R., Kandoth C., Bahceci I., Dervishi L., Dogrusoz U., Zhou W., Shen H., Laird P.W., Way G.P., Greene C.S., Liang H., Xiao Y., Wang C., Iavarone A., Berger A.H., Bivona T.G., Lazar A.J., Hammer G.D., Giordano T., Kwong L.N., McArthur G., Huang C., Tward A.D., Frederick M.J., McCormick F., Meyerson M.; Cancer Genome Atlas Research Network, Van Allen E.M., Cherniack A.D., Ciriello G., Sander C., Schultz N. (2019) Pathways, oncogenic signaling cancer, the atlas, genome. Cell. 173, 321–337.
Yeh H.H., Tseng Y.F., Hsu Y.C., Lan S.H., Wu S.Y., Raghavaraju G., Cheng D.E., Lee Y.R., Chang T.Y., Chow N.H., Hung W.C., Liu H.S. (2015) Ras induces experimental lung metastasis through up-regulation of RbAp46 to suppress RECK promoter activity. BMC Cancer. 15, 1–14.
Sasahara R.M., Takahashi C., Noda M. (1999) Involvement of the Sp1 site in Ras-mediated downregulation of the RECK metastasis suppressor gene. Biochem. Biophys. Res. Commun. 264, 668–675.
Cleynen I., Huysmans C., Sasazuki T., Shirasawa S., Van De Ven W., Peeters K. (2007) Transcriptional control of the human high mobility group A1 gene: basal and oncogenic Ras-regulated expression. Cancer Res. 67, 4620–4629.
Ye J., Xu R.H., Taylor-Papadimitriou J., Pitha P.M. (1996) Sp1 binding plays a critical role in Erb-B2- and v-ras-mediated downregulation of alpha2-integrin expression in human mammary epithelial cells. Mol. Cell. Biol. 16, 6178–6189.
Nair V., Sreevalsan S., Basha R., Abdelrahlm M., Abudayyeh A., Hoffman A.R., Safe S. (2014) Mechanism of metformin-dependent inhibition of mammalian target of rapamycin (mTOR) and Ras activity in pancreatic cancer. J. Biol. Chem. 289, 27692–27701.
Park S., Yeung M.L., Beach S., Shields J.M., Yeung K.C. (2005) RKIP downregulates B-Raf kinase activity in melanoma cancer cells. Oncogene. 24, 3535–3540.
Lee H.C., Tian B., Sedivy J.M., Wands J.R., Kim M. (2006) Loss of Raf kinase inhibitor protein promotes cell proliferation and migration of human hepatoma cells. Gastroenterology. 131, 1208–1217.
Hagan S., Al-Mulla F., Mallon E., Oien K., Ferrier R., Gusterson B., Curto García J.J., Kolch W. (2005) Reduction of Raf-1 kinase inhibitor protein expression correlates with breast cancer metastasis. Clin. Cancer Res. 11, 7392–7397.
Zhang B., Wang O., Qin J., Liu S., Sun S., Liu H., Kuang J., Jiang G., Zhang W. (2013) Cis-acting elements and trans-acting factors in the transcriptional regulation of Raf kinase inhibitory protein expression. PLoS One. 8, 1–10.
Hill C.S., Treisman R. (1995) Transcriptional regulation by extracellular signals: mechanisms and specificity. Cell. 80, 199–211.
Merchant J.L., Du M., Todisco A. (1999) Sp1 phosphorylation by Erk 2 stimulates DNA binding. Biochem. Biophys. Res. Commun. 254, 454–461.
Karkhanis M., Park J.I. (2015) Sp1 regulates Raf/MEK/ERK-induced p21CIP1 transcription in TP53-mutated cancer cells. Cell Signal. 27, 479–486.
Benasciutti E., Pagès G., Kenzior O., Folk W., Blasi F., Crippa M.P. (2004) MAPK and JNK transduction pathways can phosphorylate Sp1 to activate the uPA minimal promoter element and endogenous gene transcription. Blood. 104, 256–262.
Chien S.Y., Tsai C.H., Liu S.C., Huang C.C., Lin T.H., Yang Y.Z., Tang C.H. (2020) Noggin inhibits IL-1β and BMP-2 expression, and attenuates cartilage degeneration and subchondral bone destruction in experimental osteoarthritis. Cells. 9, 1–21.
Zhang Y., Chen H., Zhou S., Wang S., Zheng K., Xu D., Liu Y., Wang X., Wang X., Yan H., Zhang L., Liu Q., Chen W., Wang Y. (2015) Sp1 and c-Myc modulate drug resistance of leukemia stem cells by regulating survivin expression through the ERK-MSK MAPK signaling pathway. Mol. Cancer. 14, 56.
Kim J.E., Ko A.R., Hyun H.W., Min S.J., Kang T.C. (2018) P2RX7-MAPK1/2-SP1 axis inhibits mTOR independent HSPB1-mediated astroglial autophagy article. Cell Death Dis. 9, 1–16.
Tobiume K., Matsuzawa A., Takahashi T., Nishitoh H., Morita K.-I., Takeda K., Minowa O., Miyazono K., Noda T., Ichijo H. (2001) ASK1 is required for sustained activations of JNK/p38 MAP kinases and apoptosis. EMBO Rep. 2, 222–228.
Tedeschi A., Bradke F. (2013) The DLK signalling pathway – a double-edged sword in neural development and regeneration. EMBO Rep. 14, 605–614.
Sorrentino A., Thakur N., Grimsby S., Marcusson A., von Bulow V., Schuster N., Zhang S., Heldin C.H., Landström M. (2008) The type I TGF-β receptor engages TRAF6 to activate TAK1 in a receptor kinase-independent manner. Nat. Cell Biol. 10, 1199–1207.
Cuadrado A., Nebreda A.R. (2010) Mechanisms and functions of p38 MAPK signalling. Biochem. J. 429, 403–417.
Johnson G.V.W., Bailey C.D.C. (2003) The p38 MAP kinase signaling pathway in Alzheimer’s disease. Exp. Neurol. 183, 263–268.
Huang Z.N., Chen J.M., Huang L.C., Fang Y.H., Her L.S. (2021) Inhibition of p38 mitogen-activated protein kinase ameliorates HAP40 depletion-induced toxicity and proteasomal defect in Huntington’s disease model. Mol. Neurobiol. 58, 2704–2723.
He J., Zhong W., Zhang M., Zhang R., Hu W. (2018) P38 mitogen-activated protein kinase and Parkinson’s disease. Transl. Neurosci. 9, 147–153.
Gibbs K.L., Kalmar B., Rhymes E.R., Fellows A.D., Ahmed M., Whiting P., Davies C.H., Greensmith L., Schiavo G. (2018) Inhibiting p38 MAPK alpha rescues axonal retrograde transport defects in a mouse model of ALS. Cell Death Dis. 9, 596.
Krementsov D.N., Thornton T.M., Teuscher C., Rincon M. (2013) The emerging role of p38 mitogen-activated protein kinase in multiple sclerosis and its models. Mol. Cell. Biol. 33, 3728–3734.
Korb A., Tohidast-Akrad M., Cetin E., Axmann R., Smolen J., Schett G. (2006) Differential tissue expression and activation of p38 MAPK and isoforms in rheumatoid arthritis. Arthritis Rheum. 54, 2745–2756.
Whittall T., Wang Y., Kelly C.G., Thompson R., Sanderson J., Lomer M., Soon S.Y., Bergmeier L.A., Singh M., Lehner T. (2006) Tumour necrosis factor-a production stimulated by heat shock protein 70 and its inhibition in circulating dendritic cells and cells eluted from mucosal tissues in Crohn’s disease. Clin. Exp. Immunol. 143, 550–559.
Yu X.J., Li C.Y., Dai H.Y., Cai D.X., Wang K.Y., Xu Y.H., Chen L.M., Zhou C.L. (2007) Expression and localization of the activated mitogen-activated protein kinase in lesional psoriatic skin. Exp. Mol. Pathol. 83, 413–418.
Lam C.W.K., Ip W.K., Wong C.K., Lam C.W.K. (2006) Interleukin (IL)-4 and IL-13 up-regulate monocyte chemoattractant protein-1 expression in human bronchial epithelial cells: involvement of p 38 mitogen-activated protein kinase, extracellular signal-regulated kinase 1/2 and Janus kinase-2 but not c-Jun. Clin. Exp. Immunol. 145, 162–172.
Lim A.K.H., Nikolic-Paterson D.J., Ma F.Y., Ozols E., Thomas M.C., Flavell R.A., Davis R.J., Tesch G.H. (2009) Role of MKK3-p38 MAPK signalling in the development of type 2 diabetes and renal injury in obese db/db mice. Diabetologia. 52, 347–358.
Coulthard L.R., White D.E., Jones D.L., Mcdermott M.F. (2009) Europe PMC Funders Group p38 MAPK: stress responses from molecular mechanisms to therapeutics. Trends Mol. Med. 15, 369–379.
Ma W., Lim W., Gee K., Aucoin S., Nandan D., Kozlowski M., Diaz-Mitoma F., Kumar A. (2001) The p38 mitogen-activated kinase pathway regulates the human interleukin-10 promoter via the activation of Sp1 transcription factor in lipopolysaccharide-stimulated human macrophages. J. Biol. Chem. 276, 13664–13674.
Guida N., Laudati G., Mascolo L., Valsecchi V., Sirabella R., Selleri C., Di Renzo G., Canzoniero L.M.T., Formisano L. (2017) p38/Sp1/Sp4/HDAC4/BDNF axis is a novel molecular pathway of the neurotoxic effect of the methylmercury. Front. Neurosci. 11, 1–10.
Lee Y.C., Oslund K.L., Thai P., Velichko S., Fujisawa T., Duong T., Denison M.S., Wu R. (2011) 2,3,7,8-tetrachlorodibenzo-p-dioxin-induced MUC5AC expression: aryl hydrocarbon receptor-independent/EGFR/ERK/p38-dependent SP1-based transcription. Am. J. Respir. Cell Mol. Biol. 45, 270–276.
Pan W., Chang M.J., Booyse F.M., Grenett H.E., Bradley K.M., Wolkowicz P.E., Shang Q., Tabengwa E.M. (2008) Quercetin induced tissue-type plasminogen activator expression is mediated through Sp1 and p38 mitogen-activated protein kinase in human endothelial cells. J. Thromb. Haemost. 6, 976–985.
Hu H., Han T., Zhuo M., Wu L.L., Yuan C., Wu L., Lei W., Jiao F., Wang L.W. (2017) Elevated COX-2 expression promotes angiogenesis through EGFR/p38-MAPK/Sp1-dependent signalling in pancreatic cancer. Sci. Rep. 7, 1–10.
D’Addario M., Arora P.D., McCulloch C.A. (2006) Role of p38 in stress activation of Sp1. Gene. 379, 51–61.
Lee S.J., Kim C.E., Seo K.W., Kim C.D. (2010) HNE-induced 5-LO expression is regulated by NF-κB/ERK and Sp1/p38 MAPK pathways via EGF receptor in murine macrophages. Cardiovasc. Res. 88, 352–359.
Dasari A., Bartholomew J.N., Volonte D., Galbiati F. (2006) Oxidative stress induces premature senescence by stimulating caveolin-1 gene transcription through p38 mitogen-activated protein kinase/Sp1-mediated activation of two GC-rich promoter elements. Cancer Res. 66, 10805–10814.
Lin C.Y., Chang S.L.Y., Fong Y.C., Hsu C.J., Tang C.H. (2013) Apoptosis signal-regulating kinase 1 is involved in brain-derived neurotrophic factor (bdnf)-enhanced cell motility and matrix metalloproteinase 1 expression in human chondrosarcoma cells. Int. J. Mol. Sci. 14, 15459–15478.
Hou S.X., Zheng Z., Chen X., Perrimon N. (2002) The JAK/STAT pathway in model organisms: emerging roles in cell movement. Dev. Cell. 3, 765–778.
Bousoik E., Montazeri Aliabadi H. (2018) “Do we know Jack” about JAK? A closer look at JAK/STAT signaling pathway. Front. Oncol. 8, 1–20.
Harrison D.A. (2012) The JAK / STAT pathway. Cold Spring Harb. Perspect. Biol. 4, 1–3.
Kamran G., Arian L. O’Shea J.J. (2009) Janus kinases in immune cell signaling. Immunol. Rev. 228, 273–287.
Daw S., Chatterjee R., Law A., Law S. (2016) Analysis of hematopathology and alteration of JAK1/STAT3/ STAT5 signaling axis in experimental myelodysplastic syndrome. Chem. Biol. Interact. 260, 176–185.
Simon L.S., Taylor P.C., Choy E.H., Sebba A., Quebe A., Knopp K.L., Porreca F. (2021) The Jak/STAT pathway: a focus on pain in rheumatoid arthritis. Semin. Arthritis Rheum. 51, 278–284.
Shao F., Pang X., Baeg G.H. (2021) Targeting the JAK/STAT signaling pathway for breast cancer. Curr. Med. Chem. 28, 5137–5151.
Akahane K., Li Z., Etchin J., Berezovskaya A., Gjini E., Masse C.E., Miao W., Rocnik J., Kapeller R., Greenwood J.R., Tiv H, Sanda T., Weinstock D.M., Look A.T. (2017) Anti-leukaemic activity of the TYK2 selective inhibitor NDI-031301 in T-cell acute lymphoblastic leukaemia. Br. J. Haematol. 177, 271–282.
Hong D., Kurzrock R., Kim Y., Woessner R., Younes A., Nemunaitis J., Fowler N., Zhou T., Schmidt J., Jo M., Lee S.J., Yamashita M., Hughes S.G., Fayad L., Piha-Paul S., Nadella M.V.P., Mohseni M., Lawson D., Reimer C., Blakey D.C., Xiao X., Hsu J., Revenko A., Monia B.P., MacLeod A.R. (2015) AZD9150, a next-generation antisense oligonucleotide inhibitor of STAT3 with early evidence of clinical activity in lymphoma and lung cancer. Sci. Transl. Med. 7, 1–13.
Takeuchi T., Tanaka Y., Iwasaki M., Ishikura H., Saeki S., Kaneko Y. (2016) Efficacy and safety of the oral Janus kinase inhibitor peficitinib (ASP015K) monotherapy in patients with moderate to severe rheumatoid arthritis in Japan: a 12-week, randomised, double-blind, placebo-controlled phase IIb study. Ann. Rheum. Dis. 75, 1057–1064.
Yamada O., Kawauchi K. (2013) The role of the JAK-STAT pathway and related signal cascades in telomerase activation during the development of hematologic malignancies. Jak-Stat. 2, e25256.
Cieslik K., Abrams C.S., Wu K.K. (2001) Up-regulation of endothelial nitric-oxide synthase promoter by the phosphatidylinositol 3-kinase γ/Janus kinase 2/MEK-1-dependent pathway. J. Biol. Chem. 276, 1211–1219.
Shuai K. (2000) Modulation of STAT signaling by STAT-interacting proteins. Oncogene. 19, 2638–2644.
Look D.C., Pelletier M.B., Tidwell R.M., Roswit W.T., Holtzman M.J. (1995) Stat1 depends on transcriptional synergy with Sp1. J. Biol. Chem. 270, 30264–30267.
Yang X.P., Irani K., Mattagajasingh S., Dipaula A., Khanday F., Ozaki M., Fox-Talbot K., Baldwin W.M., Becker L.C. (2005) Signal transducer and activator of transcription 3α and specificity protein 1 interact to upregulate intercellular adhesion molecule-1 in ischemic-reperfused myocardium and vascular endothelium. Arterioscler. Thromb. Vasc. Biol. 25, 1395–1400.
Loeffler S., Fayard B., Weis J., Weissenberger J. (2005) Interleukin-6 induces transcriptional activation of vascular endothelial growth factor (VEGF) in astrocytes in vivo and regulates VEGF promoter activity in glioblastoma cells via direct interaction between STAT3 and Sp1. Int. J. Cancer. 115, 202–213.
Cantwell C.A., Sterneck E., Johnson P.F. (1998) Interleukin-6-specific activation of the C/EBPδ gene in hepatocytes is mediated by Stat3 and Sp1. Mol. Cell. Biol. 18, 2108–2117.
Wei M., Liu B., Gu Q., Su L., Yu Y., Zhu Z. (2013) Stat6 cooperates with Sp1 in controlling breast cancer cell proliferation by modulating the expression of p21Cip1/WAF1 and p27Kip1. Cell. Oncol. 36, 79–93.
Wei M., He Q., Yang Z., Wang Z., Zhang Q., Liu B., Gu Q., Su L., Yu Y., Zhu Z., Zhang G. (2014) Integrity of the LXXLL motif in Stat6 is required for the inhibition of breast cancer cell growth and enhancement of differentiation in the context of progesterone. BMC Cancer. 14, 1–17.
Qiu L., Ge L., Hu Q. (2020) Dexmedetomidine protects SK-N-SH nerve cells from oxidative injury by maintaining iron homeostasis. Biol. Pharm. Bull. 43, 424–431.
Martino A., Holmes J.H., Lord J.D., Moon J.J., Nelson B.H. (2001) Stat5 and Sp1 regulate transcription of the cyclin D2 gene in response to IL-2. J. Immunol. 166, 1723–1729.
Hsu Y.A., Huang C.C., Kung Y.J., Lin H.J., Chang C.Y., Lee K.R., Wan L. (2016) The anti-proliferative effects of type I IFN involve STAT6-mediated regulation of Sp1 and BCL6. Cancer Lett. 375, 303–312.
Cabrera-Ortega A.A., Feinberg D., Liang Y., Rossa C., Graves D.T. (2017) The role of forkhead box 1 (FOXO1) in the immune system: dendritic cells, T cells, B cells, and hematopoietic stem cells. Crit. Rev. Immunol. 37, 1–13.
Pandey M.K., DeGrado T.R. (2016) Glycogen synthase kinase-3 (GSK-3)-targeted therapy and imaging. Theranostics. 6, 571–593.
Wei X., Luo L., Chen J. (2019) Roles of mTOR signaling in tissue regeneration. Cells. 8, 1–23.
Cretella D., Digiacomo G., Giovannetti E., Cavazzoni A. (2019) PTEN alterations as a potential mechanism for tumor. Cancers (Basel). 11, 1–17.
Chalhoub N., Baker S.J. (2009) PTEN and the PI3-kinase pathway in cancer. Annu. Rev. Pathol. Mech. Dis. 4, 127–150.
Nakatani K., Thompson D.A., Barthel A., Sakaue H., Liu W., Weigel R.J., Roth R.A. (1999) Up-regulation of Akt3 in estrogen receptor-deficient breast cancers and androgen-independent prostate cancer lines. J. Biol. Chem. 274, 21528–21532.
Madhunapantula S.R. V., Mosca P.J., Robertson G.P. (2011) The Akt signaling pathway: an emerging therapeutic target in malignant melanoma. Cancer Biol. Ther. 12, 1032–1049.
Yu J., Luo H., Li N., Duan X. (2015) Suppression of type I collagen expression by miR-29b via PI3K, Akt, and Sp1 pathway, part II: an in vivo investigation. Investig. Ophthalmol. Vis. Sci. 56, 6019–6028.
Li N., Cui J., Duan X., Chen H., Fan F. (2012) Suppression of type I collagen expression by miR-29b via PI3K, Akt, and Sp1 pathway in human Tenon’s fibroblasts. Investig. Ophthalmol. Vis. Sci. 53, 1670–1678.
Lee J.Y., Chung J., Byun Y., Kim K.H., An S.H., Kwon K. (2021) Molecular sciences mesenchymal stem cell-derived small extracellular vesicles protect cardiomyocytes from doxorubicin-induced cardiomyopathy by upregulating survivin expression via the miR-199a-3p-Akt-Sp1/p53 signaling pathway. Int. J. Mol. Sci. 22, 7102.
Yi R., Yang S., Lin X., Zhong L., Liao Y., Hu Z., Huang T., Long H., Lin J., Wu Z., Xie C., Ding S., Luo J., Luo Q., Song Y. (2020) miR-5188 augments glioma growth, migration and invasion through an SP1-modulated FOXO1-PI3K/AKT-c-JUN-positive feedback circuit. J. Cell. Mol. Med. 24, 11800–11813.
Schüle M., Butto T., Dewi S., Schlichtholz L., Strand S., Gerber S., Endres K., Schweiger S., Winter J. (2021) mTOR driven gene transcription is required for cholesterol production in neurons of the developing cerebral cortex. Int. J. Mol. Sci. 22, 1–18.
Ravi V., Jain A., Khan D., Ahamed F., Mishra S., Giri M., Inbaraj M., Krishna S., Sarikhani M., Maity S., Kumar S., Shah R.A., Dave P., Pandit A.S., Rajendran R., Desingu, P.A., Varshney U., Das S., Kolthur-Seetharam U., Rajakumari S., Singh M., Sundaresan N.R. (2019) SIRT6 transcriptionally regulates global protein synthesis through transcription factor Sp1 independent of its deacetylase activity. Nucl. Acids Res. 47, 9115–9131.
Saxton R.A., Sabatini D.M. (2017) mTOR signaling in growth, metabolism, and disease. Cell. 168, 960–976.
Morita M., Gravel S.P., Hulea L., Larsson O., Pollak M., St-Pierre J., Topisirovic I. (2015) mTOR coordinates protein synthesis, mitochondrial activity. Cell Cycle. 14, 473–480.
Astrinidis A., Kim J., Crystal M.K., Olofsson B.A., Torabi B., Sorokina E.M., Azizkhan-Clifford J. (2010) The transcription factor Sp1 regulates centriole function and chromosomal stability through a functional interaction with the mammalian target of rapamycin/raptor complex. Genes. Chromosomes Cancer. 49, 282–297.
Yin P., Zhao C., Li Z., Mei C., Yao W., Liu Y., Li N., Qi J., Wang L., Shi Y., Qiu S., Fan J., Zha X. (2012) Sp1 is involved in regulation of cystathionine γ-lyase gene expression and biological function by PI3K/Akt pathway in human hepatocellular carcinoma cell lines. Cell. Signal. 24, 1229–1240.
Mireuta M., Darnel A., Pollak M. (2010) IGFBP-2 expression in MCF-7 cells is regulated by the PI3K/AKT/mTOR pathway through Sp1-induced increase in transcription. Growth Factors. 28, 243–255.
Reisinger K., Kaufmann R., Gille J. (2003) Increased Sp1 phosphorylation as a mechanism of hepatocyte growth factor (HGF/SF)-induced vascular endothelial growth factor (VEGF/VPF) transcription. J. Cell Sci. 116, 225–238.
Brunet S., Sardon T., Zimmerman T., Wittmann T., Pepperkok R., Karsenti E., Vernos I. (2004) Characterization of the TPX2 domains involved in microtubule nucleation and spindle assembly in Xenopus nucleation around chromatin and functions in a network of other molecules, some of which also are regulated by. Mol. Biol. Cell. 15, 5318–5328.
Beishline K., Azizkhan-Clifford J. (2015) Sp1 and the “hallmarks of cancer.” FEBS J. 282, 224–258.
Tang S.W., Yang T.C., Lin W.C., Chang W.H., Wang C.C., Lai M.K., Lin J.Y. (2011) Nicotinamide N-methyltransferase induces cellular invasion through activating matrix metalloproteinase-2 expression in clear cell renal cell carcinoma cells. Carcinogenesis. 32, 138–145.
Jiang J., Gu J. (2010) β1,4-Galactosyltransferase V: a growth regulator in glioma. Methods Enzymol. 479, 3–23.
Koshikawa N., Hayashi J.I., Nakagawara A., Takenaga K. (2009) Reactive oxygen species-generating mitochondrial DNA mutation up-regulates hypoxia-inducible factor-1α gene transcription via phosphatidylinositol 3-kinase-Akt/protein kinase C/histone deacetylase pathway. J. Biol. Chem. 284, 33185–33194.
Jin H.O., An S., Lee H.C., Woo S.H., Seo S.K., Choe T.B., Yoo D.H., Lee S.B., Um H.D., Lee S.J., Park M.J., Kim J.I., Hong S.I., Rhee C.H., Park I.C. (2007) Hypoxic condition- and high cell density-induced expression of Redd1 is regulated by activation of hypoxia-inducible factor-1α and Sp1 through the phosphatidylinositol 3-kinase/Akt signaling pathway. Cell. Signal. 19, 1393–1403.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология