

Молекулярная биология, 2022, T. 56, № 5, стр. 764-773
Блокирование двух альтернативных путей выживания клеток КАСУМИ-1 с помощью рибонуклеазы биназы и ингибитора киназ AKT1/2
В. А. Митькевич a, b, *, И. Ю. Петрушанко a, М. Г. Энгельгардт a, О. И. Кечко a, А. А. Макаров a
a Институт молекулярной биологии им. В.А. Энгельгардта Российской академии наук
119991 Москва, Россия
b Институт фундаментальной медицины и биологии, Казанский (Приволжский) федеральный университет
420008 Казань, Россия
* E-mail: mitkevich@gmail.com
Поступила в редакцию 11.03.2022
После доработки 01.04.2022
Принята к публикации 01.04.2022
- EDN: YECYYJ
- DOI: 10.31857/S002689842205010X
Аннотация
Лечение злокачественных новообразований зачастую требует использования комбинаций химиотерапевтических средств. Однако, чтобы подобрать комбинации, эффективные против конкретных опухолевых клеток, необходимо понимать механизмы действия препаратов, входящих в состав комбинации. Рибонуклеаза Bacillus pumilus (биназа) рассматривается в качестве адъювантного противоопухолевого средства, причем чувствительность злокачественных клеток к апоптогенному действию биназы зависит от определенных онкогенов. В линии клеток острого миелогенного лейкоза Касуми-1 биназа блокирует путь пролиферации, опосредованный мутантной тирозинкиназой KIT, что, как показано в нашей работе, активирует альтернативный путь пролиферации через AKT-киназу. В клетках Касуми-1 биназа в сочетании с ингибитором Akt1/2 индуцирует апоптоз, при этом их токсические эффекты складываются: ингибитор Akt1/2 блокирует путь, индуцируемый биназой, после подавления KIT-зависимого пути. Таким образом комбинация биназы и ингибиторов AKT-киназ может эффективно блокировать различные пути пролиферации опухолевых клеток и использоваться для их элиминации.
ВВЕДЕНИЕ
Монотерапия злокачественных заболеваний зачастую не позволяет полностью элиминировать раковые клетки из организма. Поэтому необходимо подбирать комбинации химиотерапевтических препаратов, эффективные для конкретной опухоли. Как правило, такой подбор основан на эмпирических закономерностях, он занимает достаточно много времени, отнимает силы у пациента и является дорогостоящей процедурой. Для разработки схем лечения злокачественных заболеваний требуется понимание механизмов действия химиотерапевтических препаратов на конкретные опухолевые клетки [1].
Считается, что рибонуклеазы могут использоваться в качестве средств адьювантной терапии опухолей, воздействующих как на сами злокачественные клетки, так и на продукты их “зловредной” деятельности – циркулирующие РНК [2–4]. Ранее мы показали эффективность РНКазы Bacillus pumilus (биназы) в направленной элиминации различных опухолевых клеток человека и в терапии опухолей у мышей [5–9]. В основе молекулярного механизма действия биназы лежит запуск апоптоза в опухолевых клетках, причем чувствительность злокачественных клеток к апоптогенному действию биназы определяется определенными онкогенами – KIT, RAS, FLT3, AML1-ETO, E6, E7 [5, 7, 10–12]. Присутствие этих онкогенов можно рассматривать как фактор, предсказывающий эффективное уничтожение опухолевых клеток под действием РНКазы биназы [4, 13]. Кроме того, повысить эффективность биназы можно, используя ее в сочетании с другими терапевтическими средствами [7, 14].
Мишенью токсического действия биназы в клеточной линии Касуми-1 острого миелогенного лейкоза служит онкоген KIT, кодирующий рецепторную тирозинкиназу [5]. Точечные мутации в гене KIT приводят к конститутивной активации белка KIT и к неконтролируемой пролиферации и выживанию клеток острого миелогенного лейкоза [15]. Биназа блокирует путь пролиферации, обусловленный экспрессией этого онкогена, и запускает таким образом апоптоз в опухолевых клетках [13]. При этом блокирование активности тирозинкиназы KIT в клетках Касуми-1 специфическим ингибитором иматинибом снижает токсическое действие биназы [5]. Мы предположили, что выживаемость клеток в условиях блокирования сигнального пути KIT-JNK может быть связана с активацией альтернативного, независимого от KIT пути пролиферации, опосредованного AKT-киназой, поскольку известно, что в клетках Касуми-1 AKT-киназа конститутивно активирована и эта активация не связана с PI3K [16, 17]. В представленной работе изучено токсическое действие ингибитора киназ Akt1/2 и его сочетания с биназой на клетки Касуми-1.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Фермент и клеточная культура. В работе использовали гуанилспецифичную РНКазу B. pumilis дикого типа (молекулярная масса 12.3 кДа, 109 аминокислотных остатков, pI 9.5), полученную согласно [18]. Ингибитор киназ Akt1/2 (“Merck”, США) растворяли в ДМСО в концентрации 20 мМ и использовали в концентрации от 2.5 до 60 мкМ. Суспензионную клеточную культуру Касуми-1 выращивали на среде RPMI-1640, содержащей 10% эмбриональной телячьей сыворотки, 2 мМ глутамина, 100 Ед./мл пенициллина и 100 мкг/мл стрептомицина, при температуре 37°C в атмосфере 5% СО2.
Оценка жизнеспособности клеток. Жизнеспособность клеток оценивали с помощью теста WST-1 (“Roche Diagnostics”, Швейцария). Тест основан на расщеплении водорастворимой соли тетразолия митохондриальными дегидрогеназами в живых клетках. Клетки Касуми-1 рассаживали в 96-луночные планшеты на 24 ч, затем обрабатывали биназой и/или ингибитором Akt1/2 в течение 24 ч. После этого клетки инкубировали в течение 60 мин при 37°C в присутствии реагента WST-1. Поглощение образцов измеряли с помощью микропланшетного ридера Multiscan FC (“Thermo Fisher Scientific”, США) на длине волны 450 нм. Референсные значения определяли на длине волны 620 нм. В качестве контроля использовали смесь среды без клеток с реагентом WST-1. Жизнеспособность необработанных клеток принимали за 100%. Приведены средние значения, вычисленные из трех независимых измерений.
Проточная цитометрия. Апоптоз. Клетки исследовали методом проточной цитометрии на цитофлуориметре BD LSRFortessa (“Becton Dickinson“, США). При проведении цитометрических опытов клетки с поврежденной мембраной выявляли с помощью йодида пропидия (PI, “Sigma”, США) (Ex/Em = 535/617 нм). PI добавляли к клеточной суспензии до конечной концентрации 10 мкг/мл за 1 мин до начала измерений. Окрашиваемые PI (PI-положительные) клетки считали некротическими. Апоптотические клетки регистрировали с помощью аннексина V (“Invitrogen”, США), меченного флуоресцеинизотиоцианатом (FITC, Ex/Em = 494/518 нм). Клетки, которые окрашивались как PI, так и аннексином V, считали находящимися на поздних стадиях апоптоза. Для определения апоптоза суспензию клеток центрифугировали (1000 об./мин, 10 мин), отбирали супернатант и ресуспендировали в буфере для связывания аннексина (10 мM HEPES, 140 мM NaCl, 2.5 мM CaCl2, pH 7.4). Затем к 100 мкл суспензии клеток (106 клеток/мл) добавляли 5 мкл раствора конъюгата аннексина. Пробы инкубировали в течение 15 мин в темноте при комнатной температуре, после чего добавляли 400 мкл буфера для связывания аннексина и ставили на лед.
Изменение митохондриального потенциала. Митохондриальный потенциал (Ψ) определяли с помощью красителя MitoProbeDilC1(5) (Ex/Em = = 638/658 нм, “Invitrogen”, США). Клетки (1 × 106) инкубировали с 0.5 мкМ DilC1(5) в течение 30 мин при 37°C в темноте. Затем клетки промывали фосфатно-солевым буфером (PBS) при 4°C и ресуспендировали в PBS.
Уровень активных форм кислорода, кальция и восстановленного глутатиона. Уровень активных форм кислорода (АФК) оценивали с помощью дигидрородамина 123 (DHR123, Ex/Em = 507/525 нм, “Invitrogen”), который позволил определять уровень АФК не только в цитозоле, но и в митохондриях. Клетки окрашивали, добавляя DHR123 до конечной концентрации 10 мкМ, и инкубировали в течение 30 мин при 37°C в темноте. Уровень восстановленного глутатиона определяли с использованием ThiolTracker (Ex/Em = 405/525 нм, “Invitrogen”). Для окрашивания клеток ThiolTracker добавляли до конечной концентрации 7.5 мкМ и инкубировали в течение 30 мин при 37°C в темноте. Уровень Ca2+ определяли, окрашивая клетки fluo-4 (Ex/Em = 494/516 нм, “Invitrogen”). Клетки инкубировали с красителем в концентрации 2.5 мкМ в течение 30 мин при 37°C. Затем клетки отмывали и ресуспендировали в PBS. Данные представлены как среднее из трех независимых экспериментов в трех повторностях со стандартными отклонениями.
Вестерн-блотинг. Клетки собирали, отмывали в холодном PBS, а затем лизировали при +4°С в течение 30–60 мин при постоянном перемешивании в RIPA-буфере (25 мМ Трис-HCl pH 7.6, 150 мМ NaCl, 1% NP-40, 1% дезоксихолата натрия, 0.1% додецилсульфата Na (ДДС-Na), 0.2 мМ фенилметансульфонилфторид (PMSF), коктейль ингибиторов протеаз (“Roсhe”, Швейцария, одна таблетка на 10 мл буфера) из расчета 30 мкл буфера на 1 млн. клеток. Полученные лизаты центрифугировали (10 мин, 16 000 g), надосадочный раствор отбирали и определяли в нем концентрацию белка. Далее образцы разделяли с помощью одномерного электрофореза в полиакриламидном геле и переносили на мембраны PVDF. Мембраны блокировали 5%-ным раствором обезжиренного молока в буфере PBSТ (10 мМ KH2PO4, 150 мМ NaCl, 0.1% Tween-20) в течение 2 ч при комнатной температуре на качалке и инкубировали с первичными антителами к AKT (#4691, разбавление 1 : 5000) и Phospho-Akt (Ser473) (#9271, разбавление 1 : 2000) от (“Cell Signaling Technol”, США) в течение ночи при 4°C. Затем блоты инкубировали в течение 30 мин при комнатной температуре с вторичными антителами (“ИМТЕК”, Россия), конъюгированными с пероксидазой хрена, которые брали в разведении 1 : 10 000. Для визуализации использовали Bio-Rad ChemiDoc MP. Результаты обрабатывали в программе Image Lab. Перед визуализацией мембрану инкубировали в течение 2 мин с хемилюминесцентным субстратом SuperSignal™ West Femto Maximum Sensitivity Substrate kit (“Thermo Scientific”). Данные нормировали по актину, детектированному с помощью антител AM4302 (“Ambion”, США). В работе приведены средние значения, вычисленные из трех независимых измерений со стандартным отклонением.
Статистический анализ. Приведены средние данные со стандартным отклонением (±SD), вычисленные не менее чем из трех независимых экспериментов. Сравнение групп данных проводили с использованием t-критерия Стьюдента. Данные считали статистически значимыми при р < 0.05.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ
Действие биназы и ингибитора киназ Akt1/2 на жизнеспособность клеток Касуми-1
Для оценки влияния биназы и ингибитора Akt1/2 на жизнеспособность клеток Касуми-1 мы обработали клетки биназой в концентрации 0.8 мкM и ингибитором Akt1/2 (2.5, 5, 10, 25, 40 и 60 мкM) по отдельности и в комбинациях (рис. 1). Через 24 ч жизнеспособность клеток под действием биназы снижалась на 29%. Действие ингибитора Akt1/2 в использованных концентрациях приводило к снижению жизнеспособности клеток на 41, 48, 55, 85, 92 и 95%, а в комбинации с биназой – на 64, 69, 86, 98, 98 и 98% соответственно.
Рис. 1.
Действие биназы (0.8 мкМ), ингибитора Akt1/2 (2.5, 5, 10, 25, 40 и 60 мкМ) и их комбинаций на жизнеспособность клеток Касуми-1 после 24 ч обработки. Жизнеспособность клеток в контроле взята за 100%. Представлены средние значения, вычисленные из трех независимых экспериментов в трипликатах ± SD; *p < 0.05, **p < 0.01, ***p < 0.001, ***p < 0.0001.
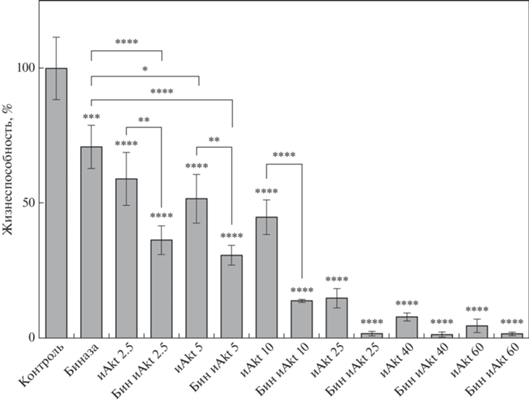
Биназа усиливает чувствительность клеток Касуми-1 к ингибитору киназ Akt1/2
Чтобы проверить гипотезу об активации независимого от KIT пути пролиферации, мы оценили уровень активации AKT-киназы в клетках Касуми-1 в ответ на обработку биназой (0.8 мкM) и ингибитором Akt1/2 (5 мкM). Киназа AKT конститутивно активирована в клетках этой линии, поэтому соотношение фосфо-AKT к общему количеству AKT в контрольных клетках приняли за базальный уровень. Под действием биназы уровень активации AKT-киназы увеличивается на 25% (рис. 2). Это свидетельствует о том, что в клетках Касуми-1 биназа активирует путь пролиферации, опосредуемый AKT-киназой. Ингибитор Akt1/2 снижал уровень активации киназы на 22%, тогда как комбинация биназы и ингибитора Akt1/2 снижала уровень активации AKT-киназы на 90% (рис. 2). Таким образом, в присутствии биназы ингибитор Akt1/2 более эффективно блокирует активацию AKT-киназы в клетках Касуми-1.
Рис. 2.
Влияние биназы и ингибитора Akt1/2 на активацию AKT-киназы в клетках Касуми-1. Репрезентативный Вестерн-блот, показывающий уровни белков AKT, р-AKT Ser473 и актина в клетках Касуми-1, обработанных 0.8 мкМ биназы, 5 мкМ ингибитора Akt1/2 и их комбинацией в течение 24 ч, относительно необработанных клеток. Гистограмма показывает уровень активации AKT-киназы в клетках, определенный как отношение уровней р-AKT Ser473 к общему уровню внутриклеточной AKT-киназы. Каждый столбик представляет среднее значение ± SD, вычисленное из трех независимых экспериментов. *p < 0.05, ***p < 0.01.
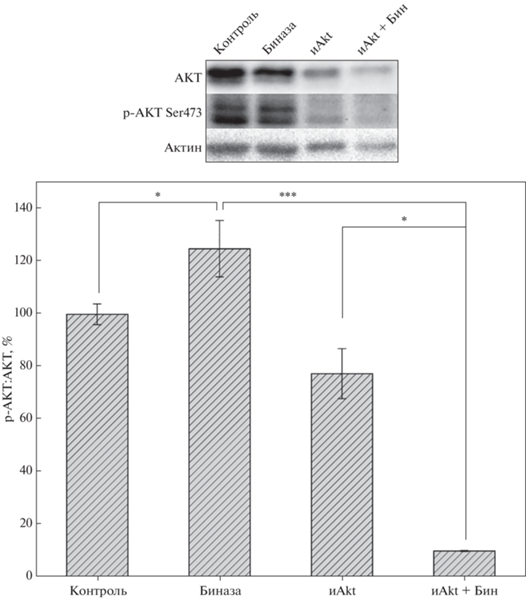
Биназа и ингибитор Akt1/2 оказывают проапоптическое действие на клетки Касуми-1
С помощью проточной цитометрии установлено, что под действием биназы (0.8 мкМ) количество мертвых клеток в популяции увеличивалось в 2.1 раза, тогда как под влиянием ингибитора Akt1/2 (5 мкM) доля PI-положительных клеток возросла в 2.4 раза по сравнению с контролем (рис. 3а). Применение комбинации этих агентов привело к увеличению количества мертвых клеток в популяции в 3.4 раза. Применение биназы и ингибитора AKT-киназ по отдельности приводило в обоих случаях к повышению количества апоптотических клеток в популяции в 1.7 раза, а совместное воздействие этих веществ вызывало двукратное увеличение количества таких клеток.
Рис. 3.
Влияние биназы (0.8 мкМ), ингибитора Akt1/2 (5 мкM) и их комбинации на клетки Касуми-1 через 24 ч инкубации. Количество мертвых клеток (пустые колонки) и апоптотических клеток (серые колонки) (а), и клеток с низким митохондриальным потенциалом ψ (б) представлено как процент от общего числа клеток в каждом варианте. Уровни АФК (в), GSH (г) и внутриклеточного Ca2+ (д) представлены относительно уровня в необработанном контроле. Приведены средние значения, вычисленные из трех независимых экспериментов в трипликатах ± SD; *p < 0.05, **p < 0.02, ***p < 0.005, ***p < 0.0003.
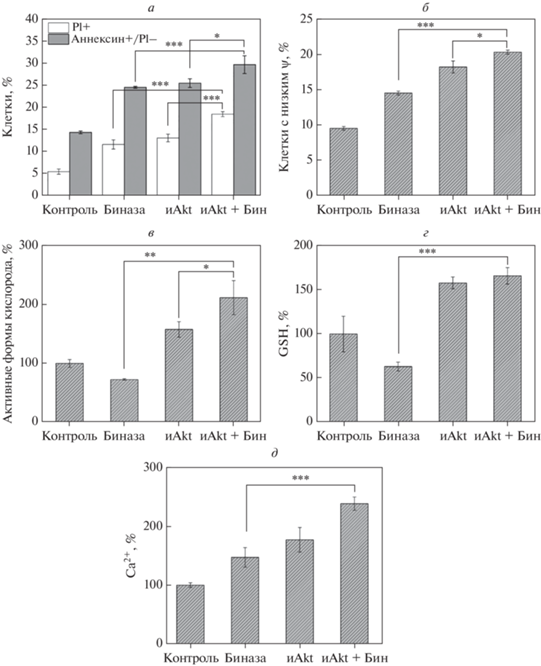
Апоптоз, вызываемый биназой и ингибитором Akt1/2, сопровождается увеличением количества клеток со сниженным мембранным потенциалом митохондрий (рис. 3б). Так, в присутствии биназы, ингибитора Akt1/2 и их комбинации количество таких клеток в популяции возрастает в 1.5, 1.7 и 2 раза соответственно. Под действием биназы уровень активных форм кислорода (АФК) в клетках Касуми-1 снижается на 27% (рис. 3в). Напротив, ингибитор Akt1/2 приводил к повышению уровня АФК в клетках на 58%, а сочетанное действие двух агентов увеличило содержание АФК на 112%. Сходным было и изменение уровня восстановленного глутатиона (рис. 3г). В присутствии биназы, ингибитора Akt1/2 и их комбинации уровень глутатиона составил 65, 158 и 167% от уровня в контроле соответственно. Уровень внутриклеточного Ca2+ в клетках Касуми-1, обработанных биназой и ингибитором Akt1/2, возрастает на 50 и 80% соответственно, а при их совместном применении – на 245%.
ОБСУЖДЕНИЕ РЕЗУЛЬТАТОВ
Нами показано, что совместное применение биназы и ингибитора Akt1/2 индуцирует гибель клеток Касуми-1, при этом токсические эффекты этих соединений складываются. Под действием биназы в клетках Касуми-1, как показано ранее, блокируется пролиферативный путь, опосредуемый тирозинкиназой KIT [5]. Однако из полученных данных следует, что в условиях блокады антиапоптотического сигнала через KIT под действием биназы в клетках Касуми-1 активируется AKT-киназа (рис. 2), что приводит к выживанию части клеток.
Известно, что конститутивно активированная в клетках Касуми-1 AKT-киназа может дополнительно активироваться в ответ на различные стимулы [19, 20]. Путь PI3K/AKT конститутивно активирован у 50–80% больных острыми миелогенными лейкозами [20], хотя активирующих мутаций самой киназы AKT при миелогенных лейкозах до сих пор не обнаружено [21]. Имеющиеся данные указывают на то, что различные ингибиторы AKT- или PI3K-киназ могут оказывать проапоптотическое действие на разные линии миелогенных лейкозов. Например, показано значительное снижение темпов роста опухоли, а также увеличение количества апоптотических клеток при ингибировании AKT-киназы [21]. Известно, что активация AKT-киназ оказывает антиапоптотическое действие на клетки преимущественно через ингибирование апоптоз-зависимых событий в митохондриях [22]. Данные, полученные в этой работе, свидетельствуют, что апоптоз, индуцируемый ингибитором Akt1/2 в клетках Касуми-1, сопровождается падением митохондриального потенциала, увеличением уровней АФК, восстановленного глутатиона и внутриклеточного кальция.
Апоптоз, индуцируемый биназой в клетках Касуми-1, сопровождается снижением уровня АФК и восстановленного глутатиона, увеличением количества клеток со сниженным митохондриальным потенциалом и ростом уровня внутриклеточного кальция. Ранее также было показано, что индуцируемый биназой апоптоз имеет признаки как рецепторного, так и митохондриального: действие биназы приводит к активации каспазы-8 и повреждению митохондрий [23].
Повышение уровня АФК в опухолевых клетках тесно связано с их метаболизмом и увеличенной скоростью пролиферации [24, 25]. При этом повышение уровня GSH во многих опухолевых клетках человека считается важным патологическим фактором и указывает на устойчивость к противоопухолевой терапии [26, 27]. Роль редокс-баланса и, в частности, глутатиона в жизнеспособности раковых клеток активно изучается [28]. В качестве одного из средств терапии рассматривается истощение клеток по глутатиону [28]. Индукция небольшого сдвига редокс-статуса раковых клеток в сторону окислительного стресса вызывает адаптацию клеток к повышенному уровню АФК вследствие регуляции целого ряда белков антиоксидантной защиты, способствуя выживанию клеток и возникновению агрессивных, терапевтически устойчивых форм рака [29]. Несмотря на очевидное влияние АФК на развитие и прогрессию опухолей, лечение онкологических больных антиоксидантами не улучшает, а в некоторых случаях даже ухудшает исход болезни [30]. Для индукции гибели раковых клеток необходимо значительное смещение их редокс-состояния в крайнее окислительное состояние [29].
Установлена тесная связь между редокс-статусом клеток и активностью AKT-киназы. Экзогенная или эндогенная индукция АФК способствует активации AKT-киназы в клетках гепатомы через PI3K-зависимый путь [31], увеличивая пролиферацию клеток. Позже показали, что пероксид водорода или ингибирование синтеза глутатиона индуцирует фосфорилирование AKT, что приводит к эндотелиальной дифференцировке сферических, подобных стволовым, клеток рака печени из линии HEP-G2 [32]. Оказалось, что редокс-чувствительность AKT обусловлена модификациями остатков цистеина. Установлено, что AKT содержит остатки цистеина, редокс-зависимая модификация которых может вызывать изменение ее фосфорилирования в ответ на изменение редокс-статуса клетки [33]. Замены этих остатков не влияют на базовую активность AKT, но нарушают ее редокс-регуляцию и снижают способность клеток выживать в условиях окислительного стресса [33]. Обнаружено, что определяющую роль в редокс-регуляции AKT играет глутатионилирование остатков цистеина [34, 35]. Снижение активности глутаредоксина, катализирующего реакцию глутатионилирования-деглутатионилирования, в зависимости от редокс-условий приводит к снижению активации (фосфорилирования) AKT при окислительном стрессе [33, 34]. В частности, на клетках пигментного эпителия сетчатки показано, что при окислительном стрессе происходит активация AKT и одновременное повышение глутатионилирования AKT, при этом глутаредоксин снижает глутатионилирование AKT, способствуя длительному существованию ее активной фосфорилированной формы (Ser473), что способствует выживанию клеток [34]. C другой стороны, в клетках нейробластомы SH-SY5Y наблюдается увеличение глутатионилирования фосфо-AKT (Ser473) под действием липополисахаридов или ингибитора глутатионтрансферазы Omega 1-1, что предполагает клеточно-специфичную редокс-регуляцию AKT [35]. Можно заключить, что умеренный окислительный стресс приводит к активации AKT, способствуя выживаемости клеток, тогда как сильный окислительный стресс может приводить к полному окислению белка и его протеасомной деградации [33], что будет снижать жизнеспособность клеток.
Таким образом, нельзя исключить, что в активацию AKT-зависимого пути пролиферации клеток Касуми-1 [19, 20] вовлечено повышение уровня АФК. Активация AKT-зависимого пути пролиферации биназой может быть связана не только с инактивацией KIT-зависимого пути, как мы предполагали ранее [5], но и с изменением редокс-статуса клеток под действием биназы. Так, ранее мы показали, что биназа в концентрации от 1.5 до 40 мкМ приводит к дозозависимому снижению уровня АФК в клетках Касуми-1 [23]. В данной работе нами установлено, что биназа (уже в концентрации 0.8 мкМ) одновременно со снижением уровня АФК, повышенного в злокачественных клетках, приводит к снижению уровня глутатиона, тем самым приближая величину редокс-статуса клеток Касуми-1 к значениям, характерным для нормальных клеток. В условиях опухолевого роста это должно отрицательно сказаться на адаптационном потенциале клеток. Cдвиг редокс-статуса клеток может приводить к изменению глутатионилирования белков. Действительно, инкубация клеток Касуми-1 с биназой в концентрации 0.8 мкМ приводит к глутатионилированию Na,K-АТРазы, что вносит вклад в ингибирование ее активности [4]. Таким образом, можно предположить, что повышение уровня фосфорилирования AKT cвязано с изменением уровня ее глутатионилирования вследствие изменения редокс-условий под действием биназы.
В отличие от биназы, ингибирование AKT-зависимого пути пролиферации с помощью ингибитора Akt/2 приводит к существенному росту АФК и повышению уровня GSH (рис. 3в, г). Это свидетельствует о разных механизмах действия данных веществ, что, вероятно, позволяет им дополнять и усиливать действие друг друга. Повышение уровня АФК под действием ингибитора AКТ1/2 наблюдали ранее в кардиомиоцитах, однако это повышение было гораздо менее значительным [36]. Причина этого пока не установлена. Возрастание уровня кальция и содержания (%) клеток со сниженным митохондриальным потенциалом позволяет предположить, что в этот процесс вовлечено изменение состояния митохондрий, причем основной вклад в изменение пула АФК вносят митохондрии. Это предположение подтверждается тем фактом, что при использовании красителя DHR, позволяющего детектировать как цитозольные, так и митохондриальные АФК, мы наблюдали гораздо более выраженный рост АФК, чем при использовании красителя DCF [36], который выявляет только цитозольные АФК. Совместное применение биназы и ингибитора Akt1/2 приводит к существенному снижению уровня активной, фосфорилированной формы AKT-1 и более значительному росту АФК, что и определяет повышение цитотоксического действия при совместном применении двух веществ.
Описано комбинированное применение ингибиторов AKT-киназ с другими противоопухолевыми препаратами [37–39]. Нами показано, что обработка биназой делает клетки Касуми-1 более чувствительными к ингибированию AKT-киназ, что проявляется в снижении соотношения р-AKT: AKT по сравнению с клетками, обработанными ингибитором Akt1/2 без биназы. Этот подход приводит к усилению процессов апоптоза и гибели большего количества клеток, чем при использовании этих агентов по отдельности.
Ранее биназу уже рассматривали в качестве компонента комплексной терапии. Так показано, что биназа восстанавливает чувствительность HPV-16-положительных клеток SiHa (вирус папилломы человека типа 16) к интерферону [7]. Этот эффект обусловлен подавлением биназой вирусных белков E6 и E7, блокирующих проведение сигнала от рецептора интерферона. Также установлено, что биназа повышает эффективность полихимиотерапевтической комбинации доксорубицина, винкристина, циклофосфамида и преднизолона, снижая при этом гепатотоксичность химиотерапии [8].
Полученные данные свидетельствуют о взаимодополняющем действии ингибитора Akt1/2 и биназы, которое достигается за счет ингибирования различных путей пролиферации опухолевых клеток и повышения эффективности действия ингибитора Akt1/2, в присутствии биназы. Таким образом, сочетанное применение биназы и ингибитора Akt1/2 может быть перспективным для терапии лейкозов и других злокачественных заболеваний, в основе которых лежит онкотрансформация, включающая изменение функционирования KIT.
Авторы благодарят Лагуткина Дениса за помощь в проведении Вестерн-блот-гибридизации.
Работа выполнена в рамках Программы стратегического академического лидерства КФУ (Приоритет 2030) при финансовой поддержке гранта Российского фонда фундаментальных исследований (№ 18-29-08024) и гранта ICGEB № CRP/RUS20-01.
В исследовании не использованы биологические материалы, полученные от людей или животных.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Pan S.T., Li Z.L., He Z.X., Qiu J.X., Zhou S.F. (2016) Molecular mechanisms for tumour resistance to chemotherapy. Clin. Exp. Pharmacol. Physiol. 43, 723–737.
Mironova N., Vlassov V. (2019) Surveillance of tumour development: the relationship between tumour-associated rnas and ribonucleases. Front. Pharmacol. 10, 1019.
Gotte G., Menegazzi M. (2019) Biological activities of secretory RNases: focus on their oligomerization to design antitumor drugs. Front. Immunol. 10, 2626.
Mitkevich V.A., Petrushanko I.Y., Makarov A.A. (2019) RNases disrupt the adaptive potential of malignant cells: perspectives for therapy. Front. Pharmacol. 10, 922.
Mitkevich V.A., Petrushanko I.Y., Spirin P.V., Fedorova T.V., Kretova O.V., Tchurikov N.A., Prassolov V.S., Ilinskaya O.N., Makarov A.A. (2011) Sensitivity of acute myeloid leukemia Kasumi-1 cells to binase toxic action depends on the expression of KIT and AML1-ETO oncogenes. Cell Cycle. 10, 4090–4097.
Sen’kova A.V., Mironova N.L., Patutina O.A., Mitke-vich V.A., Markov O.V., Petrushanko I.Y., Burnysheva K.M., Zenkova M.A., Makarov A.A. (2014) Ribonuclease binase decreases destructive changes of the liver and restores its regeneration potential in mouse lung carcinoma model. Biochimie. 101, 256–259.
Mitkevich V.A., Burnysheva K.M., Petrushanko I.Y., Adzhubei A.A., Schulga A.A., Chumakov P.M., Makarov A.A. (2017) Binase treatment increases interferon sensitivity and apoptosis in SiHa cervical carcinoma cells by downregulating E6 and E7 human papilloma virus oncoproteins. Oncotarget. 8, 72666–72675.
Mironova N.L., Petrushanko I.Y., Patutina O.A., Sen’kova A.V., Simonenko O.V., Mitkevich V.A., Markov O.V., Zenkova M.A., Makarov A.A. (2013) Ribonuclease binase inhibits primary tumor growth and metastases via apoptosis induction in tumor cells. Cell Cycle. 12, 2120–2131.
Зеленихин П.В., Еад Мохамед И.С., Надырова А.И., Сироткина А.А., Ульянова В.В., Миронова Н.Л., Митькевич В.А., Макаров А.А., Зенкова М.А., Ильинская О.Н. (2020) Рибонуклеаза Bacillus pumilus ингибирует миграцию клеток аденокарциномы двенадцатиперстной кишки человека HuTu 80. Молекуляр. биология. 54, 146–152.
Митькевич В.А., Орлова Н.Н., Петрушанко И.Ю., Симоненко О.В., Спирин П.В., Прокофьева М.М., Stocking C., Макаров А.А., Прасолов В.С. (2013) Экспрессия онкогена FLT3-ITD сообщает предшественникам В-клеток мыши линии BAF3 чувствительность к цитотоксическому действию биназы. Молекуляр. биология. 47, 282–285.
Mitkevich V.A., Petrushanko I.Y., Kretova O.V., Spirin P.V., Zelenikhin P.V., Prassolov V.S., Tchu-rikov N.A., Ilinskaya O.N., Makarov A.A. (2010) Oncogenic c-kit transcript is a target for binase. Cell Cycle. 9, 2674–2678.
Ilinskaya O.N., Singh I., Dudkina E., Ulyanova V., Kayumov A., Barreto G. (2016) Direct inhibition of oncogenic KRAS by Bacillus pumilus ribonuclease (binase). Biochim. Biophys. Acta. 1863, 1559–1567.
Митькевич В.А., Макаров А.А., Ильинская О.Н. (2014) Клеточные мишени противоопухолевых рибонуклеаз. Молекуляр. биология. 48, 214–222.
Зеленихин П.В., Макеева А.В., Нгуен Т.Н., Сирадж Е.А., Ильинская О.Н. (2016). Сочетанное действие биназы и блеомицина на клетки аденокарциномы легких человека. Биомед. химия. 62, 279–282.
Wang Y.-Y., Zhou G.-B., Tong Y.T., Chen B., Shi J.Y., Liang W.-X., Jin X.-L., You J.-H., Yang G., Shen Z.-X., Chen J., Xiong S.-M., Chen G.-Q., Feng X., Liu Y.-W., Chen Z., Chen S.-J. (2005) AML1-ETO and C-KIT mutation/overexpression in t(8;21) leukemia: implication in stepwise leukemogenesis and response to Gleevec. Proc. Natl. Acad. Sci. USA. 102, 1104–1109.
Larizza L., Magnani I., Beghini A. (2005) The Kasumi-1 cell line: a t(8;21)-kit mutant model for acute myeloid leukemia. Leuk. Lymphoma. 46, 247–255.
Martelli A.M., Evangelisti C., Chiarini F., McCubrey J.A. (2010) The phosphatidylinositol 3-kinase/Akt/mTOR signaling network as a therapeutic target in acute myelogenous leukemia patients. Oncotarget. 1, 89–103.
Schulga A., Kurbanov F., Kirpichnikov M., Protasevich I., Lobachev V., Ranjbar B., Chekhov V., Polyakov K., Engelborghs Y., Makarov A. (1998) Comparative study of binase and barnase: experience in chimeric ribonucleases. Protein Eng. 11, 775–782.
Blume-Jensen P., Janknecht R., Hunter T. (1998) The kit receptor promotes cell survival via activation of PI 3-kinase and subsequent Akt-mediated phosphorylation of Bad on Ser136. Curr. Biol. CB. 8, 779–782.
Grandage V.L., Gale R.E., Linch D.C., Khwaja A. (2005) PI3-kinase/Akt is constitutively active in primary acute myeloid leukaemia cells and regulates survival and chemoresistance via NF-kappa B, MAP kinase and p53 pathways. Leukemia. 19, 586–594.
Pei R., Si T., Lu Y., Zhou J.X., Jiang L. (2018) Salvianolic acid A, a novel PI3K/Akt inhibitor, induces cell apoptosis and suppresses tumor growth in acute myeloid leukemia. Leuk. Lymphoma. 59, 1959–1967.
Ramos A.M., Fernández C., Amrán D., Sancho P., de Blas E., Aller P. (2005) Pharmacologic inhibitors of PI3K/Akt potentiate the apoptotic action of the antileukemic drug arsenic trioxide via glutathione depletion and increased peroxide accumulation in myeloid leukemia cells. Blood. 105, 4013–4020.
Mitkevich V.A., Kretova O.V., Petrushanko I.Y., Burnysheva K.M., Sosin D.V., Simonenko O.V., Ilinskaya O.N., Tchurikov N.A., Makarov A.A. (2013) Ribonuclease binase apoptotic signature in leukemic Kasumi-1 cells. Biochimie. 95, 1344–1349.
Moloney J.N., Cotter T.G. (2018) ROS signalling in the biology of cancer. Semin. Cell Dev. Biol. 80, 50–64.
Galadari S., Rahman A., Pallichankandy S., Thayyullathil F. (2017) Reactive oxygen species and cancer paradox: to promote or to suppress? Free Radic. Biol. Med. 104, 144–164.
Traverso N., Ricciarelli R., Nitti M., Marengo B., Furfaro A.L., Pronzato M.A., Marinari U.M., Domenicotti C. (2013) Role of glutathione in cancer progression and chemoresistance. Oxid. Med. Cell. Longev. 2013, 972913.
Chen H.H., Kuo M.T. (2010) Role of glutathione in the regulation of cisplatin resistance in cancer chemotherapy. Met. Based Drugs. 2010, 430939.
Lv H., Zhen C., Liu J., Yang P., Hu L., Shang P. (2019) Unraveling the potential role of glutathione in multiple forms of cell death in cancer therapy. Oxid. Med. Cell. Longev. 2019, 3150145.
Chaiswing L., St Clair W.H., St Clair D.K. (2018) Redox paradox: a novel approach to therapeutics-resistant cancer. Antioxid. Redox Signal. 29, 1237–1272.
DeNicola G.M., Karreth F.A., Humpton T.J., Gopinathan A., Wei C., Frese K., Mangal D., Yu K.H., Yeo C.J., Calhoun E.S., Scrimieri F., Winter J.M., Hruban R.H., Iacobuzio-Donahue C., Kern S.E., Blair I.A., Tuveson D.A. (2011) Oncogene-induced Nrf2 transcription promotes ROS detoxification and tumorigenesis. Nature. 475, 106–109.
Shi D.-Y., Deng Y.-R., Liu S.-L., Zhang Y.-D., Wei L. (2003) Redox stress regulates cell proliferation and apoptosis of human hepatoma through Akt protein phosphorylation. FEBS Lett. 542, 60–64.
Zhao Z., Gao J., Li C., Xu X., Hu Y., Huang S. (2020) Reactive oxygen species induce endothelial differentiation of liver cancer stem-like sphere cells through the activation of Akt/IKK signaling pathway. Oxid. Med. Cell Longev. 2020, 1621687.
Ahmad F., Nidadavolu P., Durgadoss L., Ravindranath V. (2014) Critical cysteines in Akt1 regulate its activity and proteasomal degradation: implications for neurodegenerative diseases. Free Radic. Biol. Med. 74, 118–128.
Liu X., Jann J., Xavier C., Wu H. (2015) Glutaredoxin 1 (Grx1) protects human retinal pigment epithelial cells from oxidative damage by preventing AKT glutathionylation. Invest. Ophthalmol. Vis. Sci. 56, 2821–2832.
Saisawang C., Wongsantichon J., Robinson R.C., Ketterman A.J. (2019) Glutathione transferase Omega 1-1 (GSTO1-1) modulates Akt and MEK1/2 signaling in human neuroblastoma cell SH-SY5Y. Proteins. 87, 588–595.
Yang Z., Wu Y., Wang L., Qiu P., Zha W., Yu W. (2020) Prokineticin 2 (PK2) Rescues cardiomyocytes from high glucose/high palmitic acid-induced damage by regulating the AKT/GSK3β pathway in vitro. Oxid. Med. Cell Longev. 2020, 3163629.
Jilkova Z.M., Kuyucu A.Z., Kurma K., Ahmad Pour S.T., Roth G.S., Abbadessa G., Yu Y., Schwartz B., Sturm N., Marche P.N., Hainaut P., Decaens T. (2018) Combination of AKT inhibitor ARQ 092 and sorafenib potentiates inhibition of tumor progression in cirrhotic rat model of hepatocellular carcinoma. Oncotarget. 9, 11145–11158.
Hudis C., Swanton C., Janjigian Y.Y., Lee R., Sutherland S., Lehman R., Chandarlapaty S., Hamilton N., Gajria D., Knowles J., Shah J., Shannon K., Tetteh E., Sullivan D.M., Moreno C., Yan L., Han H.S. (2013) A phase 1 study evaluating the combination of an allosteric AKT inhibitor (MK-2206) and trastuzumab in patients with HER2-positive solid tumors. Breast Cancer Res.: BCR. 15, R110.
Tolcher A.W., Patnaik A., Papadopoulos K.P., Rasco D.W., Becerra C.R., Allred A.J., Orford K., Aktan G., Ferron-Brady G., Ibrahim N., Gauvin J., Motwani M., Cornfeld M. (2015) Phase I study of the MEK inhibitor trametinib in combination with the AKT inhibitor afuresertib in patients with solid tumors and multiple myeloma. Cancer Chemother. Pharmacol. 75, 183–189.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология