

Молекулярная биология, 2023, T. 57, № 1, стр. 24-46
Регуляторный потенциал SNP-маркеров в генах, кодирующих белки систем репарации ДНК
Н. П. Бабушкина a, *, А. Н. Кучер a
a Научно-исследовательский институт медицинской генетики,
Томский национальный исследовательский медицинский центр Российской академии наук
634050 Томск, Россия
* E-mail: nad.babushkina@medgenetics.ru
Поступила в редакцию 11.05.2022
После доработки 16.08.2022
Принята к публикации 16.08.2022
- EDN: AXUPQD
- DOI: 10.31857/S0026898423010032
Аннотация
Выявление широчайшего спектра локализованных в некодирующих участках генома однонуклеотидных полиморфизмов (SNP), ассоциированных с заболеваниями человека и патогенетически значимыми признаками, остро поставило вопрос по идентификации механизмов, объясняющих эти связи. Ранее нами выявлен ряд ассоциаций полиморфных вариантов генов, кодирующих белки репарации ДНК, с многофакторными заболеваниями. Для выяснения возможных механизмов, лежащих в их основе, нами проведена подробная аннотация регуляторного потенциала изучаемых маркеров с использованием ряда on-line ресурсов (GTXPortal, VannoPortal, Ensemble, RegulomeDB, Polympact, UCSC, GnomAD, ENCODE, GeneHancer, EpiМap Epigenomics 2021, HaploReg, GWAS4D, JASPAR, ORegAnno, DisGeNet, OMIM). В статье охарактеризован регуляторный потенциал следующих полиморфных вариантов: rs560191 (в гене TP53BP1), rs1805800 и rs709816 (NBN), rs473297 (MRE11), rs189037 и rs1801516 (ATM), rs1799977 (MLH1), rs1805321 (PMS2), rs20579 (LIG1). Приведена как общая характеристика изученных маркеров, так и информация по их влиянию на экспрессию “своего” и корегулируемых генов, на аффинность связывания факторов транскрипции. Рассмотрены опубликованные данные по адаптогенному и патологическому потенциалу этих SNP и о колокализованных с ними модификациях гистонов. Потенциальная вовлеченность на различных уровнях в регуляцию функционирования не только генов, в состав которых входят исследованные маркеры, но и близлежащих генов может объяснять ассоциированность изученных SNP с заболеваниями и их клиническими фенотипами.
ВВЕДЕНИЕ
Смещение акцентов в ассоциативных исследованиях от анализа генов-кандидатов к полногеномным ассоциативным исследованиям (GWAS) привело к открытию большого числа новых маркеров, ассоциированных с заболеваниями и количественными (в том числе и патогенетически значимыми) признаками. Оказалось, что среди ассоциированных однонуклеотидных полиморфизмов (SNP) бóльшая часть локализована в некодирующих участках генов и межгенных регионах [1]. В то же время ассоциированные с заболеваниями генетические варианты, выявленные при проведении GWAS, необязательно “указывают” на наличие функциональной значимости для развития патологии именно этих вариантов, не всегда понятно, как эти варианты изменяют функциональное состояние клетки и в итоге работу организма в целом, определяя риск развития патологии. В этой связи в последние годы особенно активно стали развиваться биоинформатические подходы, позволяющие оценить функциональную значимость полиморфных вариантов не напрямую, а через существующие блоки сцепления, эпистатические взаимодействия с различными генами, “взаимоотношения” различных уровней регуляции.
К числу относительно новых генов, структурная вариабельность которых может вносить вклад в формирование предрасположенности к различным многофакторным заболеваниям, относятся гены, кодирующие белки систем репарации ДНК. Помимо вовлеченности в канцерогенез (чему посвящены многочисленные исследования), накапливаются данные о значимости этих генов при развитии заболеваний сердечно-сосудистой, пищеварительной, мочеполовой, скелетно-мышечной, гемопоэтической и других систем (см. обзор [2]). Ранее нами изучена вовлеченность 9 SNP в генах, кодирующих белки различных репарационных систем: TP53BP1 (rs560191), NBN (rs1805800, rs709816), MRE11 (rs473297), ATM (rs189037, rs1801516), MLH1 (rs1799977), PMS2 (rs1805321), LIG1 (rs20579), ‒ в развитие многофакторной патологии различной этиологии. Мы проанализировали около 1.5 тыс. образцов ДНК из банка ДНК НИИ медицинской генетики Томского НИМЦ РАН, полученных от пациентов с различными заболеваниями: изолированная аллергическая бронхиальная астма (БА) и смешанная БА в сочетании с артериальной гипертензией (БА_АГ), хронический вирусный гепатит С (ХВГС), туберкулез (ТБ), сердечно-сосудистые заболевания (ишемическая болезнь сердца (ИБС) и аутопсийный материал умерших в результате сердечно-сосудистого события (АУТ_ИБС)); а также от жителей г. Томска (популяционная выборка – К) и долгожителей (Гер) [3‒8]. При незначительной подразделенности изученной популяции на субгруппы в соответствии с основными диагнозами пациентов (попарные FST < 1%) выявлен неплохой дифференцирующий потенциал маркеров. При визуализации матрицы генетических расстояний видно (рис. 1), что первая координата отделяет контроль (К) и группы пациентов с ИБС и умерших в раннем возрасте от сердечно-сосудистых катастроф (АУТ_ИБС) от групп пациентов с другими заболеваниями. Это может свидетельствовать о малом вкладе изученных маркеров генов, кодирующих белки систем репарации ДНК, в развитие сердечно-сосудистой патологии, что согласуется с результатами ассоциативного анализа: прямых ассоциаций с патологией не выявлено, ассоциации зарегистрированы только с патогенетически значимыми признаками [3]. Вторая координата от кластера сердечно-сосудистой патологии и контроля отделяет группу умерших от сердечно-сосудистого заболевания, из чего можно предполагать вклад изученных маркеров в раннее развитие или в быструю прогрессию таких заболеваний и, как следствие, в ранние неблагоприятные исходы. От большого кластера прочих патологий вторая координата отделяет БА – изолированную форму и сочетанную с артериальной гипертензией, ‒ что предполагает наличие вклада изученных маркеров в аллергическую компоненту этих заболеваний. И это вполне возможно, так как белки репарационных систем вовлечены в V(D)J-рекомбинацию и переключение синтеза классов иммуноглобулинов. Результаты ассоциативного анализа, в свою очередь, также подтверждают этот вывод [4, 5, 8].
Рис. 1.
Расположение изученных групп патологий в поле главных координат на основании генетических дистанций, рассчитанных по маркерам, локализованным в генах, кодирующих белки систем репарации ДНК. Расчет генетических дистанций по методу Nei и их визуализация выполнены с помощью приложения GenAlEx 6.503 [9]. ИБС – ишемическая болезнь сердца; АУТ_ИБС – аутопсийный материал умерших в результате сердечно-сосудистого события; ТБ – туберкулез; ХВГС – хронический вирусный гепатит С; БА – бронхиальная астма; БА_АГ – бронхиальная астма в сочетании с артериальной гипертензией; Гер – выборка долгожителей; К – средневозрастная популяционная выборка.

Таким образом, вклад изученных маркеров в развитие многофакторной патологии очевиден, хотя не все полученные ассоциации можно логично объяснить с точки зрения патогенеза анализируемых заболеваний. Для выяснения возможных механизмов фенотипической реализации установленных ассоциаций нами проведена подробная аннотация регуляторного потенциала изучаемых маркеров. Аннотация выполнена с использованием таких открытых ресурсов, как GTXPortal, VannoPortal, Ensemble, RegulomeDB, Polympact, UCSC, GnomAD, ENCODE, GeneHancer, EpiМap Epigenomics 2021, HaploReg, GWAS4D, JASPAR, ORegAnno, DisGeNet и OMIM.
ОБЩАЯ ХАРАКТЕРИСТИКА ИССЛЕДОВАННЫХ МАРКЕРОВ
Маркеры для анализа изначально выбирали с учетом их вероятной функциональной значимости, соответственно они располагались либо в кодирующей (rs560191, rs709816, rs1801516, rs1799977, rs1805321), либо в промоторной (rs473297, rs1805800, rs189037, rs20579) части генов (табл. 1), что предполагает влияние замены на изменения либо структуры белка, либо уровня экспрессии. Согласно рангу по RegulomeDB, наибольшим регуляторным потенциалом обладают rs560191 и rs473297, которые представляют собой eQTL-варианты, а также расположены в регионах связывания транскрипционных факторов (TF) или в регионах чувствительности к ДНКазе. Наименьший регуляторный потенциал предполагается для rs709816 (изменение мотивов связывания TF) и rs1801516 (ранг – “Другое”).
Таблица 1.
Исследованные маркеры: локализация и функциональный класс
| SNP ID: нуклеотидная замена | Хромосомная локализация | Ген/локализация в гене/заменаa | RegulomeDB,b ранг/шкала |
MAFc (min‒max) |
|---|---|---|---|---|
| rs560191: G>C | 15q15.3 | TP53BP1/ex 9/Asp358Glu | 1f/0.554 | 0.526 (0.231‒0.973) |
| rs473297: T>G | 11q21 | MRE11/5'UTRd (in1/ANCRD) |
1f/0.223 | 0.539 (0.346‒0.652) |
| rs1805800: C>T | 8q21.3 | NBN/5'UTR | 4/0.609 | 0.353 (0.167‒0.500) |
| rs709816: G>A | 8q21.3 | NBN/ex10/Asp399 | 6/0.288 | 0.609 (0.319‒0.925) |
| rs189037: G>A | 11q22.3 | ATM/5'UTR | 2a/0.98 | 0.467 (0.138‒0.703) |
| rs1801516: G>A | 11q22.3 | ATM/ex37/Asp1853Asn | 7/0.184 | 0.067 (0.0004‒0.237) |
| rs1799977: A>G | 3p22.2 | MLH1/ex8/Ile219Val | 5/0.611 | 0.170 (0.006‒0.360) |
| rs1805321: G>A | 7p22.1 | PMS2/ex11/Pro470Ser | 5/0.135 | 0.358 (0.206‒0.505) |
| rs20579: G>A | 19q13.33 | LIG1/5'UTR | 5/0.304 | 0.173 (0.059‒0.343) |
[i] a Замена аминокислотного остатка в указанной позиции кодируемого белка; in ‒ интрон, ex ‒ экзон.
[ii] b Функциональный класс по классификации RegulomeDB (https://regulomedb.org/regulome-search/): 1f – вариант экспрессионного локуса количественных признаков (eQTL) в мотиве связывания TF или в регионе гиперчувствительности к Д-НКазе; 2a – вариант локализован в мотиве связывания TF и изменяет эти мотивы, а также в регионе гиперчувствительности к ДНКазе; 4 – вариант локализован в мотиве связывания TF и в регионе гиперчувствительности к ДНКазе; 5 – вариант локализован в мотиве связывания TF или в регионе гиперчувствительности к ДНКазе; 6 – нарушение мотивов связывания TF; 7 – другое.
Вариант rs560191 в гене TP53BP1 представляет собой миссенс-замену G/C в экзоне 9, приводящую к замене Asp358Glu в кодируемом белке (табл. 1). Средняя частота аллеля С составляет 0.526, варьируя от 0.213 у финнов до 0.973 у афроамериканцев. У африканцев и афроамериканцев в том же геномном положении зарегистрирована также замена G/T, частота аллеля Т ‒ около 6 × 10‒5 (http://www.ensembl.org/index.html, https://gnomad. broadinstitute.org/). Данный SNP локализован между двумя дистальными энхансерподобными регуляторными элементами: E1757875/enhD и E1757876/enhD (https://genome.ucsc.edu/, https:// www.encodeproject.org/).
В гене NBN изучено две нуклеотидные замены: rs1805800 и rs709816 (табл. 1). rs1805800 представляет собой замену C/T в 5`UTR гена NBN. Среднепопуляционная частота аллеля T rs1805800 составляет 0.353, варьируя в пределах от 0.167 (африканские популяции Карибов и Барбадоса) до 0.500 у индийцев-гуджарати (http://www.ensembl. org/index.html, https://gnomad.broadinstitute.org/). Данный маркер находится на расстоянии 395 п.н. от промотора гена NBN; входит в состав промотора GH08J089980 и проксимального энхансерподобного элемента E2646296/enhP (https:// g-enome.ucsc.edu/, https://www.encodeproject.org/, https://www.genecards.org/). Замена A/G (rs709816) в экзоне 10 гена NBN синонимичная (Asp399). В референсной последовательности описан аллель A, хотя фактически это производный аллель. Среднепопуляционная частота аллеля G составляет 0.609, наименьшая частота зарегистрирована у жителей Англии и Шотландии (0.319), наибольшая – у населения Гамбии (0.925) (http://www. mulinlab.org/vportal/index.html, http://www.ensembl. org/index.html, https://gnomad.broadinstitute.org/). Эта сайленс-замена локализована менее чем в 5000 п.н. от дистального энхансерподобного элемента E2646270/enhD и энхансера GH06J089948 (https://genome.ucsc.edu/, https://www.encodeproject. org/, https://www.genecards.org/).
Два SNP изучено также в гене ATM (табл. 1). rs189037 представляет собой замену G/A в 5'UTR гена, локализованную в CpG-островке, в 29 п.н. от промотора гена ATM, внутри промотора GH11J108219, между промоторподобными элементами E1567620/prom (в 73 п.н.) и E1567621/prom (в 128 п.н.) (https://genome.ucsc.edu/, https://www. encodeproject.org/, https://www.genecards.org/). Среднепопуляционная частота составляет 0.467, варьируя от 0.138 у гамбийцев до 0.703 у населения Пакистана (http://www.ensembl.org/index.html, https://gnomad.broadinstitute.org/). rs1801516 – миссенс-вариант G/A в 37 экзоне, приводящий к замене Asp1853Asn. Этот вариант локализован в мультирегионе взаимодействия KDELC2/GH11J108219, расположен на расстоянии 893 п.н. от дистального энхансерподобного элемента E1567660/enhD (https://genome.ucsc.edu/, https://www.encodeproject.org/, https://www.genecards.org/). Среднепопуляционная частота аллеля А составляет 0.0669, варьируя от 0.0004 у японцев до 0.2367 у финнов (http://www.ensembl.org/index.html, https://gnomad. broadinstitute.org/).
rs473297 в гене MRE11 (табл. 1) представляет собой замену T/G в 5'UTR, локализованную в 79 п.н. от промотора GH11J094492, в 550 п.н. от проксимального энхансерподобного элемента E1561900/enhP, в регионе взаимодействия MRE11/GH11J094492 (https://genome.ucsc.edu/, https://www.genecards.org/). Среднепопуляционная частота аллеля G составляет 0.539, варьируя от 0.346 у британцев и шотландцев до 0.652 у ишан из Нигерии. В этой же точке генома у жителей Восточной Азии описана замена T/А, частота аллеля A составляет менее 3 × 10‒4 (http://www.ensembl.org/index.html, https://gnomad.broadinstitute.org/).
В экзоне 8 гена MLH1 изучена несинонимичная замена A/G ‒ rs1799977 (табл. 1), ‒ ведущая к замене Ile219Val в кодируемом белке. Этот вариант локализован в мультирегионе взаимодействия LRRFIP2/GH03J036988 (https://genome.ucsc.edu/, https://www.genecards.org/). Среднепопуляционная частота аллеля G составляет 0.170, варьируя от 0.006 (менде, Сьерра-Леоне) до 0.360 (тосканцы, Италия). В редких случаях в этой точке генома регистрируют замену A/T (описана в базе TOPMed) с частотой аллеля T 1.5 × 10‒5 (http://www.ensembl. org/index.html, https://gnomad.broadinstitute.org/).
rs1805321 (табл. 1) в гене PMS2 представляет собой нуклеотидную замену G/A в экзоне 11, приводящую к замене Pro470Ser в кодируемом белке. Этот SNP локализован в 1512 п.н. от дистального энхансерподобного элемента E2531915/enhD (https:// genome.ucsc.edu/, https://www.encodeproject.org/). Среднепопуляционная частота замены – 0.358, варьирует в пределах от 0.206 (у менде, Сьерра-Леоне) до 0.505 (у британцев Англии и Шотландии) (http://www.ensembl.org/index.html, https://gnomad.broadinstitute.org/).
Замена G/A (rs20579) расположена в 5'UTR гена LIG1 (табл. 1), в некодирующей части экзона 2, она локализована в мультирегионах взаимодействия LIG1/GH19J048121 и PLA2G4C/GH19J048253; в 158 п.н. от замены заканчивается проксимальный энхансерподобный элемент E1959701/enhP (https:// genome.ucsc.edu/, https://www.encodeproject.org/). Среднепопуляционная частота аллеля A составляет 0.173, варьирует от 0.0594 (у японцев) до 0.343 (у йоруба и ишан в Нигерии) (http://www.ensembl. org/index.html, https://gnomad.broadinstitute.org/).
Таким образом, все анализируемые нуклеотидные замены локализованы в регионах (или рядом с ними), непосредственно регулирующих транскрипцию генов (промоторов, энхансеров и подобных им элементов), и, следовательно, могут обладать регуляторным потенциалом.
МОДИФИКАЦИИ ГИСТОНОВ, КОЛОКАЛИЗОВАННЫЕ С АНАЛИЗИРУЕМЫМИ МАРКЕРАМИ
Известно, что в определении функционального состояния отдельно взятых фрагментов генома важнейшую роль играют посттрансляционные модификации гистонов (“гистоновый код”) [10]. Для анализа гистоновых модификаций в регионах локализации исследуемых SNP были использованы данные репозитория EpiМap Epigenomics 2021, представляющего собой интегральный р-есурс, обобщающий эпигеномные карты по 869 биообразцам, отнесенным к 33 категориям тканей (http://compbio.mit.edu/epimap/#chromatin-states) [11]. Под биообразцами при этом понимаются как клетки различных тканей (в норме на разных стадиях развития и при патологии), так и различные клеточные линии и их дериваты.
Для кодирующих регионов генов, в которых локализовано пять привлеченных к рассмотрению полиморфных вариантов, отмечается небольшое количество гистоновых модификаций, характерных для активно транскрибируемого хроматина. Так, для всех пяти локусов выявлено триметилирование лизина в позиции 36 гистона H3 (H3K36me3), типичное для открытого хроматина и способствующее элонгации транскрипции (табл. 2) (http://www.mulinlab.org/vportal/index.html, http://compbio.mit.edu/epimap/#chromatin-states). Другие эпигенетические метки (также характеризующие активную транскрипцию) встречаются редко, в отдельных типах клеток. Так, с регионом гиперчувствительности к ДНКазе колокализованы четыре из исследованных замен в кодирующих участках генов: rs560191 (в быстрорастущем клоне линии LNCaP, звездчатых клетках печени, гепатоцитах, лимфобластоидной клеточной линии GM19239, линии фибробластов легких AG08396), rs709816 (в эмбриональных клетках желудка), rs1801516 (в гематопоэтических стволовых и B-клетках при миелоидном лейкозе), rs560191 (в быстрорастущем клоне линии LNCaP, звездчатых клетках печени, гепатоцитах, лимфобластоидной клеточной линии GM19239, линии фибробластов легких AG08396), rs1799977 (в клетках аденокрциномы простаты) (http://www. mulinlab.org/vportal/index.html, http://compbio.mit.edu/ epimap/#chromatin-states). Кроме того, rs560191 в гене TP53BP колокализован с регионом связывания РНК-полимеразы II (в клетках влагалища), с ацетилированным лизином в позиции 23 гистона H3 – H3K23ac (в клетках трофобласта, дериватах H1-hESC), с H3K79me1 (в мезенхимальных и мезодермальных стволовых клетках, дериватах H1-hESC; в эмбриональных стволовых клетках линии H9; в эмбриональных фибробластах легкого линии IMR-90), с H3K27ac и H3K9ac (в гепатоцитах), H4K20me1 (в эмбриональных клетках, подобных стволовым, ‒ линия H1-hESC) (http://www. mulinlab.org/vportal/index.html, http://compbio.mit. edu/epimap/#chromatin-states). Нуклеотидная последовательность, содержащая rs709816 в гене NBN, колокализована с H3K27ac (большеберцовые нервы), с H3K79me2 (нейробластома) (http:// www.mulinlab.org/vportal/index.html, http://compbio. mit.edu/epimap/#chromatin-states). rs1801516 в гене АТМ колокализован с H4K20me1 (колоректальная аденокарцинома) (табл. 2) (http://www. mulinlab. org/vportal/index.html, http://compbio.mit.edu/epimap/#chromatin-states).
Таблица 2.
Колокализованные с анализируемыми маркерами модификации хроматина
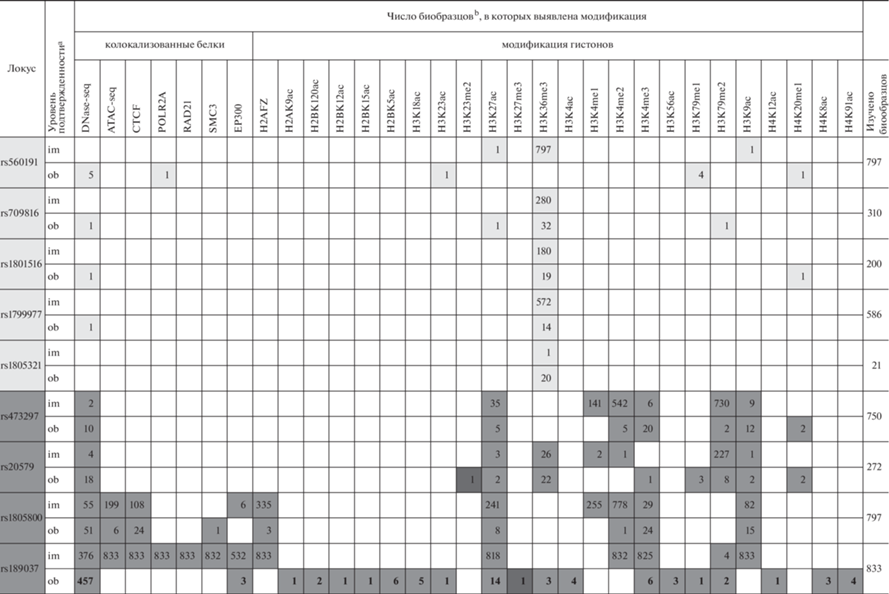
[i] a im (imputed) – предполагаемая (вмененная), ob (observed) – наблюдаемая колокализация с модифицированным гистоном/белком хроматина.
[ii] b Биообразцы представляют собой отдельные ткани/клеточные линии/клетки/индуцированные плюрипотентные стволовые клетки/дериваты и т.д. (данные EpiМap Epigenomics, http://compbio.mit.edu/epimap/#chromatin-states). Светло-серая заливка – способствующие транскрипции модификации, колокализованные с заменами в кодирующих регионах, темно-серая – с заменами в 5'UTR; черная заливка – модификации-репрессоры.
Иной паттерн модификаций гистонов колокализуется с маркерами в 5'UTR генов (табл. 2). Каждый из этих SNP колокализован с пятью модификациями хроматина: регионом гиперчувствительности к ДНКазе, H3K27ac и H3K4me2 (характеризующими энхансерные последовательности), H3K4me3 и H3K9ac (способствующими активации транскрипции). Три маркера колокализованы с модификацией H3K4me1 (также характерной для энхансерных последовательностей): rs473297 в 141, rs1805800 в 255 биообразцах и rs20579 в эпителиальных клетках меланомы и нейроэпителиоме. С модификацией, поддерживающей активацию транскрипции и элонгацию, H3K79me2, колокализованы следующие маркеры: rs473297 в 732, rs20579 в 235 и rs189037 в 6 биообразцах (в различных субпопуляциях T-лимфоцитов, а также в T-лимфоцитах при остром лимфобластном лейкозе и в лимфобластоидной клеточной линии). Модификация H3K36me3, поддерживающая элонгацию транскрипции, колокализуется с rs20579 (в 48 образцах) и rs189037 (в T-лимфоцитах, гладкомышечных клетках двенадцатиперстной кишки, фибробластах крайней плоти).
Два локуса (rs1805800 и rs189037) находятся в открытом хроматине с сайтами связывания таких белков, как CTCF, SMC3, EP300, H2AFZ. rs189037, кроме этого, колокализован с сайтом связывания РНК-полимеразы II и RAD21. Известно, что транскрипционный репрессор CTCF связывается с инсуляторами генов [12]. Кроме того, у млекопитающих CTCF совместно с когезиновым комплексом (в который входят в том числе SMC3 и RAD21) и гистоном H2AFZ принимает участие в организации хромосом в топологически ассоциированные домены [13, 14]. Следовательно, нуклеотидные последовательности, в которых находятся rs1805800 и rs189037, имеют важное топологическое значение и любые изменения в их структуре могут способствовать развитию патологических состояний.
Два маркера колокализованы с H3K79me1 (активация элонгации): rs20579 (в эмбриональных фибробластах легкого, дериватах линии H1-hESC – мезенхимальных и мезодермальных стволовых клетках), rs189037 (в клетках трофобласта ‒ дериваты из H1-hESC). С монометилированным лизином в положении 20 гистона H4 (H4K20me1), также поддерживающим активацию транскрипции [15], колокализованы rs473297 (в двух клеточных линиях Т-лимфоцитов при остром лимфобластном лейкозе) и rs20579 (в Т-лимфоцитах при остром лимфобластном лейкозе и в эндотелиальных клетках пупочной вены новорожденного).
rs189037 в гене АТМ в различных эмбриональных тканях/дериватах колокализован с целым рядом гистоновых модификаций, также поддерживающих активацию транскрипции: H2AK9ac, H2BK120ac, H2BK12ac, H2BK15ac, H3K23ac, H3K4ac, H2BK5ac, H3K18ac, H3K56ac, H4K12ac, H4K8ac и H4K91ac. Такое множество модификаций в единичных эмбриональных клеточных линиях может свидетельствовать о важной роли данного локуса на ранних этапах развития.
Все перечисленные выше модификации гистонов запускают/усиливают/поддерживают транскрипцию. И лишь две из колокализованных с анализируемыми маркерами модификации приводят к транскрипционному сайленсингу – это H3K27me3 [16] и H3K23me2 [17]. Среди изученных маркеров с H3K27me3 колокализован rs189037 в клетках зародышевого матрикса; а с H3K23me2 – rs20579 в эмбриональных стволовых клетках линии H1-hESC. Вероятно, более тонкая регуляция функций этих локусов в эмбриональном развитии может играть важную роль.
Таким образом, с учетом характера модификаций гистонов, колокализованных с привлеченными к анализу SNP генов систем репарации ДНК, можно заключить, что, во-первых, все эти регионы в большинстве изученных тканей активно транскрибируются; во-вторых, два региона (колокализованные с rs1805800 и rs189037) участвуют в организации топологических доменов, причем один из них (колокализованный с rs180937), вероятно, играет важную роль в эмбриональном развитии.
ЗАВИСИМОСТЬ ЭКСПРЕССИОННОГО СТАТУСА ГЕНОВ ОТ АНАЛИЗИРУЕМЫХ ПОЛИМОРФНЫХ ВАРИАНТОВ
Репарация ДНК ‒ один из базовых процессов живой клетки, а экспрессия генов, белковые продукты которых задействованы в его реализации и контроле, идет во всех типах тканей на всех стадиях онтогенеза. Однако, в отличие от генов “домашнего хозяйства”, уровень экспрессии генов, кодирующих белки систем репарации ДНК варьирует в зависимости от потребности клетки; и чем шире сфера компетенции этих генов, тем выше наблюдаемый уровень экспрессии. Для всех рассматриваемых здесь генов минимальный уровень экспрессии составлял от 1‒2 TPM (Transcripts Per Million – транскриптов на миллион): АТМ в скелетных мышцах, MRE11 в тканях головного мозга, TP53BP1 в цельной крови, PMS2 в миокарде, LIG1 в левом желудочке ‒ до 5‒6 TPM (NBN в коре почек, MLH1 в цельной крови) (https://gtexportal.org/home/). Интересно, что максимальный уровень этих транскриптов в большинстве случаев детектировали в лимфоцитах, стимулированных вирусом Эпштейна‒Барр (АТМ – 21, MRE11 –22, NBN – 67, MLH1 – 48, PMS2 – 22, LIG1 – 51 TPM), и только для гена TP53BP1 максимальный уровень экспрессии регистрировали в гипофизе – 50 TPM (https://gtexportal.org/home/). Все вышеперечисленные маркеры (табл. 1) относятся к cys-eQTL-вариантам (то есть могут влиять на экспрессию “своего” и расположенных рядом генов) и бóльшая часть – к sQTL-вариантами (влияют на сплайсинг).
В подавляющем большинстве случаев изменение экспрессии подчиняется линейной зависимости: либо RR > RP > PP, либо RR < RP < PP (где R – референсный аллель, P – производный аллель), – поэтому далее мы преимущественно обсудим различия между экспрессией гомозиготных генотипов, подразумевая, что гетерозиготы занимают промежуточное положение. Те случаи, когда наблюдаются отклонения от указанной тенденции, будут рассмотрены особо.
Влияние rs560191 в гене TP53BP1 на экспрессию и сплайсинг
rs560191 (TP53BP1) находится в пределах большого блока корегулируемых генов (рис. 2). Уровень сцепления, близкий к единице, выявлен для SNP, расположенных вокруг rs560191 и охватывающих регион больше 87 млн.п.н. Этот блок сцепления включает маркеры, которые представляют собой eQTL для 20 генов и sQTL для 11 генов (http://www.mulinlab.org/vportal/index.html).
Рис. 2.
Взаимодействия между регуляторными элементами и генами (согласно GeneHancer) в регионе локализации гена TP53BP1. Разными цветами выделены отдельные гены и их регуляторные взаимодействия (воспроизведено из онлайн-браузера UCSC: https://genome.ucsc.edu/).

Для большинства генов, регулируемых rs560191 (табл. S1 , см. Дополнительные материалы на сайте http://www.molecbio.ru/downloads/2023/1/supp_ Бабушкина_rus.zip), характерно однонаправленное изменение экспрессии в различных тканях в зависимости от генотипа. Так, для AC011330.13 (4 ткани), AC011330.5 (40 тканей), ADAL (37 тканей), CATSPER2 (1 ткань), CCNDBP1 (4 ткани), CKMT1A (4 ткани), CKMT1B (1 ткань), PDIA3 (3 ткани), TP53BP1 (15 тканей), TTBK2 (2 ткани) у гомозиготных носителей анцестрального аллеля (GG) уровень экспрессии выше, чем у гомозигот по производному аллелю (CC), а для гетерозигот характерны промежуточные значения. В то же время обратная ситуация (GG < CC) наблюдается для генов CATSPER2P1 (14 тканей), MAP1A (2 ткани), RNU6-554P (1 ткань), STRC (27 тканей), STRCP1 (25 тканей), TGM7 (3 ткани), TUBGCP4 (2 ткани). Для трех генов (LCMT2, TGM5, ZSCAN29) отмечается межтканевая вариабельность характера экспрессии. Так, максимальный уровень экспрессии гена LCMT2 в слизистой оболочке пищевода наблюдается у гомозигот СС, в то время как в четырех других тканях (большеберцовые нервы, культуры фибробластов, семенники, гладкомышечные клетки пищевода) – у гомозигот GG. В трех тканях (в слизистой оболочке и гладкомышечных клетках пищевода, ободочной кишке) характер экспрессии гена TGM5 изменяется в направлении GG > GC > CC, а в трансформированных вирусом Эпштейна‒Барр лимфоцитах – в обратном направлении (GG < GC < CC). Экспрессия гена ZSCAN29 выше у гомозигот по предковому аллелю в 25 исследованных тканях, кроме тканей мозга (хвостатое и прилежащее ядро базальных ганглиев, фронтальная кора, передняя поясная кора), в которых экспрессия этого гена наиболее выражена у носителей производного аллеля в гомозиготном состоянии (https://gtexportal.org/home/).
Показано влияние генотипов rs560191 на сплайсинг 11 генов, из них 10 совпадают с генами, для которых этот полиморфный вариант является eQTL, еще один ген – PPIP5K1 – не входит в этот список. Данный регион характеризуется сложной регуляцией транскрипции. В частности, происходит образование общего транскрипта при считывании генов CATSPER2P1, AC011330.5, CATSPER2. Для них показаны общие эффекты сплайсинга: производный аллель в дозозависимой манере увеличивает эффективность вырезания интрона 11 гена CATSPER2, что оказывает влияние на сплайсинг мРНК генов AC011330.5 (в 20 тканях) и CATSPER2 (в 37 тканях). Эффективность вырезания интрона 10 гена CATSPER2 из общего транскрипта снижается в скелетных мышцах, шейном отделе спинного мозга, мозжечке и черной субстанции головного мозга (оказывая влияние на сплайсинг мРНК гена CATSPER2) и повышается в семенниках (влияя на сплайсинг CATSPER2P1 и CATSPER2). Повышение эффективности вырезания области, включающей экзон 1–интрон 4 гена CATSPER2, влияет на сплайсинг мРНК гена AC011330.5 (в мозжечке). Кроме этого, носительство производного аллея rs560191 приводит к снижению эффективности вырезания отдельных интронов в генах AC011330.5, CATSPER2, PPIP5K1, STRCP1, TGM5, TGM7, TP53BP1 и ADAL (показано в одной‒трех тканях для каждого транскрипта).
Повышение эффективности вырезания показано для экзона 2 (с прилегающими интронами) гена ADAL (в 9 тканях), экзона 19 (с прилегающими интронами) гена STRC (в мозжечке), интрона 2 гена ZSCAN29 (в сигмовидной кишке) (https://gtexportal.org/home/).
Соотношение изоформ белка, синтезирующихся в результате альтернативного сплайсинга, зависит в том числе от того, насколько эффективно будет происходить вырезание тех или иных последовательностей из пре-мРНК. В случае, когда зарегистрирована зависимость и уровня экспрессии, и сплайсинга от одного и того же варианта одного и того же гена в одной и той же ткани, можно проанализировать, каким образом меняется синтез зрелой мРНК в зависимости от анализируемой нуклеотидной замены. Можно предположить, что при однонаправленном изменении, например при усилении экспрессии в направлении RR < RP < PP и повышении эффективности вырезания какого-либо фрагмента мРНК, общее увеличение продукции гена происходит, главным образом, за счет того/тех вариантов, в которых вырезаемый фрагмент отсутствует. И напротив, если эффективность снижается (в соответствии с примером выше, это RR > RP > PP), то усиление экспрессии происходит за счет относительного увеличения частоты тех вариантов, в которых этот фрагмент остается.
Анализ данных по влиянию rs560191 на экспрессию и сплайсинг показывает, что повышение эффективности вырезания интрона 11 гена CATSPER2 сопряжено со снижением уровня транскрипции генов CATSPER2 (в слизистой оболочке желудка) и AC011330.5 (в 21 ткани). Снижение экспрессии AC011330.5 в мозжечке идет на фоне увеличения эффективности вырезания экзона 1–интрона 4 гена CATSPER2, а в семенниках ‒ интрона 25 гена AC011330.5. Снижение экспрессии гена ADAL происходит на фоне повышения эффективности вырезания экзона 2 с прилегающими интронами (в 7 тканях) и понижения эффективности вырезания интрона 2 (в гипофизе). Общее снижение экспрессии генов TGM5 и TP53BP1 сопряжено со снижением эффективности вырезания их интронов (9 и 23 соответственно). Снижение эффективности вырезания интрона 11 в гене TGM7 и интрона 24 гена STRCP1, а также повышение эффективности вырезания экзона 19 с прилегающими интронами в гене STRC сопряжены с увеличением уровня экспрессии соответствующих генов в отдельных тканях (https://gtexportal.org/home/).
Влияние rs1805800 и rs709816 в гене NBN на экспрессию и сплайсинг
Изученные маркеры в гене NBN находятся на расстоянии 29 628 п.н.: rs1805800 локализован в регионе рядом с 5'UTR гена NBN, rs709816 – в экзоне 10. Вместе с тем, они достаточно тесно сцеплены. Так, для популяции г. Томска значения показателей сцепления: коэффициента неравновесия по сцеплению (D'), логарифма отношения шансов (LOD-score) и коэффициента корреляции Пирсона (r2) ‒ равны соответственно 0.942, 85.11 и 0.712; по данным проекта “1000 Genomes” у европеоидов D' = 0.995, r2 = 0.846. Исходя из данных проекта “1000 Genomes” по сцеплению маркеров, SNP, расположенные в пределах региона размером 142 т.п.н., включающего полностью ген NBN, сцеплены достаточно тесно (D' в пределах 0.945‒1.00) и образуют единый регуляторный блок eQTL- и sQTL-вариантов (http://www.mulinlab.org/vportal/index.html, https://genome.ucsc.edu/).
По данным ресурса GTExPortal (https://gtexportal.org/home/), указанные нуклеотидные замены относятся к eQTL-вариантам, влияющим на экспрессию как “своего”, так и близкорасположенных генов: CALB1, DECR1, OSGIN2 ‒ и, кроме того, на уровень экспрессии гена RP11-662G23.1, локализованного более чем в 821 т.п.н. от промотора гена NBN (табл. S1 , см. Дополнительные материалы на сайте http://www. molecbio.ru/downloads/2023/1/supp_Бабушкина rus.zip) (https://gtexportal.org/home/, https://genome. ucsc.edu/).
Для большинства изученных тканей характерно однонаправленное изменение экспрессии ряда генов в зависимости от генотипов по rs1805800. Так, при увеличении дозы альтернативного аллеля (T) возрастает уровень экспрессии гена CALB1 (в 6 из 6 изученных тканей), NBN (в 15 из 18 тканей), OSGIN2 (в трех тканях из четырех изученных). Из общей тенденции есть несколько исключений. Для RP11-662G23.1 в единственной изученной ткани (мышечной ткани пищевода) отмечается сходная в отношении гомозиготных генотипов тенденция (TT>CC), однако минимальный уровень экспрессии зарегистрирован у носителей гетерозиготного генотипа. Самая высокая экспрессия наблюдается у гомозигот по референсному аллелю в четырех тканях из пяти изученных для гена DECR1 (за исключением скелетных мышц, в которых наблюдается обратная зависимость). Для гена NBN к исключениям относятся цельная кровь и ткань пищеводно-желудочного соединения, в которых наиболее высокий уровень экспрессии детектируют у гомозигот с референсным генотипом (СС), а также кора головного мозга, где, при общей тенденции CC<TT, уровень экспрессии максимален у гетерозигот. Для ткани пищеводно-желудочного соединения показаны также отличия в экспрессии гена OSGIN2: у гомозигот по производному аллелю уровень э-кспрессии ниже, чем по анцестральному (https://gtexportal.org/home/).
В зависимости от генотипов по маркеру rs709816 в экзоне 10 гена NBN меняется уровень экспрессии тех же генов, хотя характер изменений несколько иной. У гомозигот по альтернативному аллелю (AA) экспрессия гена NBN выше в 14 из 14 изученных тканей (как показано и для rs1805800), однако более низкий уровень экспрессии зарегистрирован для генов CALB1 (в 7 изученных тканях) и RP11-662G23.1 (в мышечной ткани пищевода). Ген DECR1 экспрессируется интенсивнее у носителей производного аллеля в четырех изученных тканях, еще в двух (в скелетных мышцах и ушке предсердия) зависимость экспрессии обратная. Уровень экспрессии гена OSGIN2 в мышечной ткани пищевода выше у гомозигот по альтернативному аллелю, тогда как в цельной крови и ткани пищеводно-желудочного соединения – у гомозигот по референсному аллелю (https://gtexportal.org/home/).
Анализируемые маркеры в гене NBN относятся к sQTL-вариантам для генов DECR1 и NBN. При процессинге мРНК гена DECR1 эффективность вырезания интрона 1 выше у носителей производных аллелей обоих изученных маркеров: для генотипа ТТ rs1805800 в легких и коже, не подвергающейся солнечной экспозиции; для генотипа GG rs709816 – в легких, коже, не подвергающейся солнечной экспозиции, и тканях молочной железы (https://gtexportal.org/home/).
Для гена NBN отмечено тканезависимое изменение соотношений вариантов сплайсинга в зависимости от генотипов по изученным маркерам. У носителей гомозиготных генотипов обоих исследованных маркеров повышается эффективность вырезания интрона 4 в семенниках, скелетных мышцах, большеберцовых артериях, ткани пищеводно-желудочного соединения, ткани молочной железы и снижается в большеберцовых нервах и в лимфоцитах, трансформированных вирусом Эпштейна–Барр. Аналогично снижается эффективность вырезания интрона 2 в культурах фибробластов и в подкожной жировой клетчатке (https://gtexportal.org/home/).
В базах данных для пяти тканей приведена информация по изменениям и уровням экспрессии гена NBN, а также вариантам его сплайсинга (в зависимости от изученных SNP). В отношении обоих маркеров (rs1805800 и rs709816) выявленная изменчивость носит одинаковый характер: в трех тканях носительство альтернативных аллелей приводит к увеличению экспрессии гена в целом при увеличении эффективности вырезания интрона 4 (в семенниках, скелетных мышцах, большеберцовых артериях); в двух тканях общее увеличение уровня экспрессии происходит на фоне снижения эффективности вырезания интрона 4 (в большеберцовых нервах) и интрона 2 (в подкожной жировой клетчатке) (https://gtexportal.org/home/).
Влияние rs189037 и rs1801516 в гене ATM на экспрессию и сплайсинг
Исследованные маркеры в гене АТМ в популяции г. Томска находятся в неравновесии по сцеплению. Так, показатели сцепления для rs189037 и rs1801516 в гене АТМ следующие: D' = 0.869, LOD = 11.44, r2 = 0.109. В научных публикациях и базах данных (см., например, Ensemble: http:// www.ensembl.org/index.html) их сцепление не анализируется ввиду значительной удаленности друг от друга – изученные маркеры rs189037 (промоторный регион) и rs1801516 (экзон 37) находятся на расстоянии 81.5 т.п.н. Тем не менее для каждого из анализируемых SNP идентифицирован обширный регион, маркеры в котором тесно сцеплены и являются eQTL для одних и тех же генов. Для rs189037 размер этого региона не менее 167 т.п.н., для rs1801516 – не менее 192 т.п.н. (http://www.mulinlab.org/vportal/index.html).
rs189037 представляет собой eQTL-вариант для “своего” и близлежащих генов: ACAT1, NPAT, C11orf65, KDELC2 (POGLUT3) (табл. S1 , см. Дополнительные материалы на сайте http://www.molecbio.ru/downloads/2023/1/supp_Бабушкина_rus.zip). В большинстве случаев альтернативный аллель (А) приводит к снижению уровня экспрессии корегулируемых генов. Исключения отмечены в отдельных тканях для генов NPAT (в цельной крови) и АТМ (в щитовидной железе), а также для C11orf65 в коре надпочечников (единственная ткань, для которой в настоящее время показано влияние rs189037 на уровень экспрессии рассматриваемого гена). Кроме того, для гена ATM в трех тканях (ушке предсердия, сальнике и слизистой оболочке пищевода) наименьший уровень экспрессии зарегистрирован у гетерозигот (при сохранении общей тенденции: GG>AA) (https://gtexportal.org/home/).
rs1801516 также выступает как eQTL-вариант для “cвоего” и близлежащих генов: ACAT1, NPAT, C11orf65 (табл. S1 , см. Дополнительные материалы на сайте http://www.molecbio.ru/downloads/2023/ 1/supp_Бабушкина_rus.zip). У гомозигот по альтернативному аллелю (AA) регистрируют пониженные уровни экспрессии генов ACAT1 (в щитовидной железе, скелетных мышцах, легких, культурах фибробластов) и ATM (мышечная ткань пищевода) и повышенные для генов NPAT (большеберцовые нервы и аорта) и C11orf65 (в гипофизе, щитовидной железе и коже, подвергающейся солнечной экспозиции) (https://gtexportal.org/home/).
К sQTL относится только rs189037, причем только для гена ATM: в культуре фибробластов происходит дозозависимое снижение эффективности вырезание интрона 40, в то время как в легких, поджелудочной железе и сальнике эффективность вырезания повышается, как и в отношении интрона 26 в большеберцовых нервах (https:// gtexportal.org/home/).
Для двух тканей можно соотнести eQTL- и sQTL-влияние rs189037 на синтез зрелой мРНК белка АТМ. Общее снижение уровня экспрессии гена ATM в культуре фибробластов происходит параллельно с увеличением эффективности вырезания интрона 40, а в клетках сальника – на фоне снижения эффективности вырезания этого интрона (https://gtexportal.org/home/).
Влияние rs473297 в гене MRE11 на экспрессию и сплайсинг
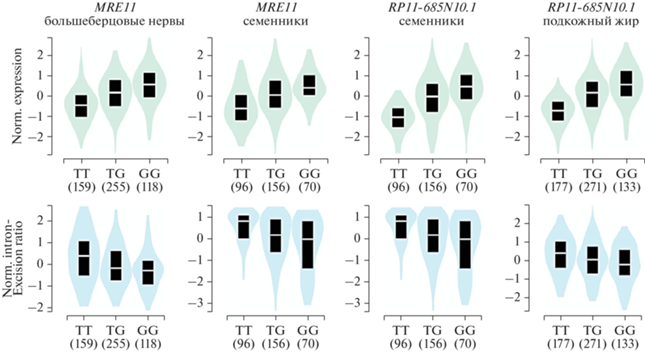
rs473297 находится в большом блоке сцепления (не менее 123 т.п.н.), в пределах которого D' между анализируемым маркером и другими eQTL-SNP составляет 0.95‒1.00. rs473297 служит eQTL-вариантом для генов MRE11 (49 тканей), GPR83 (4 ткани), IZUMO1R (1 ткань) и RP11-685N10.1 (37 тканей) (табл. S1 , см. Дополнительные материалы на сайте http://www.molecbio.ru/ downloads/2023/1/supp_Бабушкина_rus.zip). Во всех изученных тканях наличие альтернативного варианта приводит к возрастанию уровня экспрессии генов IZUMO1R, MRE11, RP11-685N10.1 и к снижению экспрессии гена GPR83 (https:// g-texportal.org/home/). Влияние на уровень сплайсинга показано для генов MRE11 (семенники и большеберцовые нервы) и RP11-685N10.1 (8 тканей). Наличие замены приводит к снижению эффективности вырезания интрона 7, внутри которого локализован ген RP11-685N10.1. В большеберцовых нервах происходит также снижение эффективности вырезания интрона 1 мРНК гена MRE11 (рис. 3) (https://gtexportal.org/home/).
Рис. 3.
Изменение уровня экспрессии (вверху) и эффективности сплайсинга (внизу) генов MRE11 (в большеберцовых нервах и семенниках) и RP11-685N10.1 (в семенниках и подкожной жировой ткани) в зависимости от rs473297 в гене MRE11. Ось Х – генотипы по rs473297, ось Y – нормализованные уровни экспрессии (вверху), нормализованный уровень вырезания интрона (по данным GTXPortal: https://gtexportal.org/home/).
Влияние rs1799977в гене MLH1 на экспрессию и сплайсинг
Для rs1799977 в гене MLH1 показано полное сцепление (и участие в регуляции одних и тех же генов) с маркерами, находящимися от него на значительном удалении ‒ более 78 т.п.н. в направлении к 3'-концу гена. Тесное сцепление (D' = 0.912) показано и с маркером, лежащим на расстоянии более 80 т.п.н. по направлению к 5'‑концу (интронный вариант гена TRANK1); в то же время этот маркер фактически относится к другому регуляторному блоку (http://www.mulinlab.org/vportal/index.html, https://genome.ucsc.edu/). По данным проектов ENCODE и GeneHancer, весь этот регион обогащен регуляторными последовательностями, наблюдается значительная корегуляция колокализованных генов (https:// g-enome.ucsc.edu/, https://www.genecards.org/). rs1799977 ‒ eQTL-вариант для десяти и sQTL для трех генов (табл. S1 , см. Дополнительные материалы на сайте http://www.molecbio.ru/downloads/ 2023/1/supp_Бабушкина_rus.zip). Для пяти генов есть информация об изменениях уровней экспрессии в зависимости от данной замены в одной ткани. Показано, что у гомозигот по вариантному аллелю уровни экспрессии MLH1 (в скелетных мышцах), EPM2AIP1 (в щитовидной железе), RP11-259K5.2 (в коже, подвергавшейся солнечной экспозиции) повышены, а ITGA9 (в цельной крови) и PRADC1P1 (в большеберцовых нервах) снижены. Для гена RP11-129K12.1 показано снижение уровня экспрессии в трех исследованных тканях (в подкожной жировой клетчатке, сальнике, коже, подвергающейся солнечной экспозиции). Для остальных генов зарегистрирован тканезависимый характер изменения экспрессии. Так, при наличии производного аллеля (G) выявлен более низкий уровень экспрессии генов RP11-285J16.1 и UBE2FP1 (в коже, подвергающейся солнечной экспозиции), а также GOLGA4 (в слизистой оболочке пищевода) и LRRFIP2 (в коже вне зависимости от солнечной экспозиции и в цельной крови). Напротив, у носителей генотипа GG выше уровень экспрессии генов RP11-285J16.1 и UBE2FP1 (в щитовидной железе), GOLGA4 (в скелетных мышцах и коже, подвергающейся солнечной экспозиции), LRRFIP2 (в 14 тканях) (https://gtexportal.org/home/).
Влияние rs1799977 на сплайсинг показано для трех генов. В подавляющем большинстве случаев замена приводит к снижению эффективности вырезания интронов: интрон 12 в гене MLH1 (в большеберцовых артериях), интрон 1 в LRRFIP2 (в 31 ткани), интрон 22 в GOLGA4 (в слизистой оболочке пищевода). Повышение эффективности вырезания показано только для интрона 2 гена GOLGA4 (в скелетных мышцах) и интрона 1 гена LRRFIP2 (в левом желудочке) (https://gtexportal. org/home/). Несмотря на сходный тип “сплайсингового ответа” на альтернативный аллель, в результате тканеспецифичного изменения характера экспрессии LRRFIP2 имеет место тканеспецифичное изменение соотношений сплайсинговых вариантов этого гена. Так, на фоне снижения эффективности вырезания интрона 1 из мРНК гена LRRFIP2 в трех тканях (кожа вне зависимости от солнечной экспозиции и цельная кровь) происходит снижение экспрессии этого гена, а в 11 тканях ‒ усиление (https://gtexportal.org/home/).
Влияние rs1805321 в гене PMS2 на экспрессию и сплайсинг
rs1805321 в гене PMS2 находится в блоке сцепления eQTL-SNP, охватывающем не менее 76 т.п.н. Эта замена ‒ eQTL-вариант для 7 генов (табл. S1 , см. Дополнительные материалы на сайте http:// www.molecbio.ru/downloads/2023/1/supp_Бабушкина_rus.zip). В подавляющем большинстве случаев альтернативный аллель (A) приводит к возрастанию уровня экспрессии. Такой характер изменчивости показан для генов ANKRD61 (в 6 тканях), CCZ1B (в 11 тканях), EIF2AK1 (в 17 тканях), PMS2 (в 28 тканях), SNORA42 (в трех тканях). В ряде тканей уровень экспрессии этих генов носит несколько иной характер: при сохранении общей тенденции (GG<AA) максимальный уровень наблюдается у гетерозигот. Такая зависимость зарегистрирована для гена ANKRD61 в большеберцовых нервах, для EIF2AK1 в адипоцитах сальника, для PMS2 в коже, не подвергавшейся солнечной экспозиции, и в базальных ганглиях головного мозга; для SNORA42 во фронтальной коре головного мозга. И, напротив, показано снижение экспрессии при наличии производного аллеля для гена CCZ1 (в 8 тканях). Для гена RAC1 влияние данной замены выявлено в одной ткани (щитовидная железа), уровень максимален у гомозигот по референсному аллелю, но минимален у гетерозигот (https://gtexportal.org/home/).
Влияние rs1805321на сплайсинг показано для одного гена – AIMP2 – в двух тканях: эффективность вырезания интрона 1 при наличии производного аллеля снижается в скелетных мышцах и возрастает в коже, подвергающейся солнечной экспозиции (https://gtexportal.org/home/).
Влияние rs20579 в гене LIG1 на экспрессию и сплайсинг
rs20579 в гене LIG1 находится внутри региона сцепления eQTL-SNP, охватывающего около 28 т.п.н. Это eQTL-вариант для 6 генов (табл. S1 , см. Дополнительные материалы на сайте http://www.molecbio.ru/downloads/2023/1/supp_ Бабушкина_rus.zip). Производный аллель приводит к возрастанию уровня экспрессии генов PLA2G4C (в 22 тканях), PLA2G4C-AS1 (в легких), CTC-453G23.5 (в коже, подвергавшейся солнечной экспозиции). Понижение экспрессии наблюдается для генов LIG1 (в большеберцовых нервах) и AC022154.7 (в скелетных мышцах). Тканезависимое изменение уровня экспрессии зарегистрировано для гена CARD8: у носителей производного аллеля в путамене (базальные ганглии головного мозга) наблюдается повышение уровня экспрессии, а в прилежащем ядре (базальные ганглии головного мозга), напротив, снижение (https://gtexportal.org/home/). Влияния rs20579 на сплайсинг к настоящему времени не показано.
Таким образом, все рассматриваемые маркеры изменяют транскрипцию как своего, так и близлежащих генов, будучи для них cys-QTL-вариантами. Тканезависимый характер изменений может определять различия степени поражения тканей и органов при развитии патологического фенотипа; при этом поражения органов-мишеней могут быть обусловлены изменениями (как уровня экспрессии в целом, так и альтернативного сплайсинга) спектра корегулируемых генов. Например, ген ACAT1, корегулируемый и колокализованный с геном ATM (табл. S1 , см. Дополнительные материалы на сайте http:// www. molecbio.ru/downloads/2023/1/supp_Бабушкина_ rus.zip), кодирует митохондриальную ацетил-КоА-ацетилтрансферазу ‒ один из ключевых ферментов этерефикации холестерина. Следовательно, вовлеченность этого фермента в развитие атеросклероза и его патогенетически значимых признаков логически обоснована; блокаторы этого фермента используют для лечения атеросклероза [18]. Одним из возможных объяснений полученных нами ранее ассоциаций гена ATM с липидными показателями у больных ИБС [7] может быть именно изменение уровня экспрессии ACAT1 (и уже его влияние на развитие патологии) в результате нуклеотидных замен в гене ATM.
ИЗМЕНЕНИЯ МОТИВОВ СВЯЗЫВАНИЯ ТРАНСКРИПЦИОННЫХ ФАКТОРОВ, ПРОИСХОДЯЩИЕ В РЕЗУЛЬТАТЕ АНАЛИЗИРУЕМЫХ НУКЛЕОТИДНЫХ ЗАМЕН
В результате изменения нуклеотидной последовательности могут меняться сайты связывания TF, что позволяет определять статус нуклеотидных замен в качестве cys-eQTL-SNP. В настоящее время накоплена обширная информация о взаимодействии хроматина и различных TF. Имеющиеся данные получены как экспериментальным путем (методами иммунопреципитации хроматина, например ChIP-Seq), так и биоинформатическими методами. Существует множество инструментов для биоинформатического анализа, позволяющих выявить в нуклеотидной последовательности вероятные мотивы связывания TF и предсказать, каким образом будет меняться их аффинность в результате всевозможных структурных изменений. Однако следует учитывать, что различные инструменты анализа не всегда дают согласующуюся информацию.
Мы проанализировали изменения мотивов связывания TF с помощью ресурсов HaploReg (https://pubs.broadinstitute.org/mammals/haploreg/ haploreg.php), Poplympact (https://regulomedb.org/ regulome-search/), GWAS4D (http://mulinlab.tmu. edu.cn/gwas4d/gwas4d/gwas4d/gwas4d_server) (рис. 4, круговые диаграммы Венна). Наименьшее число изменяющихся мотивов предсказывает HaploReg (https://pubs.broadinstitute.org/mammals/haploreg/ haploreg.php). Согласно полученным данным, в результате проанализированных вариантов меняются сайты связывания TF в генах ATM (rs189037, rs1801516), TP53BP1 (rs560191), NBN (rs1805800). Ресурс Polympact не определяет изменений мотивов в результате замены в гене TP53BP1, но показывает их наличие в генах ATM, NBN, MRE11, PMS2 (рис. 4). Согласно GWAS4D, все изученные нуклеотидные замены влияют на аффинность связывания TF с их сайтами. Так, rs560191 в гене TP53BP1 изменяет только 8 сайтов, а rs189037 в гене ATM ‒ 33; всего для 9 изученных SNP меняется 168 мотивов для 99 TF (https://bcglab.cibio. unitn.it/polympact/, https://pubs.broadinstitute.org/ mammals/haploreg/haploreg.php, http://mulinlab. tmu.edu.cn/gwas4d/gwas4d/gwas4d /gwas4d_server).
Рис. 4.
Влияние изученных SNP-маркеров на сайты связывания транскрипционных факторов. Круговые диаграммы – теоретически рассчитанные мотивы связывания TF с изменяющейся в результате нуклеотидной замены аффинностью (по данным HaploReg, Poplympact, GWAS4D). Овальные диаграммы ‒ экспериментально подтвержденные ДНК-белковые взаимодействия в регионах локализации нуклеотидной замены (по данным, приведенным в JASPAR, ORegAnno, ENCODE, VannoPortal). Цифрами указано число мотивов в разных базах данных; названия совпадающих по данным различных ресурсов TF приведены рядом.
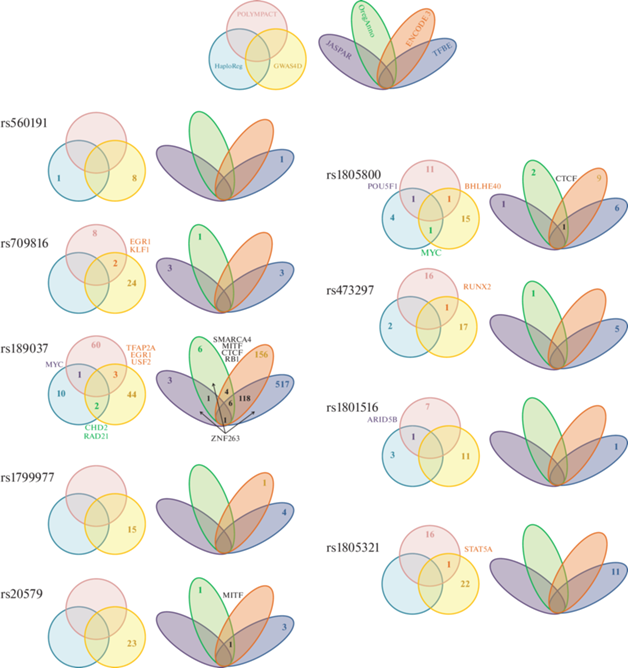
Для перекрывающихся мотивов все алгоритмы дают согласующуюся информацию по изменению аффинности связывания TF при изменении нуклеотидной последовательности, хотя перекрывание полученных списков в целом довольно слабое. Так, и HaploReg, и GWAS4D предсказывают изменения аффинности сайтов связывания MYC (rs1805800 в гене NBN), а также CHD2 и RAD21 (rs189037 в гене ATM). HaploReg и Polympact дают согласующиеся результаты по отношению к POU5F1 (rs1805800, ген NBN), MYC (rs189037, ген ATM) и ARID5B (rs1801516, ген ATM). Наибольшее число совпадающих результатов получено между данными GWAS4D и Polympact: rs1805800 (NBN) приводит к изменению сайта связывания BHLHE40; rs709816 (NBN) – EGR1 и KLF1; rs473297 (MRE11) – RUNX2; rs189037 (ATM) – USF2, EGR1, TFAP2A; rs1805321 (PMS2) – STAT5A (https://regulomedb. org/regulome-search/, https://pubs.broadinstitute.org/ mammals/haploreg/haploreg.php, http://mulinlab. tmu.edu.cn/gwas4d/gwas4d/gwas4d/gwas4d_server) (рис. 4). Большинство из этих TF экспрессируются с разным уровнем в широком спектре тканей, за исключением TFAP2A, который экспрессируется в тканях пищеварительной системы (малая слюнная железа, слизистая оболочка пищевода), мочевыводящей системы (кора и мозговое вещество почки, мочевой пузырь), репродуктивной системы (семенники, влагалище), коже (вне зависимости от солнечной экспозиции) (https://genome. ucsc.edu/, https://www.encodeproject.org/).
Чтобы проанализировать, связывание каких TF подтверждено экспериментально, мы привлекли данные ChIP-Seq-анализа из ресурсов JASPAR (https://jaspar.genereg.net/) [19], ORegAnno (http:// www.oreganno.org/) [20], ENCODE (https://www. encodeproject.org/) [21], а также Vann-oPortal (http://www.mulinlab.org/vportal/index. html), на котором представлены объединенные данные CistromeDB 20181120, DeepBlueR V1.0, GTRD 2020-06, EpiMap 2021-01-11, включающие информацию о связывании TF и других взаимодействующих с хроматином белков [22‒24] (рис. 4, овальные диаграммы Венна).
На основании данных по иммунопреципитации хроматина можно сделать вывод, что регионы локализации анализируемых SNP в кодирующей последовательности минимально связаны с TF. Так, по данным JASPAR, ORegAnno и ENCODE, не обнаружено сайтов связывания TF для rs560191 в гене TP53BP1, rs1801516 в гене ATM, rs1805321 в гене PMS2. Однако по данным VannoPortal, для этих нуклеотидных последовательностей показано связывание в отдельных тканях/клеточных линиях с TF либо иными белками: модификаторами хроматина, ферментативными комплексами и другими, также влияющими на уровень экспрессии белков. Например, в регионе локализации rs560191 выявлено связывание лизиновой метилтрансферазы KMT2A (в клеточной линии RS4 острого лимфобластного лейкоза) и POLR2A – субъединицы А РНК-полимеразы II (в клетках влагалища); с регионом локализации rs1801516 в гене ATM связывается BRD4 (в клетках крови); в области гена PMS2, содержащей rs1805321, обнаружено связывание 13 белков (каждый в 1‒3 типах клеток). В регионах еще двух SNP в кодирующей последовательности и одного маркера в 5'UTR факты связывания с единичными TF зарегистрированы в нескольких базах данных, но эти результаты не перекрываются. Так, с регионом локализации rs1799977 в гене MLH1 по данным ENCODE связывается CTCF (в клетках аденомы паращитовидной железы), по данным VannoPortal – FOXA1 (в клетках простаты), SPIB (в костном мозге), GATA4 и FOXA2 (в коже). С регионом локализации rs709816 в гене NBN связывается ряд белков: ZSCAN4, CEBPD, CEBPA (JASPAR); E2F1 (ORegAnno); AR, MYC, POLR2A, BRD4 (VannoPortal). Область локализации rs473297 в гене MRE11 совпадает с сайтом связывания SMARCA4 (ORegAnno); ARID1A, FOXA2, GATA3, GATA4, GATA6 (VannoPortal) (http:// www.mulinlab.org/vportal/index.html, https://www. encodeproject.org/, https://jaspar.genereg.net/, http:// www.oreganno.org/).
Только для трех изученных маркеров (5'UTR генов NBN, LIG1, ATM) списки связываемых с областью их локализации белков частично пересекаются (рис. 4). Для rs1805800 выявлено связывание нуклеотидной последовательности с TFEB (JASPAR); SMARCA4 и CTCF (ORegAnno); CTCF (в 150 тканях), H2AZ, EP300, SNAI2, SMC3, NR3C1 (VannoPortal); CTCF (в 187 тканях) и еще 8 TF (в 1‒5 тканях) (ENCODE). Таким образом, в трех из проанализированных ресурсов содержится информация о связывании этой нуклеотидной последовательности с транскрипционным репрессором CTCF. В регионе rs20579 (в гене LIG1) зарегистрировано связывание MITF (ORegAnno); H2AZ, MITF, FOXP1 (VannoPortal). В двух базах в этом регионе зарегистрировано связывание MITF.
Наибольшее число ДНК-белковых взаимодействий зарегистрировано для региона, в котором находится rs189037 (ген ATM). В ресурсе JASPAR для этой области показано взаимодействие с факторами ZNF263, SP5 и ZNF148. Согласно ORegAnno, ДНК в этом регионе ассоциирована с SMARCA4, RBL2, RB1, ZNF263, CTCF и MITF. С этой областью взаимодействует 156 TF по данным ENCO-DE, 517 ‒ по данным VannoPortal; для двух последних ресурсов 118 белков общие. В списках сразу трех ресурсов встречаются SMARCA4, RB1, CTCF, MITF (ORegAnno, ENCODE, VannoPortal) и ZNF263 (ORegAnno, JASPER, VannoPortal); из них к TF, связываемым в широком спектре тканей, относится только CTCF (рис. 4).
Как можно заметить, для двух промоторных регионов (rs1805800 и rs189037) характерно связывание с CTCF. Это CCCTC-связывающий фактор, играющий важную мультифункциональную роль в ремоделировании хроматина [12, 25]. Хорошо известна роль CTCF в связывании инсуляторных последовательностей, поэтому можно предположить, что именно эта функция CTCF наиболее значима в данных локусах.
В то время как результаты, полученные с помощью методов биоинформатического анализа, указывают на наличие возможности связывания с ДНК определенных TF, методы иммунопреципитации хроматина показывают реально существующие связи, но в отдельных клетках/тканях на определенной стадии развития, в определенном физиологическом состоянии и с определенной нуклеотидной последовательностью. В связи с этим отсутствие экспериментального подтверждения теоретических расчетов нельзя рассматривать как доказательство отсутствия такого взаимодействия в принципе ‒ просто оно не зарегистрировано в проанализированных биообразцах. Мы сопоставили результаты биоинформатического анализа и данные ChIP-seq для исследованных маркеров. Только для rs189037 выявлено совпадение TF и других белков хроматина, аффинность которых к сайтам связывания теоретически изменяется в результате нуклеотидной замены и связывание которых с этим регионом имеет экспериментальное подтверждение. Для аннотированных в базе JASPAR TF не выявлено изменений аффинности к мотивам связывания, а для ряда TF, аннотированных в трех других базах, такие мотивы идентифицированы (табл. 3).
Таблица 3.
Экспериментально подтвержденные ДНК-белковые взаимодействия, для которых теоретически предсказано изменение аффинности связывания транскрипционных факторов с их сайтами в результате нуклеотидной замены rs189037
| Биоинформатический ресурс | Ресурсы с данными иммунопреципитации хроматинаа | |||
|---|---|---|---|---|
| JASPAR | ORegAnno | ENCODE | VannoPortal | |
| HaploReg | 0 | 0 | 4 MYC ↑ RAD21 ~ |
5 MYC ↑ RAD21 ~ ELF1 ↓ ZNF143 ↓ |
| GWAS4D | 0 | 1 CTFC ↑ |
17 CTCF ↑ RAD21 ↑ |
26 CTCF ↑ RAD21 ↑ SMC3 ↑ GABPA ↑ NRF1 ↑ HDAC2 ↓ SIN3A ↑ CDK8 ↑ USF1 ↑ |
| Polympact | 0 | 0 | 9 MYC ↑ |
22 MYC ↑ REST ↑ E2F1 ↓ (7 сайтов) TRIM28 ↓ USF1 ↑ (6 сайтов) |
а Указано число TF, общих для разных ресурсов; приведены названия только тех TF, наличие которых в данной точке зарегистрировано более чем в 10 тканях. Стрелками ↑ и ↓ указано соответственно увеличение и снижение аффинности TF к мотиву при наличии альтернативного аллеля; знак “~” указывает на незначительное изменение (по биоинформатическим данным). Отдельные мотивы в пределах одного региона в различных ресурсах могут быть сдвинуты вправо или влево относительно друг друга.
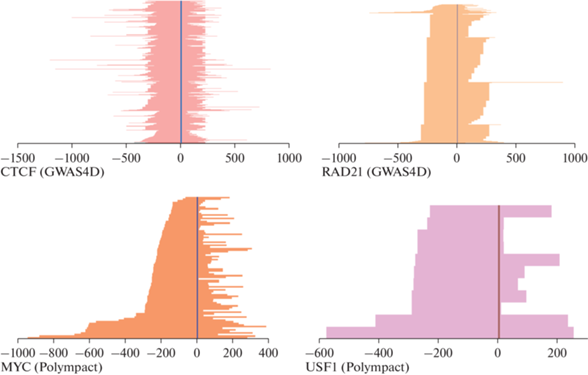
Экспериментально выявляемые регионы ДНК-белковых взаимодействий достаточно протяженны (среди проанализированных их длина составила 152‒1975 п.н.). Длина рассчитанных теоретически мотивов значительно короче (8‒20 п.н.). Для всех потенциально изменяющихся мотивов подтверждена их локализация внутри региона взаимодействия (на рис. 5 в качестве примера приведены данные для мотивов четырех TF). Таким образом, нуклеотидная замена в ДНК может приводить к изменению аффинности связывания соответствующих TF и тем самым определять эффективность их функционирования. Кроме того, при наличии альтернативного аллеля биоинформатические ресурсы предсказывают появление de novo сайтов связывания TF, предполагающих появление новых регуляторных контуров. Появление таких сайтов предсказано для всех изученных SNP. Непонятно, реализуется ли эта возможность, но с учетом тканеспецифичности широкого спектра TF можно определить, какие ткани могут быть мишенями в этом случае.
Рис. 5.
Локализация изменяемых мотивов связывания транскрипционных факторов в регионах иммунопреципитации соответствующих белков в различных тканях (составлено по данным Polympact и VannoPortal). Вертикальная полоса представляет собой теоретически рассчитанный изменяемый мотив, цветные горизонтальные полосы – экспериментально выявленные регионы ДНК-белковых взаимодействий в различных тканях. Ось Х представляет собой длину мотивов связывания TF (в нуклеотидах); за нулевую отметку принято положение теоретически рассчитанного изменяемого мотива. Ось Y – ткани, в которых с помощью иммунопреципитации хроматина показано связывание этих TF (число тканей составляет 1457, 912, 89 и 11 для CTCF, RAD21, MYC и USF1 соответственно).
Таким образом, разнообразие изменяемых мотивов связывания TF может объяснить роль исследованных SNP в качестве QTL-вариантов. Так, например, с регионом локализации rs1189037 (в гене АТМ) связывается фактор USF1 (http://www. mulinlab.org/vportal/index.html), регулирующий экспрессию широкого спектра генов метаболизма липидов и глюкозы [26, 27]. В литературе описана мутация в гене USF1, приводящая к семейной гиперлипидемии [28], из чего можно сделать вывод о его важной роли в детерминации уровня липидов. rs1189037 меняет аффинность связывания USF1. Так, согласно ресурсу GWAS4D, при наличии альтернативного аллеля появляется новый, ранее не существовавший мотив связывания этого TF с локализацией 11:108093828–108093837; согласно ресурсу Polympact, увеличивается аффинность пяти вероятных мотивов и появляется один новый (с точкой старта в 11:108093826). Таким образом, при наличии альтернативного аллеля сродство TF к последовательности ДНК усиливается, что может влиять на эффективность его функционирования. Следовательно, этот путь может быть еще одним механизмом, объясняющим ассоциированность rs1189037 с липидными показателями у больных ИБС [7]. Вместе с тем вышеприведенная информация указывает на то, что рассматриваемые маркеры при ассоциативных исследованиях могут отражать участие в патологическом процессе не только “своего” гена, но и тех, в регуляции функционирования которых они принимают участие.
АДАПТОГЕННЫЙ И ПАТОЛОГИЧЕСКИЙ ПОТЕНЦИАЛ АНАЛИЗИРУЕМЫХ ПОЛИМОРФНЫХ ВАРИАНТОВ
Межвидовые сравнения свидетельствуют о высоком уровне консервативности четырех из девяти проанализированных локусов. В первую очередь это касается SNP в кодирующем регионе гена ATM – rs1801516, для которого консервативность у позвоночных определена с помощью всех анализируемых алгоритмов (табл. 4). Немногим ниже уровень консервативности rs1799977 – замены в кодирующем регионе гена MLH1: методом PhyloP (измеряет эволюционную консервативность отдельных последовательностей ДНК) выявлена высокая консервативность у млекопитающих и позвоночных в целом, но не у приматов. Методами PhastCons, GERP++ консервативность этого локуса на разных эволюционных уровнях подтверждена (табл. 4). Можно предположить, что данный регион в ходе эволюционной истории стал изменяться именно у приматов.
Таблица 4.
Эволюционная роль изученных полиморфных вариантов генов, кодирующих белки систем репарации ДНК
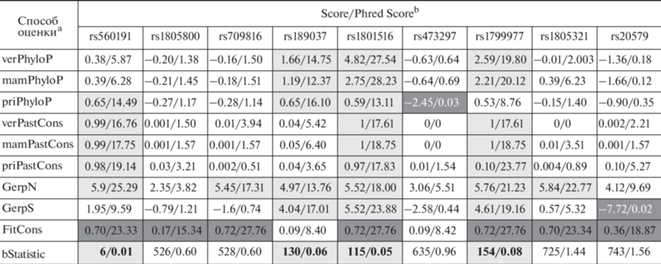
[i] a Показатели PhyloP и PastCons оценивают эволюционную консервативность на основании межвидовых сравнений, за исключением человека (приставки обозначают классификационные ранги: ver – позвоночные, mam – млекопитающие, pri – приматы). Балл PhyloP соответствует ‒lg p-value нулевой гипотезы нейтральной эволюции; положительные значения (до 3) указывают на очищающий отбор, а отрицательные значения (до ‒ 14) свидетельствуют об ускоренной эволюции консервативных (и, следовательно, вероятно функциональных) элементов [29, 30]. Показатели PhastCons оценивают вероятность того, что локус содержится в консервативном элементе, варьируют от 0 до 1 и отражают вероятность отрицательного отбора [30‒32]. Оценки GerpN и GerpS основаны на анализе отдельных нуклеотидов: высокое значение GerpN указывает на высокую гомологию локуса у разных видов; положительные значения GerpS ‒ на дефицит замен, отрицательные – на их избыточность ‒ и оценивают уровень “ненейтральности” локуса [30, 33]. FitCons оценивает вероятность того, что нуклеотид в данном положении влияет на приспособленность (на основании метода INSIGHT); показатель варьирует от 0 до 1 [34]. bStatistic ‒ оценка фонового отбора, указывающая на ожидаемую долю нейтрального разнообразия, присутствующего на соответствующем участке: значения, близкие к 0, означают почти полное удаление разнообразия в результате отбора; значения, близкие к 1, указывают на незначительный эффект отбора на разнообразие.
b Score – оценка, полученная для каждого варианта с помощью использованного алгоритма; Phred Score – показатель качества полученной оценки (значение ≥20 соответствует вероятности ошибки 1%). Цифры в светло-серой заливке отображают вероятно консервативные точки генома; в темно-серой заливке – высокое влияние на приспособленность; светло-серая заливка с жирным шрифтом указывает на сильный фоновый отбор, темно-серая с белым шрифтом – локус находится под положительным отбором, черная – локус высоконейтрален.
Несколько менее выражена консервативность промоторной последовательности гена ATM – для rs189037 устойчивое сохранение последовательности выявлено с помощью анализов PhyloP и GERP++, но не PhastCons (определяющего вероятность того, что каждый нуклеотид принадлежит консервативному элементу) (табл. 4). Напротив, для rs560191 в гене TP53BP1 консервативность определена методом PhastCons на различных уровнях, но PhyloP подтверждает ее наличие только у приматов, а оценки GERP++ ‒ наличие высокой гомологии у разных видов (табл. 4). Статистика GerpN (оценивающая сохранение последовательности у видов) определяет как консервативные наибольшее число проанализированных локусов (rs560191, rs709816, rs189037, rs1801516, rs1799977, rs1805321), причем только этот метод считает “вероятно консервативными” rs709816 (в гене NBN) и rs1805321 (в гене PMS2) (табл. 4). Согласно всем видам анализа, к консервативным не относятся промоторные регионы генов NBN (rs1805800), MRE11 (rs473297) и LIG1 (rs20579).
Не все полученные оценки хорошо согласуются между собой. Так, показано, что 7 из 9 изученных SNP оказывают выраженное влияние на приспособленность (согласно оценкам пригодности FitCons), в том числе и “высоконейтральный” rs20579. В то же время локус rs473297, находящийся под действием позитивного отбора (то есть замена в этом локусе, вероятно, имеет некоторые адаптивные преимущества), оценки FitCons определяют как не оказывающий влияния на приспособленность. Интересно, что действие фонового отбора зарегистрировано для четырех локусов: обеих замен в гене ATM и несинонимичных замен в генах TP53BP1 и MLH1 (табл. 4). Фоновый отбор показывает воздействие не на сами анализируемые локусы, а на тесно сцепленные с ними, то есть консервативность этих регионов может быть связана не столько (или не исключительно) с важной ролью именно анализируемых последовательностей, но и быть следствием сцепления. В пользу этого свидетельствует тот факт, что почти во всех генах, в которых локализованы рассматриваемые SNP, описаны мутации, приводящие к моногенным заболеваниям (за исключением TP53BP1). Это такие патологии, как атаксия-телеангиэктазия и сходное с ней заболевание (Ataxia-telangiectasia-like disorder 1 ‒ заболевание, подобное атаксии-телеангиэктазии, типа 1), вызываемые мутациями в генах AТМ и MRE11 соответственно; синдром хромосомных поломок Ниймеген (мутации в гене NBN); синдром Линча и синдром Туркота (причина которых ‒ мутации и в MLH1, и в PMS2), синдром Мюир‒Торре (мутации в гене MLH1); аутосомно-рецессивный иммунодефицит-96 (мутации в LIG1) (https://www.ncbi. nlm.nih.gov/omim/?term).
С позиций “менделевского кода” полиморфные варианты в генах менделевских заболеваний могут оказаться значимыми для многофакторных патологий, в которых задействованы те же биохимические пути. Замечено, что по результатам полногеномных ассоциативных исследований наиболее значимые ассоциации нередко выявляют с маркерами, локализованными вблизи генов менделевских заболеваний, причем наблюдается некоторое перекрывание фенотипических проявлений изучаемых моногенных и сложнонаследуемых заболеваний [35]. Считается, что до четверти генов, мутации в которых приводят к менделевским заболеваниям, ассоциировано и с многофакторной патологией [36]. С этой точки зрения изучаемые маркеры перспективны для ассоциативного анализа широкого спектра патологий.
Оценки патогенности исследованных SNP неоднозначны. С одной стороны, данные по оценкам патогенности несинонимичных вариантов, полученные на основании широкого спектра наиболее часто используемых алгоритмов, подтверждают отсутствие патологически значимых нарушений белковых молекул вследствие указанных нуклеотидных замен (табл. 5). С другой стороны, геномная оценка патогенности (по regBase [37]) указывает на вероятность вовлеченности в патологические процессы всех анализируемых вариантов (табл. 6). В наибольшей степени это касается SNP в генах ATM (rs189037 в 5'UTR, rs1801516 в экзоне 37) и MLH1 (rs1799977 в экзоне 8). Можно предположить, что геномные оценки патогенности отображают вероятность вовлеченности маркера в развитие многофакторной патологии. Оценки онкогенности также указывают на вероятность “драйверного” эффекта (likely cancer driver) данных нуклеотидных замен для развития онкопатологии (табл. 6).
Таблица 5.
Оценка патогенности несинонимичных нуклеотидных заменa
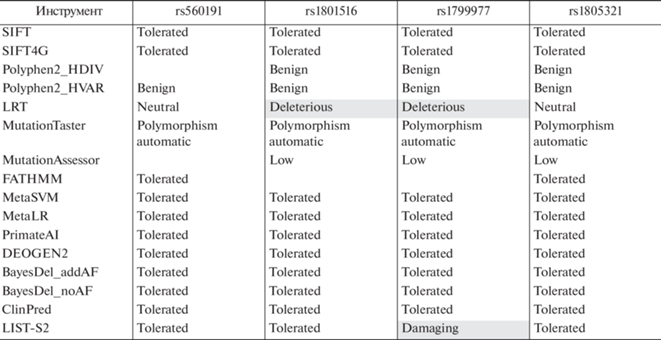
a Представлены данные по статусу патогенности миссенс-вариантов, полученные с помощью различных алгоритмов оценки, аккумулированные в dbNSFP (составлено по данным VannoPortal, http://www.mulinlab.org/vportal/index.html). В зависимости от алгоритмов и решаемых с их помощью задач статус патогенности оценивали в разных терминах. В таблице сохранена исходная терминология: к благоприятным вариантам относятся Tolerated, Benign, Neutral, Polymorphism automatic, Low; к неблагоприятным ‒ Deleterious, Damaging.
Таблица 6.
Геномная оценка патогенности и онкогенности нуклеотидных замена
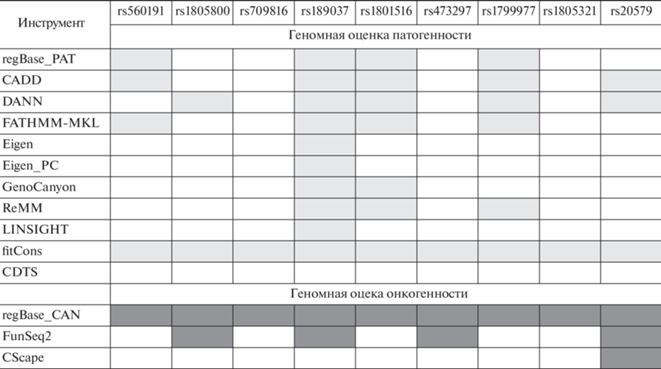
а Суммированы данные по геномной оценке патогенности и онкогенности, полученные с помощью различных часто используемых алгоритмов оценки некодирующих вариантов, заархивированных в базе данных regBase (RegBase_PAT и RegBase_CAN) [24, 37]. Составлено на основании информации в VannoPortal (http://www.mulinlab.org/vportal/index.html). Светло-серые блоки – оценка “вероятно патогенный”, темно-серые – “вероятный фактор развития рака”.
Следует отметить, что вовлеченность данных маркеров в развитие многофакторной патологии в литературе обсуждается мало. Так, в базе DisGeNet (https://www.disgenet.org/) отсутствует информация относительно rs1805800, rs473297, rs1805321 в контексте привлечения их к ассоциативному анализу при любых патологиях. Только с точки зрения вовлеченности в развитие онкопатологии и/или ее осложнений изучают rs560191, rs709816, rs1801516 и rs20579. Преимущественно при онкопатологии изучают также rs189037 и rs1799977, хотя анализировали вовлеченность rs1799977 в развитие язвенного колита [38, 39], rs189037 ‒ в развитие сердечно-сосудистой патологии, азооспермии, когнитивных нарушений [40‒43]. В отношении всех рассматриваемых маркеров результаты ассоциативных исследований противоречивы, в некоторых случаях обсуждается вероятность этноспецифических особенностей ассоциаций (https://www.disgenet.org/).
Таким образом, всесторонний анализ изучаемых локусов дает большой объем дополнительной информации, которая может быть полезной для объяснения результатов ассоциативных исследований и более адекватной оценки патологического и адаптогенного потенциала изучаемых локусов. Результаты проведенного анализа указывают на высокий регуляторный потенциал рассматриваемых локусов. Так, показано, что большинство из них находится в составе крупных блоков сцепления QTL-SNP, оказывающих влияние на уровни экспрессии широкого спектра генов (например, rs560191 является eQTL для 20 генов), а влияние этих замен может сказаться не только на изменении генной экспрессии в целом, но и на сплайсинге. Выявлен возможный эффект варианта rs189037 на усиление топологической роли включающего его хромосомного региона. Показано, что рассматриваемые маркеры при ассоциативных исследованиях могут отражать участие в патологическом процессе не только “своего” гена, но и тех генов, в регуляции которых они принимают участие.
Работа выполнена при частичном финансировании Госзадания Министерства науки и высшего образования (№ 122020300041-7).
Этические нормы соблюдены. Обзор написан с использованием открытых публикаций.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Zhang F., Lupski J.R. (2015) Non-coding genetic variants in human disease. Hum. Mol. Genet. 24(R1), R102‒R110. https://doi.org/10.1093/hmg/ddv259
Бабушкина Н.П., Постригань А.Е., Кучер А.Н. (2021) Вовлеченность генов белков BRСA1-ассоциированного комплекса наблюдения за геномом (BASC) в развитие многофакторной патологии. Молекуляр. биология. 55(2), 318–337. https://doi.org/10.31857/S0026898421020038
Бабушкина Н.П., Постригань А.Е., Кучер А.Н. (2018) Вовлеченность генов систем репарации ДНК в развитие сердечно-сосудистой патологии. Сб.: Молекулярно-биологические технологии в медицинской практике. Ред. А.Б. Масленников. Новосибирск: Академиздат, с. 48‒62.
Бабушкина Н.П., Постригань А.Е., Хитринская Е.Ю., Кучер А.Н. (2019) Средовые эффекты на ассоциации генов белков систем репарации ДНК с бронхиальной астмой. VII Съезд Вавиловского общества генетиков и селекционеров (ВОГиС) (2019), Санкт-Петербург, Россия. Сборник тезисов, с. 788.
Бабушкина Н.П., Постригань А.Е., Хитринская Е.Ю., Кучер А.Н. (2019) Вовлеченность полиморфных вариантов генов систем репарации ДНК в развитие многофакторных заболеваний. Сб.: Генетика человека и патология: актуальные проблемы клинической и молекулярной цитогенетики. Ред. В.А. Степанов. Томск: Литературное бюро, с. 5–6.
Бабушкина Н.П., Постригань А.Е., Кучер А.Н. (2020) Гены белков репарации ДНК и продолжительности жизни. Медицинская генетика. 19(5), 99–100. https://doi.org/10.25557/2073-7998.2020.05.99-100
Бабушкина Н.П., Постригань А.Е., Кучер А.Н., Кужелева Е.А., Гарганеева А.А. (2020) Ассоциации полиморфизма генов систем репарации ДНК с показателями липидного обмена. Кардиология 2020 – новые вызовы и новые решения, Казань. Сборник тезисов, с. 811.
Постригань А.Е., Бабушкина Н.П., Кучер А.Н. (2020) Вовлеченность полиморфизма гена NBN в формирование предрасположенности к дистропным заболеваниям. Медицинская генетика. 19(8), 98–99. https://doi.org/10.25557/2073-7998.2020.08.98-99
Peakall R., Smouse P.E. (2012) GenAlEx 6: genetic analysis in Excel. Population genetic software for teaching and research ‒ an update. Bioinformatics. 28(19), 2537‒2539. https://doi.org/10.1093/bioinformatics/bts460
Taverna S.D., Li H., Ruthenburg A.J., Allis C.D., Patel D.J. (2007) How chromatin-binding modules interpret histone modifications: lessons from professional pocket pickers. Nat. Struct. Mol. Biol. 14(11), 1025‒1040. https://doi.org/10.1038/nsmb1338
Hoon D.S.B., Rahimzadeh N., Bustos M.A. (2021) EpiMap: fine-tuning integrative epigenomics maps to understand complex human regulatory genomic circuitry. Signal. Transduct. Target Ther. 6(1), 179. https://doi.org/10.1038/s41392-021-00620-5
Fu Y., Sinha M., Peterson C.L., Weng Z. (2008) The insulator binding protein CTCF positions 20 nucleosomes around its binding sites across the human genome. PLoS Genet. 4(7), e1000138. https://doi.org/10.1371/journal.pgen.1000138
Rubio E.D., Reiss D.J., Welcsh P.L., Disteche C.M., Filippova G.N., Baliga N.S., Aebersold R., Ranish J.A., Krumm A. (2008) CTCF physically links cohesin to chromatin. Proc. Natl. Acad. Sci. USA. 105(24), 8309‒8314. https://doi.org/10.1073/pnas.0801273105
Mishiro T., Ishihara K., Hino S., Tsutsumi S., Aburatani H., Shirahige K., Kinoshita Y., Nakao M. (2009) Architectural roles of multiple chromatin insulators at the human apolipoprotein gene cluster. EMBO J. 28(9), 1234‒1245. https://doi.org/10.1038/emboj.2009.81
Shoaib M., Chen Q., Shi X., Nair N., Prasanna C., Yang R., Walter D., Frederiksen K.S., Einarsson H., Svensson J.P., Liu C.F., Ekwall K., Lerdrup M., Nordenskiöld L., Sørensen C.S. (2021) Histone H4 lysine 20 mono-methylation directly facilitates chromatin openness and promotes transcription of housekeeping genes. Nat. Commun. 12(1), 4800. https://doi.org/10.1038/s41467-021-25051-2
Hansen K.H., Bracken A.P., Pasini D., Dietrich N., Gehani S.S., Monrad A., Rappsilber J., Lerdrup M., Helin K. (2008) A model for transmission of the H3K27me3 epigenetic mark. Nat. Cell Biol. 10(11), 1291‒1300. https://doi.org/10.1038/ncb1787
Vandamme J., Sidoli S., Mariani L., Friis C., Christensen J., Helin K., Jensen O.N., Salcini A.E. (2015) H3K23me2 is a new heterochromatic mark in Caenorhabditis elegans. Nucleic Acids Res. 43(20), 9694‒9710. https://doi.org/10.1093/nar/gkv1063
Yang W., Bai Y., Xiong Y., Zhang J., Chen S., Zheng X., Meng X., Li L., Wang J., Xu C., Yan C., Wang L., Chang C.C., Chang T.Y., Zhang T., Zhou P., Song B.L., Liu W., Sun S.C., Liu X., Li B.L., Xu C. (2016) Potentiating the antitumour response of CD8+ T cells by modulating cholesterol metabolism. Nature. 531(7596), 651‒655. https://doi.org/10.1038/nature17412
Fornes O., Castro-Mondragon J.A., Khan A., van der Lee R., Zhang X., Richmond P.A., Modi B.P., Correard S., Gheorghe M., Baranasic D., Santana-Garcia W., Tan G., Cheneby J., Ballester B., Parcy F., Sandelin A., Lenhard B., Wasserman W.W., Mathelier A. (2020) JASPAR 2020: update of the open-access database of transcription factor binding profiles. Nucleic A-cids Res. 48(D1), D87‒D92. https://doi.org/10.1093/nar/gkz1001
Lesurf R., Cotto K.C., Wang G., Griffith M., Kasaian K., Jones S.J., Montgomery S.B., Griffith O.L.; Open Regulatory Annotation Consortium. (2016) ORegAnno 3.0: a community-driven resource for curated regulatory annotation. Nucleic Acids Res. 44(D1), D126‒D132. https://doi.org/10.1093/nar/gkv1203
Kazachenka A., Bertozzi T.M., Sjoberg-Herrera M.K., Walker N., Gardner J., Gunning R., Pahita E., Adams S., Adams D., Ferguson-Smith A.C. (2018) Identification, characterization, and heritability of murine metastable epialleles: implications for non-genetic inheritance. Cell. 175(5), 1259‒1271.e13. https://doi.org/10.1016/j.cell.2018.09.043
Li M.J., Wang L.Y., Xia Z., Sham P.C., Wang J. (2013) GWAS3D: detecting human regulatory variants by integrative analysis of genome-wide associations, chromosome interactions and histone modifications. Nucleic Acids Res. 41(Web Server issue), W150‒W158. https://doi.org/10.1093/nar/gkt456
Huang D., Yi X., Zhang S., Zheng Z., Wang P., Xuan C., Sham P.C., Wang J., Li M.J. (2018) GWAS4D: multidimensional analysis of context-specific regulatory variant for human complex diseases and traits. Nucleic Acids Res. 46(W1), W114‒W120. https://doi.org/10.1093/nar/gky407
Huang D., Zhou Y., Yi X., Fan X., Wang J., Yao H., Sham P.C., Hao J., Chen K., Li M.J. (2022) VannoPortal: multiscale functional annotation of human genetic variants for interrogating molecular mechanism of traits and diseases. Nucleic Acids Res. 50(D1), D1408‒D1416. https://doi.org/10.1093/nar/gkab853
Lee R., Kang M.K., Kim Y.J., Yang B., Shim H., Kim S., Kim K., Yang C.M., Min B.G., Jung W.J., Lee E.C., Joo J.S., Park G., Cho W.K., Kim H.P. (2022) CTCF-mediated chromatin looping provides a topological framework for the formation of phase-separated transcriptional condensates. Nucleic Acids Res. 50(1), 207‒226. https://doi.org/10.1093/nar/gkab1242
Putt W., Palmen J., Nicaud V., Tregouet D.A., Tahri-Daizadeh N., Flavell D.M., Humphries S.E., Talmud P.J.; EARSII group. (2004) Variation in USF1 shows haplotype effects, gene : gene and gene : environment associations with glucose and lipid parameters in the European Atherosclerosis Research Study II. Hum. Mol. Genet. 13(15), 1587‒1597. https://doi.org/10.1093/hmg/ddh168
Laurila P.P., Naukkarinen J., Kristiansson K., Ripatti S., Kauttu T., Silander K., Salomaa V., Perola M., Karhunen P.J., Barter P.J., Ehnholm C., Peltonen L. (2010) Genetic association and interaction analysis of USF1 and APOA5 on lipid levels and atherosclerosis. Arterioscler. Thromb. Vasc. Biol. 30(2), 346‒352. https://doi.org/10.1161/ATVBAHA.109.188912
Taghizadeh E., Mirzaei F., Jalilian N., Ghayour Mobarhan M., Ferns G.A., Pasdar A. (2020) A novel mutation in USF1 gene is associated with familial combined hyperlipidemia. IUBMB Life. 72(4), 616‒623. https://doi.org/10.1002/iub.2186
Pollard K.S., Hubisz M.J., Rosenbloom K.R., Siepel A. (2010) Detection of nonneutral substitution rates on mammalian phylogenies. Genome Res. 20(1), 110–121. https://doi.org/10.1101/gr.097857.109
Caron B., Luo Y., Rausell A. (2019) NCBoost classifies pathogenic non-coding variants in Mendelian diseases through supervised learning on purifying selection signals in humans. Genome Biol. 20(1), 32. https://doi.org/10.1186/s13059-019-1634-2
Siepel A. (2005) Evolutionarily conserved elements in vertebrate, insect, worm, and yeast genomes. Genome Res. 15(8), 1034‒1035. https://doi.org/10.1101/gr.3715005
Hubisz M.J., Pollard K.S., Siepel A. (2011) PHAST and RPHAST: phylogenetic analysis with space/time models. Brief. Bioinform. 12(1), 41–51. https://doi.org/10.1093/bib/bbq072
Zerbino D.R., Achuthan P., Akanni W., Amode M.R., Barrell D., Bhai J., Billis K., Cummins C., Gall A., Giron C.G., Gil L., Gordon L., Haggerty L., Haskell E., Hourlier T., Izuogu O.G., Janacek S.H., Juettemann T., To J.K., Laird M.R., Lavidas I., Liu Z., Loveland J.E., Maurel T., McLaren W., Moore B., Mudge J., Murphy D.N., Newman V., Nuhn M., Ogeh D., Ong C.K., Parker A., Patricio M., Riat H.S., Schuilenburg H., Sheppard D., Sparrow H., Taylor K., Thormann A., Vullo A., Walts B., Zadissa A., Frankish A., Hunt S.E., Kostadima M., Langridge N., Martin F.J., Muffato M., Perry E., Ruffier M., Staines D.M., Trevanion S.J., Aken B.L., Cunningham F., Yates A., Flicek P. (2018) Ensembl 2018. Nucleic Acids Res. 46(D1), D754‒D761. https://doi.org/10.1093/nar/gkx1098
Gulko B., Hubisz M.J., Gronau I., Siepel A. (2015) A method for calculating probabilities of fitness consequences for point mutations across the human genome. Nat. Genet. 47(3), 276‒283. https://doi.org/10.1038/ng.3196
Freund M.K., Burch K.S., Shi H., Mancuso N., Kichaev G., Garske K.M., Pan D.Z., Miao Z., Mohlke K.L., Laakso M., Pajukanta P., Pasaniuc B., Arboleda V.A. (2018) Phenotype-specific enrichment of Mendelian disorder genes near GWAS regions across 62 complex traits. Am. J. Hum. Genet. 103, 535‒552. https://doi.org/10.1016/j.ajhg.2018.08.017
Spataro N., Rodrıguez J. A., Navarro A., Bosch E. (2017) Properties of human disease genes and the role of genes linked to Mendelian disorders in complex disease aetiology. Hum. Mol. Genet. 26, 489–500. https://doi.org/10.1093/hmg/ddw405
Zhang S., He Y., Liu H., Zhai H., Huang D., Yi X., Dong X., Wang Z., Zhao K., Zhou Y., Wang J., Yao H., Xu H., Yang Z., Sham P.C., Chen K., Li M.J. (2019) regBase: whole genome base-wise aggregation and functional prediction for human non-coding regulatory variants. Nucleic Acids Res. 47(21), e134. https://doi.org/10.1093/nar/gkz774
Plotz G., Raedle J., Spina A., Welsch C., Stallmach A., Zeuzem S., Schmidt C. (2008) Evaluation of the MLH1 I219V alteration in DNA mismatch repair activity and ulcerative colitis. Inflamm. Bowel Dis. 14(5), 605‒611. https://doi.org/10.1002/ibd.20358
Vietri M.T., Riegler G., De Paola M., Simeone S., Boggia M., Improta A., Parisi M., Molinari A.M., Cioffi M. (2009) I219V polymorphism in hMLH1 gene in patients affected with ulcerative colitis. Genet. Test. Mol. Biomarkers. 13(2), 193‒197. https://doi.org/10.1089/gtmb.2008.0088
Li S., Zhang L., Chen T., Tian B., Deng X., Zhao Z., Yuan P., Dong B., Zhang Y., Mo X. (2011) Functional polymorphism rs189037 in the promoter region of ATM gene is associated with angiographically characterized coronary stenosis. Atherosclerosis. 219(2), 694‒697. https://doi.org/10.1016/j.atherosclerosis.2011.08.040
Li Z., Yu J., Zhang T., Li H., Ni Y. (2013) rs189037, a functional variant in ATM gene promoter, is associated with idiopathic nonobstructive azoospermia. Fertil. Steril. 100(6), 1536‒1541.e1. https://doi.org/10.1016/j.fertnstert.2013.07.1995
Ding X., Yue J.R., Yang M., Hao Q.K., Xiao H.Y., Chen T., Gao L.Y., Dong B.R. (2015) Association between the rs189037 single nucleotide polymorphism in the ATM gene promoter and cognitive impairment. Genet. Mol. Res. 14(2), 4584‒4592. https://doi.org/10.4238/2015.May.4.17
Ding X., He Y., Hao Q., Chen S., Yang M., Leng S.X., Yue J., Dong B. (2018) The association of single nucleotide polymorphism rs189037C>T in ATM gene with coronary artery disease in Chinese Han populations: a case control study. Medicine (Baltimore). 97(4), e9747. https://doi.org/10.1097/MD.0000000000009747
Дополнительные материалы
- скачать ESM.docx
- Приложение 1.
Гены, экспрессия которых регулируется анализируемыми нуклеотидными заменами
Инструменты
Молекулярная биология