

Молекулярная биология, 2023, T. 57, № 1, стр. 85-94
Линия трансгенных мышей APPswe/PS1dE9/Blg для моделирования церебрального амилоидоза при болезни Альцгеймера
Е. А. Лысикова a, *, Е. В. Кузубова b, А. И. Радченко b, Е. А. Патраханов b, К. Д. Чапров a, b, М. В. Корокин b, А. В. Дейкин b, О. С. Гудырев b, М. В. Покровский b
a Институт физиологически активных веществ Российской академии наук
142432 Черноголовка, Россия
b Белгородский государственный национальный исследовательский университет
308015 Белгород, Россия
* E-mail: lysikova.ipac@gmail.com
Поступила в редакцию 08.06.2022
После доработки 30.07.2022
Принята к публикации 30.07.2022
- EDN: AXGEVN
- DOI: 10.31857/S0026898423010081
Аннотация
Болезнь Альцгеймера – наиболее распространенная протеинопатия, при которой у пациента наблюдается неуклонное снижение когнитивных функций с одновременным накоплением в тканях мозга амилоидных бляшек – внеклеточных агрегатов амилоида β (Аβ), ассоциированных с нейровоспалением и нейродегенерацией. Крысы и мыши, Аβ которых отличаются от Аβ человека и других млекопитающих тремя аминокислотными заменами, не подвержены патологии альцгеймеровского типа. Однако в мозге трансгенных мышей со сверхэкспрессией Аβ человека наблюдается появление амилоидных бляшек, что позволяет широко использовать этих животных в биомедицине для моделирования ключевых аспектов болезни Альцгеймера. Трансгенные мыши линии APPswe/PS1dE9 часто используются для исследования молекулярных механизмов болезни Альцгеймера. В настоящей работе нами охарактеризована линия мышей APPswe/PS1dE9/Blg, полученная в результате скрещивания линии APPswe/PS1dE9 на генетическом фоне СH3 с животными С57Bl6/Chg. Нами показано, что показатели фертильности и выживаемости потомства у животных данной линии не отличаются от показателей у контрольных животных дикого типа. Гистологический анализ мозга животных APPswe/PS1dE9/Blg подтвердил сохранение нейроморфологических признаков БА с увеличением количества и размеров амилоидных бляшек в мозге по мере старения животных. Таким образом животные линии APPswe/PS1dE9/Blg являются удобной моделью при поиске терапевтических стратегий замедления прогрессии БА.
ВВЕДЕНИЕ
Болезнь Альцгеймера (БА) – нейродегенеративное заболевание и самая распространенная форма деменции [1]. БА относится к протеинопатиям – группе заболеваний, в патогенезе которых важная роль отводится изменениям структуры и/или нарушениям метаболизма склонных к агрегации белков. БА характеризуется медленной прогрессией патологического процесса и длительной продромальной стадией [2]. На сегодняшний день еще не создано эффективных методов терапии БА, а широкая распространенность делает это заболевание актуальной социальной проблемой.
В начале 20 века немецкий психиатр Алоис Альцгеймер обнаружил в патогистологических препаратах мозга пациентов с деменцией включения – сенильные бляшки. Позже было установлено, что одним из основных компонентов сенильных бляшек является агрегированный бета-амилоидный (Aβ) пептид. Aβ-пептид представляет собой продукт гидролиза белка-предшественника амилоида APP (amyloid precursor protein) [3]. Согласно принятой на сегодняшний день гипотезе амилоидного каскада, церебральный амилоидоз запускает остальные патологические процессы при прогрессии БА [4].
APP – широко экспрессируемый трансмембранный белок первого типа имеет три основные изоформы: APP695, APP751 и APP770. Изоформа APP695 является основной в нейронах, тогда как APP751 и APP770 экспрессируются преимущественно в других типах клеток [5]. Белок APP играет важную роль в разрастании нейритов и ветвлении аксонов в процессе развития мозга, он участвует в синаптогенезе и обеспечении синаптической пластичности [5]. Белок APP в норме подвергается неамилоидогенному процессингу с последовательным расщеплением α- и γ-секретазами внутри домена Aβ с образованием непатогенных фрагментов (sAPPα и C-концевых фрагментов (CTF)) [6]. Растворимая форма sAPPα обладает нейротрофической активностью и участвует в защите нейронов от эксайтотоксичности, регулируя гомеостаз кальция [5]. Последовательное протеолитическое расщепление APP β- и γ-секретазами приводит к образованию Aβ-пептида, способного к агрегации, а также непатогенных фрагментов [7]. Наиболее распространены две формы Aβ-пептида, состоящие из 40 (Aβ40) и 42 аминокислотных остатков (Aβ42), при этом форма Aβ42 более токсична для клетки [8].
БА считается многофакторным заболеванием, наиболее распространены его спорадические формы с поздним дебютом (старше 65 лет). Исследование семейных форм БА с более ранним дебютом (менее 65 лет) позволило выявить генетические факторы, ассоциированные с развитием нейродегенеративных процессов в мозге больных. На сегодняшний день известно несколько мутаций в гене APP, приводящих к существенному повышению уровня Aβ42 и ассоциированных с ранним дебютом БА [9]. Описаны также мутации в генах пресенилина 1 (PSEN1) и пресенилина 2 (PSEN2), кодирующих трансмембранные белки эндоплазматического ретикулума. Геномные исследования выявили более 170 различных мутаций в гене PSEN1 и 13 мутаций в гене PSEN2, связанных с наиболее распространенными семейными формами БА с ранним началом [7]. Белки PSEN1 и PSEN2, входящие в состав комплекса γ‑секретазы, оказывают влияние на процессинг APP [10]. Мутации в гене PSEN1 увеличивают специфичность расщепления белка APP γ-секретазой до токсичного пептида Aβ42 [11], что подтверждается повышением соотношения Aβ42/Aβ40 в плазме пациентов с мутантным геном PSEN1 по сравнению с плазмой крови пациентов со спорадическими формами БА [12].
С целью выявления молекулярных механизмов, лежащих в основе развития и прогрессии БА, созданы животные модели. К наиболее адекватным и широко используемым моделям относятся линии трансгенных мышей, в которых воспроизводится прогрессирующий церебральный амилоидоз, характерный для БА. В геном мышей были введены трансгенные кассеты, кодирующие наиболее часто встречающиеся мутации в генах, ассоциированных c развитием наследственных форм БА. Животные модели первого поколения, основанные преимущественно на сверхэкспрессии кДНК мутантных генов APP или PSEN1 человека, воспроизводят основные патологические признаки заболевания. В модельных животных второго поколения проведена замена участков (knock-in) гена App мыши на участки, кодирующие шведскую, иберийскую и/или арктическую мутации (AppNL–G–F). Трансгенная линия мышей третьего поколении содержала knock-in мутации в генах App и Psen1 (AppNL–F Psen1P117L/WT) [13]. Трансгенные животные, воспроизводящие формирование амилоидных включений – основной тип патологии при БА, представлены в табл. 1.
Таблица 1.
Наиболее часто используемые линии трансгенных мышей, моделирующие церебральный амилоидоз
| Линия (JAX mice) | Мутация | Промотор | Амилоидные бляшки (возраст) | Ссылка |
|---|---|---|---|---|
| PDAPP | hAPP (V717F) | Промотор гена фактора роста тромбоцитов-β (PDGF-β) | 6–9 мес.: кора, гиппокамп, мозолистое тело | [14] |
| APP23 (B6.Cg-Tg(Thy1-APP)3Somm/J) |
hAPP751 (K670N/M671L) | Промотор Thy1 мыши | 6–8 мес.: кора 12–24 мес.: кора, гиппокамп, таламус, амигдала |
[15] |
| Tg2576 (B6;SJL-Tg(APPSWE)2576Kha) |
hAPP695 (K670N/M671L) | Промотор гена прионного белка хомяка | 7–8 мес.: кора 11–13 мес.: гиппокамп, мозжечок |
[16] |
| Tg-SwDI (C57BL/6-Tg(Thy1-APPSwDutIowa) BWevn/Mmjax) |
hAPP770 (K670N/M671L) hAPP770 (E693Q) hAPP770 (D694N) |
Промотор Thy1 мыши | 3 мес.: кора, гиппокамп 6 мес.: обонятельные луковицы, таламус |
[17] |
| 3хTg (B6;129-Tg(APPSwe,tauP301L)1LfaPsen1tm1Mpm/Mmjax) |
hAPP (K670N/M671L) hMAPT (P301L) mPsen1 (M146V) |
Промотор Thy1 мыши (APP, MAPT), промотор Psen1 мыши |
6 мес.: кора, гиппокамп 12 мес.: гиппокамп |
[18] |
| APPswe/PS1dE9 (B6;C3-Tg(APPswe,PSEN1dE9)85Dbo/Mmjax) |
Mo/HuAPP (K670N/M671L) hPSEN1 ΔE9 |
Промотор гена прионного белка мыши | 5–6 мес.: кора, гиппокамп | [19] |
| 5xFAD (B6SJL-Tg(APPSwFILon,PSEN1*M146L*L286V)6799Vas/Mmjax) |
hAPP (K670N/M671L) hAPP (V717I) hAPP (I716V) hPSEN1 (M146L) hPSEN1(L286V) |
Промотор Thy1 мыши | 2 мес.: кора, основание гиппокампа 4 мес.: спинной мозг 6 мес.: гиппокамп, таламус, обонятельные луковицы, основание мозга |
[20] |
| J20 (B6.Cg-Zbtb20Tg(PDGFB-APPSwlnd)20Lms/2Mmjax) |
hAPP (K670N/M671L) hAPP (V717F) |
Промотор гена фактора роста тромбоцитов-β (PDGF-β) | 5–8 мес.: кора, гиппокамп | [21] |
| APPPS1 (B6.Cg-Tg(Thy1-APPSw,Thy1-PSEN1*L166P)21Jckr) |
hAPP (K670N/M671L) hPSEN1 (L166P) |
Промотор Thy1 мыши | 6 недель: кора 2–3 мес.: гиппокамп 3–5 мес.: стриатум, таламус |
[22] |
| PS/APP | hAPP (K670N/M671L) hPSEN1 (M146L) |
Промотор гена прионного белка хомяка, промотор гена фактора роста тромбоцитов-β (PDGF-β) |
6 мес.: кора, гиппокамп 12 мес.: стриатум, таламус, ствол мозга |
[23–25] |
| PS2Tg2576 | hAPP (K670N/M671L) hPSEN2 (N141I) |
Промотор гена прионного белка хомяка, промотор гена β-актина курицы |
2–3 мес.: кора, гиппокамп | [26] |
| mThy1-hAβPP751 | hAPP (K670N/M671L) hAPP (V717I) |
Промотор Thy1 мыши | 3–6 мес.: кора 5–7 мес.: гиппокамп, таламус, обонятельные луковицы |
[27] |
| PLB1-triple | hAPP (K670N/M671L) hAPP (V717I) hMAPT (P301L) hMAPT (R406W) hPSEN1 (A246E) |
Промотор CaMKII-α мыши (APP, MAPT), промотор гена прионного белка мыши (PSEN1) |
5–6 мес.: кора, гиппокамп | [28] |
| knock-in APP (AppNL–G–F) |
Mo/HuAPP (K670N/M671L) Mo/HuAPP (E693G) Mo/HuAPP (I716F) |
Промотор гена АРР мыши | 2 мес.: кора 4 мес.: гиппокамп |
[29] |
| knock-in AppNL–F Psen1P117L/WT | Mo/HuAPP (K670N/M671L) Mo/HuAPP (I716F) mPsen1 (P117L) |
Промотор гена APP мыши, промотор Psen1 мыши |
3 мес.: кора 12 мес.: гиппокамп |
[30] |
Каждая линия отличалась вариабельностью патологического фенотипа, зависящего от генетического фона, условий содержания и длительности поддержания обособленных колоний в условиях одного вивария. В нашей работе подробно охарактеризована сублиния APPswe/PS1dE9.
Геном трансгенных мышей линии APPswe/PS1dE9 содержит две трансгенные кассеты, встроенные в один локус на хромосоме 9. Первая трансгенная кассета кодирует кДНК химерного гуманизированного в домене Aβ и содержащего шведскую мутацию (K670N/M671L) гена APP под контролем прионного белка мыши. Вторая трансгенная кассета кодирует кДНК гена PSEN1 человека с делецией экзона 9 (PS1ΔE9) под контролем прионного белка мыши. Бигенная линия животных APP/PS1 создана Borchelt и соавт. путем коинъекции линеаризованной и очищенной плазмидной ДНК в пронуклеус зиготы мыши [19]. У 10% животных, полученных после коинъекции трансгенных кассет, выявлено одновременное встраивание обоих трансгенов [31]. Далее отобрали одну линию с наибольшим количеством копий трансгенной кассеты, которую использовали в дальнейших экспериментах. Высокий уровень трансгенного белка PSEN1 и трансгенного APP в мозге модельных мышей подтвержден методом иммуноблотинга [19].
При создании трансгенных животных внесли модификацию в фрагмент гена прионоподобного белка (PrP) мыши в векторе Bluescript KS+. Обозначенный phgPrP фрагмент в составе гена PrP содержал последовательность размером примерно 6 т.п.н. до сайта инициации транскрипции, состоящую из первого кодирующего экзона, первого интрона, кодирующую область PrP мыши, слитую со вторым экзоном, и примерно 3 т.п.н. 3′-нетранслируемой области. Открытую рамку считывания во фрагменте phgPrP заменили на сайт рестрикции XhoI. Полученный модифицированный вектор назвали MoPrP.Xho. В вектор MoPrP.Xho по сайтам рестрикции XhoI между экзонами 2 и 3 гена PrP мыши встроили полноразмерную кДНК гена PSEN1 человека, кодирующую белок с делецией аминокислотных остатков 290–319, фланкированную сайтами XhoI. кДНК белка-предшественника амилоида, гуманизированного в домене Аβ, содержащую шведскую мутацию (K670N/M671L), также клонировали в вектор MoPrP.Xho между экзонами 2 и 3 PrP [19]. Каждый трансген экспрессируется под контролем промотора прионоподобного белка мыши. Этот промотор активен в эмбриональном развитии, у взрослых животных он обеспечивает экспрессию трансгена преимущественно в астроцитах и нейронах центральной нервной системы [31].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Лабораторные животные. В работе использовали сублинию мышей APPswe/PS1dE9/Blg, полученную от скрещивания мышей B6;C3-Tg(APPswe,PSEN1dE9)85Dbo/Mmjax (#034829-JAX) с мышами дикого типа C57Bl6J/ChG. Линия C57Bl6J/ChG получена из питомника Charles River UK и поддерживалась в качестве обособленной колонии в условиях беспатогенного вивария ИФАВ РАН в течение 7 лет. Поддержание и размножение колонии проводили путем скрещивания трансгенных животных с животными дикого типа из тех же пометов.
Экспериментальных и контрольных животных содержали в условиях беспатогенного вивария Белгородского государственного национального исследовательского университета (НИУ БелГУ) в условиях искусственно регулируемого светового дня (12 ч день и 12 ч ночь) при температуре +22–26°C со свободным доступом к корму и воде.
Генотипирование трансгенных животных. Геномную ДНК получали из биопсийного материала уха (около 30 мг ткани). Материал помещали в лизирующий раствор, содержащий 100 мМ NaCl, 50 мМ Трис-HCl (pH 8.0), 2 мМ EDТА и 2 мг/мл протеиназы К, и инкубировали при 55°С в течение 12–16 ч после чего реакционную смесь прогревали при 85°С в течение 40 мин. Лизат центрифугировали в течение 1 мин при 10 000 g, 1 мкл супернатанта использовали в качестве матрицы в реакции ПЦР [32].
Праймеры. Использовали праймеры, предложенные Jankowsky и соавт. [19], амплифицирующие одновременно участок трансгенной кассеты (при ее наличии) и участок геномной ДНК мыши. В реакции использовали смесь из трех праймеров: общий обратный праймер PrP_rev (5'-GTG GAT ACC CCC TCC CCC AGC CTA GAC C), гомологичный последовательности гена PrP в геноме мыши и в составе трансгенной кассеты; прямой праймер PrP_for (5'-CCT CTT TGT GAC TAT GTG GAC TGA TGT CGG), гомологичный участку геномного PrP мыши, удаленного из вектора MoPrP.Xho; специфические прямые праймеры PS1 (5'-CAG GTG GTG GAG CAA GAT G) и APP (5'-CCG AGA TCT CTG AAG TGA AGA TGG ATG), гомологичные последовательностям генов мутантных белков в составе трансгенной кассеты. Реакционная смесь содержала 1× Taq Turbo буфер (“Евроген”, Россия), по 0.2 мМ каждого dNTP (“Евроген”), 0.5 мкМ каждого прямого праймера и 1 мкМ общего обратного праймера, 2 ед. HS Taq-ДНК-полимеразы (“Евроген”). Программа амплификации: 1 цикл – 95°C, 3 мин; 30 циклов – 95°C, 20 с; 55°C (АPP) или 65°C (PS1), 20 с; 72°C, 20 с; 1 цикл – 72°C, 2 мин.
Продукты амплификации разделяли с помощью электрофореза в 1.5%-ном агарозном геле в 1× ТАЕ буфере (40 мМ Трис-HCl, 20 мМ уксусная кислота, 1 мМ EDТА, 0.5 мкг/мл бромистого этидия) при 120 В в течение 30 мин.
Приготовление гистологических препаратов. Животных подвергали терминальной анестезии, мозг диссектировали и фиксировали в растворе Карнуа (6 частей 96%-ного этилового спирта, 3 части хлороформа, 1 часть ледяной уксусной кислоты) в течение ночи. Ткань дегидратировали, используя последовательное проведение в растворах этилового спирта с увеличивающейся концентрацией: 75% 1 ч; 96% (I) 5 мин; 96% (II) 45 мин; 100% (I) 5 мин; 100% (II) 10 мин. Далее последовательно инкубировали в течение 30 мин в смеси 100%-ный этиловый спирт : хлороформ (1 : 1), 1 ч в хлороформе (I), оставляли на ночь в хлороформе (II), после чего ткани пропитывали парафином (три смены по 1 ч) при 60°С. Парафиновые блоки подготавливали на станции для заливки Leica EG1160 (“Leica Biosystems”, ФРГ). Парафиновые срезы толщиной 8 мкм монтировали на предметные стекла с полилизиновым покрытием. Срезы депарафинизировали в течение 20 мин в ксилоле, регидратировали, инкубируя последовательно в этиловом спирте: 100% – 10 мин; 95% – 5 мин; 50% – 5 мин; далее трижды промывали в деионизованной воде по 5 мин. Срезы окрашивали раствором красителя Конго красный (0.5% Конго красного в 50%-ном этиловом спирте) в течение 5 мин и дифференцировали в растворе 0.2%-ного KOH в 80%-ном этиловом спирте в течение 1 мин, промывали 3 раза по 5 мин в деионизованной воде, заключали в среду на водной основе Immu-MountTM (“Thermo Scientific”, США).
РЕЗУЛЬТАТЫ И ОБСУЖДЕНИЕ
Сублиния мышей APPSWE/PS1dE9/Blg получена от скрещивания животных B6;C3-Tg(APPswe,PSEN1dE9)85Dbo/Mmjax на смешанном генетическом фоне С3H/C57Bl6 с животными C57Bl6J/ChG. Для формирования репрезентативных групп экспериментальных и контрольных животных, синхронизированных по возрасту, сначала получали нужное количество родственных производителей. Гемизиготных по трансгенной кассете мышей скрещивали с мышами дикого типа на том же смешанном генетическом фоне, полученными в предыдущих скрещиваниях. Потомство генотипировали методом конвенционной ПЦР с использованием одной пары специфичных праймеров – либо на PSEN1, либо на АРР, поскольку в геноме мыши обе трансгенные кассеты локализованы в одном локусе на хромосоме 9. Схематическое изображение положения праймеров для амплификации трансгенной кассеты представлено на рис. 1. Пример генотипирования по каждой из трансгенных кассет приведен на рис. 2.
Рис. 1.
Схема положения праймеров для детекции трансгенных кассет, содержащих кДНК гена белка-предшественника амилоида со шведской мутацией (Mo/Hu APPswe) (а) и кДНК гена пресенилина 1 человека с делецией экзона 9 (PS1ΔE9) в геноме мышей APPswe/PS1dE9 (б). Обе кассеты расположены в одном локусе на хромосоме 9 и экспрессируются каждая под собственным промотором гена прионного белка мыши (PrP). Прямой праймер PrP_for гомологичен участку эндогенного прионного белка мыши. Обратный праймер PrP_rev гомологичен последовательности эндогенного PrP и последовательности гена PrP в составе трансгенной кассеты. Прямые праймеры APP и PS1 специфично узнают последовательность внутри соответствующих трансгенных кассет. Обозначения: PrP1 – экзон 1, PrP2 – экзон 2, PrP3 – экзон 3, стрелками обозначены прямые и обратный праймеры.

Рис. 2.
Детекция трансгенных кассет в геноме мышей APPswe/PS1dE9/Blg методом ПЦР. Все образцы содержат продукт амплификации (750 п.н.) гена прионного белка мыши в качестве эндогенного контроля. Животные дикого типа (ДТ) содержат только фрагмент 750 п.н. У трансгенных животных APPswe/PS1dE9/Blg (1 и 2) появляется дополнительный фрагмент размером 1.3 т.п.н. после амплификации последовательности мутантного гена PSEN1 или фрагмент размером 400 п.н. после амплификации последовательности мутантного APP. Маркер 100+ п.н. (“Евроген”).
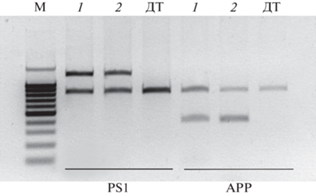
Встраивание двух трансгенных кассет в один локус в геноме мышей, показанное в [19], позволяет проводить генотипирование лишь по одной из трансгенных кассет. Результаты проведенного нами генотипирования подтверждают присутствие обеих трансгенных кассет в нескольких поколениях животных. Таким образом, для генотипирования мышей линии APPswe/PS1dE9/Blg можно проводить ПЦР с праймерами к одному из трансгенных векторов. При этом мы рекомендуем подтверждать присутствие обеих трансгенных кассет в генотипах мышей после окончательного формирования экспериментальных и контрольных групп животных в каждом третьем–четвертом поколении.
Для формирования экспериментальных групп мышей APPswe/PS1dE9/Blg скрещивали с особями дикого типа из предыдущих пометов. Нами не выявлено значительных различий в выживаемости трансгенных животных и животных дикого типа в тех же пометах. Так, анализ результатов генотипирования 315 мышей показал, что трансгенную кассету имели 52.4% потомков, 47.6% не имели модификации генома.
Нами сформированы пары производителей, состоящие из трансгенных самок и самцов дикого типа и пары из трансгенных самцов и самок дикого типа. Возраст животных-производителей 2–4 мес. Анализ фертильности самок APPswe/PS1dE9/Blg показал, что из общего числа ссаженных самок фертильными оказались 74%, тогда как среди самок дикого типа фертильными были 84%. Пересчет общего количества рожденных потомков на одну самку показал, что и самки APPswe/PS1dE9/Blg, и самки дикого типа рожают одинаковое число потомков (табл. 2).
Таблица 2.
Фертильность самок при получении трансгенных животных
| Самки-производители | Ссаженные самки, всего | Не забеременевшие самки | Потомки | Среднее число потомков в одном помете |
|---|---|---|---|---|
| APPswe/PS1dE9/blg | 35 | 9 | 140 | 5.38 |
| Дикий тип | 38 | 6 | 175 | 5.47 |
Нами показано, что показатели выживаемости и фертильности у животных линии APPswe/PS1dE9/Blg не отличаются существенно от показателей у контрольных животных дикого типа. Полученные нами данные позволяют планировать формирование пар производителей для составления экспериментальных групп животных и получения расчетного числа трансгенных животных в экспериментальных когортах.
Отличительной особенностью включений амилоидного типа является кросс-β-структура белка в их составе, обеспечивающая связывание со специфическими красителями, к которым относятся Конго красный и тиофлавин S [2]. Сроки регистрации дебюта патогистологических признаков церебрального амилоидоза в мозге мышей APPswe/PS1dE9 и динамика прогрессии нейродегенеративного процесса, полученные в разных лабораториях, несколько различаются. Наиболее часто патологию, уже широко распространенную в гиппокампе и коре головного мозга, выявляют в возрасте 6 мес. [33]. Данные о прогрессии амилоидоза различаются в зависимости от метода анализа, размера амилоидных включений (подсчет только крупных или средних и мелких включений), использования поправки Аберкромби для нивелирования возможности подсчета одного и того же включения на нескольких серийных срезах, от способов окрашивания тканей – использование специальных красителей (Конго красный или тиофлавин S) или антител к бета-амилоиду, а также от способа анализа окрашенных срезов – детекции включений только в диапазоне определенных длин волн или с дополнительным использованием поляризованного света [4, 34, 35]. Исключительно важной представляется задача подробной характеристики данной сублинии для ее использования в качестве модельной системы церебрального амилоидоза.
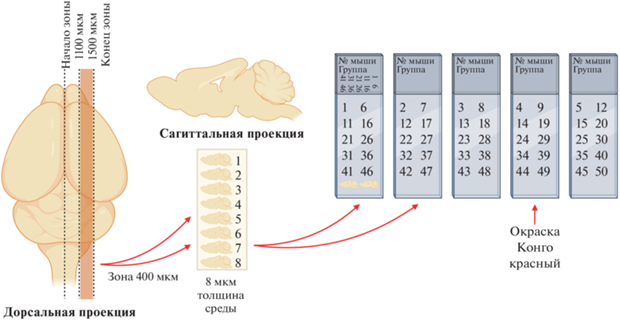
Нами проведен гистологический анализ мозга мышей APPswe/PS1dE9/Blg в возрасте 5.5 и 10 мес. Принцип формирования групп срезов для морфометрического анализа включений представлен на рис. 3.
Рис. 3.
Схема раскладки срезов мозга при подготовке гистологических препаратов. Из зоны мозга толщиной 400 мкм формировали пять стекол, состоящих из 10 срезов, на каждое предметное стекло помещали каждый пятый срез мозга.
Для оценки числа амилоидных включений в гиппокампе и коре мозга мышей APPswe/PS1dE9/Blg срезы полушария головного мозга окрашивали красителем Конго красным (рис. 4). Показано, что уже в возрасте 5.5 мес. в коре и гиппокампе трансгенных животных обнаруживаются как мелкие и средние, так и крупные амилоидные включения. В возрасте 10 мес. наблюдается увеличение числа и размера агрегатов в мозге трансгенных мышей. В мозге контрольных животных дикого типа включения амилоида не обнаруживаются. Согласно опубликованным данным, самыми старыми животными APPswe/PS1dE9, у которых изучено образование амилоидных включений, были животные в возрасте 24 мес. [33].
Рис. 4.
Гистологический анализ включений Aβ на срезах мозга трансгенных животных APPswe/PS1dE9/Blg и контрольных животных дикого типа в возрасте 5.5 и 10 мес.; окраска Конго красным. Шкала измерений 200 мкм.

Нами определена скорость образования патологических агрегатов Аβ в коре и гиппокампе трансгенных APP/PS1 и контрольных мышей в возрасте 5.5 и 10 мес. (табл. 3).
БА у человека может развиваться в течение десятилетий до момента появления первых симптомов. Нами оценена прогрессия агрегации Аβ в гиппокампе и коре у молодых трансгенных мышей (5.5 мес.) и у стареющих животных (10 мес.). Число включений в гиппокампе стареющих животных увеличивается почти в 5 раз и в 4 раза возрастает число агрегатов в коре (p < 0.0001, t-критерий Стьюдента).
Полученные нами данные соотносятся с проведенными ранее экспериментами по подсчету амилоидных бляшек в мозге трансгенных самцов линии APPswe/PS1dE9 – на срезе животных в возрасте 4 мес. в среднем обнаруживалось около 20 бляшек в коре и примерно пять бляшек в гиппокампе, тогда как у животных в возрасте 10 мес. количество включений увеличивалось примерно в 4 раза в коре и в 2 раза в гиппокампе [4].
Свою эффективность в качестве терапевтического средства, снижающего уровень агрегированной формы Aβ in vivo, показали инъекции синтетических тетрапептидов HAEE и RADD. Эти тетрапептиды специфически связываются с сайтом EVHH в пептиде Aβ, что приводит к подавлению Zn-опосредованной димеризации и позволяет снизить уровень амилоидных бляшек, обнаруживаемых в коре и гиппокампе трансгенных животных APPswe/PS1dE9 в возрасте 7 мес. [36]. Также экспериментально показано, что изомеризация остатка Asp7 и фосфорилирование Ser8 в пептиде Aβ42 может препятствовать Zn-зависимой агрегации. Инъекции синтетического пептида isoD7-pS8-Aβ42 с такими модификациями приводили к статистически значимому снижению числа амилоидных включений более чем в 3 раза в мозге 8-месячных животных APPswe/PS1dE9 (7.4 ± 2.8) по сравнению с группой трансгенных животных без инъекции (28.7 ± 4.6) [37]. Инъекции синтетического пептида pS8-Aβ42 с фосфорилированным остатком Ser8 также снижали число агрегатов амилоидного типа в 1.5 раза в гиппокампе 8-месячных трансгенных животных APPswe/PS1dE9 (18.3 ± 0.94) по сравнению с контрольными животными без инъекций (28.7 ± 4.6) [38].
Таким образом, число агрегатов Аβ в мозге трансгенных мышей линии APPswe/PS1dE9 является важным параметром, который следует учитывать при поиске терапевтических стратегий замедления прогрессии БА.
Работа выполнена при поддержке Министерства науки и высшего образования РФ, (соглашение № 075-15-2021-1346). Работы с животными финансировались из средств Государственного задания лаборатории генетических технологий и генного редактирования для биомедицины и ветеринарии FZWG-2021-0016.
Работу с животными проводили в соответствии с “Правилами лабораторной практики в Российской Федерации” от 1.04.2016 № 199н. Проведение эксперимента утверждено на заседании этического комитета НИУ БелГУ от 8.02.2021, протокол № 02.21-4.
Авторы сообщают об отсутствии конфликта интересов.
Список литературы
Konttinen H., Cabral-da-Silva M.E.C., Ohtonen S., Wojciechowski S., Shakirzyanova A., Caligola S., Giugno R., Ishchenko Y., Hernandez D., Fazaludeen M.F., Eamen S., Budia M.G., Fagerlund I., Scoyni F., Korhonen P., Huber N., Haapasalo A., Hewitt A.W., Vickers J., Smith G.C., Oksanen M., Graff C., Kanninen K.M., Lehtonen S., Propson N., Schwartz M.P., Pebay A., Koistinaho J., Ooi L., Malm T. (2019) PSEN1DeltaE9, APPswe, and APOE4 confer disparate phenotypes in human iPSC-derived microglia. Stem. Cell Rep. 13, 669–683.
Шелковникова Т.А., Куликова А.А., Цветков Ф.О., Peters O., Бачурин С.О., Бухман В.Л., Нинкина Н.Н. (2012) Протеинопатии – формы нейродегенеративных заболеваний, в основе которых лежит патологическая агрегация белков. Молекуляр. биология. 46, 402–415.
Masters C.L., Bateman R., Blennow K., Rowe C.C., Sperling R.A., Cummings J.L. (2015) Alzheimer’s disease. Nat. Rev. Dis. Primers. 1, 15056.
Kozin S.A., Cheglakov I.B., Ovsepyan A.A., Telegin G.B., Tsvetkov P.O., Lisitsa A.V., Makarov A.A. (2013) Peripherally applied synthetic peptide isoAsp7-Abeta(1-42) triggers cerebral beta-amyloidosis. Neurotoxicity Res. 24, 370–376.
Evin G., Li Q.X. (2012) Platelets and Alzheimer’s disease: potential of APP as a biomarker. W. J. Psychiatry. 2, 102–113.
Vetrivel K.S., Thinakaran G. (2006) Amyloidogenic processing of beta-amyloid precursor protein in intracellular compartments. Neurology. 66, S69–73.
De Strooper B., Annaert W. (2010) Novel research horizons for presenilins and gamma-secretases in cell biology and disease. Annu. Rev. Cell. Dev. Biol. 26, 235–260.
Manczak M., Kandimalla R., Yin X., Reddy P.H. (2018) Hippocampal mutant APP and amyloid beta-induced cognitive decline, dendritic spine loss, defective autophagy, mitophagy and mitochondrial abnormalities in a mouse model of Alzheimer’s disease. Hum. Mol. Genet. 27, 1332–1342.
Pan J.X., Sun D., Lee D., Xiong L., Ren X., Guo H.H., Yao L.L., Lu Y., Jung C., Xiong W.C. (2021) Osteoblastic Swedish mutant APP expedites brain deficits by inducing endoplasmic reticulum stress-driven senescence. Commun. Biol. 4, 1326.
Татарникова О.Г., Орлов М.А., Бобкова Н.В. (2015) Бета-амилоид и тау-белок: структура, взаимодействие и прионоподобные свойства. Успехи биол. химии. 55, 351–390.
Armstrong R.A. (2019) Risk factors for Alzheimer’s disease. Folia Neuropathol. 57, 87–105.
Jankowsky J.L., Fadale D.J., Anderson J., Xu G.M., Gonzales V., Jenkins N.A., Copeland N.G., Lee M.K., Younkin L.H., Wagner S.L., Younkin S.G., Borchelt D.R. (2004) Mutant presenilins specifically elevate the levels of the 42 residue beta-amyloid peptide in vivo: evidence for augmentation of a 42-specific gamma secretase. Hum. Mol. Genet. 13, 159–170.
Sasaguri H., Hashimoto S., Watamura N., Sato K., Takamura R., Nagata K., Tsubuki S., Ohshima T., Yoshiki A., Sato K., Kumita W., Sasaki E., Kitazume S., Nilsson P., Winblad B., Saito T., Iwata N., Saido T.C. (2022) Recent advances in the modeling of Alzheimer’s disease. Front. Neurosci. 16, 807473.
Games D., Adams D., Alessandrini R., Barbour R., Borthelette P., Blackwell C., Carr T., Clemens J., Donaldson T., Gillespie F., Guido T., Hagopian S., Johnson-Wood K., Khan K., Lee M., Leibowitz P., Lieberburg I., Little S., Masliah E., McConlogue L., Montoya-Zavala M., Mucke L., Paganini L., Penniman E., Power M., Schenk D., Seubert P., Snyder B., Soriano F., Tan H., Vitale J., Wadsworth S., Wolozin B., Zhao J. (1995) Alzheimer-type neuropathology in transgenic mice overexpressing V717F beta-amyloid precursor protein. Nature. 373, 523–527.
Kuo Y.M., Beach T.G., Sue L.I., Scott S., Layne K.J., Kokjohn T.A., Kalback W.M., Luehrs D.C., Vishnivetskaya T.A., Abramowski D., Sturchler-Pierrat C., Staufenbiel M., Weller R.O., Roher A.E. (2001) The evolution of A beta peptide burden in the APP23 transgenic mice: implications for A beta deposition in Alzheimer disease. Mol. Med. 7, 609–618.
Hsiao K., Chapman P., Nilsen S., Eckman C., Harigaya Y., Younkin S., Yang F., Cole G. (1996) Correlative memory deficits, Abeta elevation, and amyloid plaques in transgenic mice. Science. 274, 99–102.
Davis J., Xu F., Deane R., Romanov G., Previti M.L., Zeigler K., Zlokovic B.V., Van Nostrand W.E. (2004) Early-onset and robust cerebral microvascular accumulation of amyloid beta-protein in transgenic mice expressing low levels of a vasculotropic Dutch/Iowa mutant form of amyloid beta-protein precursor. J. Biol. Chem. 279, 20296–20306.
Oddo S., Caccamo A., Shepherd J.D., Murphy M.P., Golde T.E., Kayed R., Metherate R., Mattson M.P., Akbari Y., LaFerla F.M. (2003) Triple-transgenic model of Alzheimer’s disease with plaques and tangles: intracellular Abeta and synaptic dysfunction. Neuron. 39, 409–421.
Jankowsky J.L., Slunt H.H., Ratovitski T., Jenkins N.A., Copeland N.G., Borchelt D.R. (2001) Co-expression of multiple transgenes in mouse CNS: a comparison of strategies. Biomol. Engin. 17, 157–165.
Oakley H., Cole S.L., Logan S., Maus E., Shao P., Craft J., Guillozet-Bongaarts A., Ohno M., Disterhoft J., Van Eldik L., Berry R., Vassar R. (2006) Intraneuronal beta-amyloid aggregates, neurodegeneration, and neuron loss in transgenic mice with five familial Alzheimer’s disease mutations: potential factors in amyloid plaque formation. J. Neurosci. 26, 10129–10140.
Mucke L., Masliah E., Yu G.Q., Mallory M., Rockenstein E.M., Tatsuno G., Hu K., Kholodenko D., Johnson-Wood K., McConlogue L. (2000) High-level neuronal expression of abeta 1–42 in wild-type human amyloid protein precursor transgenic mice: synaptotoxicity without plaque formation. J. Neurosci. 20, 4050–4058.
Radde R., Bolmont T., Kaeser S.A., Coomaraswamy J., Lindau D., Stoltze L., Calhoun M.E., Jaggi F., Wolburg H., Gengler S., Haass C., Ghetti B., Czech C., Holscher C., Mathews P.M., Jucker M. (2006) Abeta42-driven cerebral amyloidosis in transgenic mice reveals early and robust pathology. EMBO Rep. 7, 940–946.
Holcomb L., Gordon M.N., McGowan E., Yu X., Benkovic S., Jantzen P., Wright K., Saad I., Mueller R., Morgan D., Sanders S., Zehr C., O’Campo K., Hardy J., Prada C.M., Eckman C., Younkin S., Hsiao K., Duff K. (1998) Accelerated Alzheimer-type phenotype in transgenic mice carrying both mutant amyloid precursor protein and presenilin 1 transgenes. Nat. Med. 4, 97–100.
Kurt M.A., Davies D.C., Kidd M., Duff K., Rolph S.C., Jennings K.H., Howlett D.R. (2001) Neurodegenerative changes associated with beta-amyloid deposition in the brains of mice carrying mutant amyloid precursor protein and mutant presenilin-1 transgenes. Exp. Neurol. 171, 59–71.
McGowan E., Sanders S., Iwatsubo T., Takeuchi A., Saido T., Zehr C., Yu X., Uljon S., Wang R., Mann D., Dickson D., Duff K. (1999) Amyloid phenotype characterization of transgenic mice overexpressing both mutant amyloid precursor protein and mutant presenilin 1 transgenes. Neurobiol. Dis. 6, 231–244.
Toda T., Noda Y., Ito G., Maeda M., Shimizu T. (2011) Presenilin-2 mutation causes early amyloid accumulation and memory impairment in a transgenic mouse model of Alzheimer’s disease. J. Biomed. Biotechnol. 2011, 617974.
Rockenstein E., Mallory M., Mante M., Sisk A., Masliaha E. (2001) Early formation of mature amyloid-beta protein deposits in a mutant APP transgenic model depends on levels of Abeta(1-42). J. Neurosci. Res. 66, 573–582.
Platt B., Drever B., Koss D., Stoppelkamp S., Jyoti A., Plano A., Utan A., Merrick G., Ryan D., Melis V., Wan H., Mingarelli M., Porcu E., Scrocchi L., Welch A., Riedel G. (2011) Abnormal cognition, sleep, EEG and brain metabolism in a novel knock-in Alzheimer mouse, PLB1. PloS One. 6, e27068.
Saito T., Matsuba Y., Mihira N., Takano J., Nilsson P., Itohara S., Iwata N., Saido T.C. (2014) Single App knock-in mouse models of Alzheimer’s disease. Nat. Neurosci. 17, 661–663.
Sato K., Watamura N., Fujioka R., Mihira N., Sekiguchi M., Nagata K., Ohshima T., Saito T., Saido T.C., Sasaguri H. (2021) A third-generation mouse model of Alzheimer’s disease shows early and increased cored plaque pathology composed of wild-type human amyloid beta peptide. J. Biol. Chem. 297, 101004.
Jankowsky J.L., Xu G., Fromholt D., Gonzales V., Borchelt D.R. (2003) Environmental enrichment exacerbates amyloid plaque formation in a transgenic mouse model of Alzheimer disease. J. Neuropathol. Exp. Neurol. 62, 1220–1227.
Lysikova E.A., Kukharsky M.S., Chaprov K.D., Vasilieva N.A., Roman A.Y., Ovchinnikov R.K., Deykin A.V., Ninkina N., Buchman V.L. (2019) Behavioural impairments in mice of a novel FUS transgenic line recapitulate features of frontotemporal lobar degeneration. Genes Brain Behavior. 18, e12607.
D'Angelo C., Costantini E., Salvador N., Marchioni M., Di Nicola M., Greig N.H., Reale M. (2021) nAChRs gene expression and neuroinflammation in APPswe/PS1dE9 transgenic mouse. Sci. Rep. 11, 9711.
Kuhla A., Ruhlmann C., Lindner T., Polei S., Hadlich S., Krause B.J., Vollmar B., Teipel S.J. (2017) APPswe/PS1dE9 mice with cortical amyloid pathology show a reduced NAA/Cr ratio without apparent brain atrophy: a MRS and MRI study. Neuroimage Clin. 15, 581–586.
Pezzini A., Del Zotto E., Volonghi I., Giossi A., Costa P., Padovani A. (2009) Cerebral amyloid angiopathy: a common cause of cerebral hemorrhage. Curr. Med. Chem. 16, 2498–2513.
Tsvetkov P.O., Cheglakov I.B., Ovsepyan A.A., Mediannikov O.Y., Morozov A.O., Telegin G.B., Kozin S.A. (2015) Peripherally applied synthetic tetrapeptides HAEE and RADD slow down the development of cerebral beta-amyloidosis in AbetaPP/PS1 transgenic mice. J. Alzheimer’s Dis. 46, 849–853.
Kozin S.A., Barykin E.P., Telegin G.B., Chernov A.S., Adzhubei A.A., Radko S.P., Mitkevich V.A., Makarov A.A. (2018) Intravenously injected amyloid-beta peptide with isomerized Asp7 and phosphorylated Ser8 residues inhibits cerebral beta-amyloidosis in AbetaPP/PS1 transgenic mice model of Alzheimer’s disease. Front. Neurosci. 12, 518.
Barykin E.P., Petrushanko I.Y., Kozin S.A., Telegin G.B., Chernov A.S., Lopina O.D., Radko S.P., Mitkevich V.A., Makarov A.A. (2018) Phosphorylation of the amyloid-beta peptide inhibits zinc-dependent aggregation, prevents Na,K-ATPase inhibition, and reduces cerebral plaque deposition. Front. Mol. Neurosci. 11, 302.
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология