

Молекулярная биология, 2023, T. 57, № 6, стр. 1058-1076
Транскрипционный фактор NRF2 в функционировании эндотелия
Н. Д. Кондратенко a, b, Л. А. Зиновкина c, Р. А. Зиновкин a, b, *
a Научно-исследовательский институт физико-химической биологии им. А.Н. Белозерского,
Московский государственный университет им. М.В. Ломоносова
119991 Москва, Россия
b Российский научно-исследовательский медицинский университет им. Н.И. Пирогова,
Российский геронтологический научно-клинический центр
129226 Москва, Россия
c Факультет биоинженерии и биоинформатики Московского государственного университета им. М.В. Ломоносова
119991 Москва, Россия
* E-mail: roman.zinovkin@gmail.com
Поступила в редакцию 16.04.2023
После доработки 11.05.2023
Принята к публикации 22.05.2023
- EDN: QXHFSB
- DOI: 10.31857/S0026898423060101
Аннотация
Транскрипционный фактор NRF2 – главный регулятор антиоксидантной защиты клетки, активируется под воздействием различных стимулов, таких как окислители и электрофилы, что индуцирует транскрипцию целого ряда генов, продукты которых участвуют в метаболизме ксенобиотиков и способствуют уменьшению окислительного стресса. NRF2 является одним из ключевых транскрипционных факторов, обеспечивающих функционирование клеток эндотелия – слоя клеток, выстилающих внутреннюю полость сосудов. Эндотелий выполняет множество гомеостатических функций: контролирует миграцию лейкоцитов во внутренние ткани, регулирует тромбообразование и сосудистый тонус, а также участвует в ангиогенезе. Нарушение функций эндотелия часто сопровождается воспалением и окислительным стрессом, что может приводить к клеточному старению, а также к гибели клеток путем апоптоза, некроза и ферроптоза. Эндотелиальная дисфункция вносит вклад в развитие таких распространенных сердечно-сосудистых заболеваний, как гипертензия и атеросклероз, а также сахарного диабета. Многие патофизиологические процессы в эндотелии, включая старческие изменения, сопряжены со снижением активности NRF2, что приводит к воспалительной активации и снижению активности систем антиоксидантной защиты клетки. Активация сигнального пути NRF2, как правило, способствует разрешению воспаления и устранению окислительного стресса. В данном обзоре рассмотрено значение NRF2 в осуществлении основных функций эндотелия в норме и патологии, а также преимущества и недостатки активации NRF2 как способа профилактики и лечения сердечно-сосудистых заболеваний.
ВВЕДЕНИЕ
Сердечно-сосудистые заболевания (ССЗ) являются основной причиной смертности в мире. По оценкам ВОЗ в 2019 году от ССЗ умерли около 18 млн человек, что составляет 32% всех случаев смерти. В большинстве случаев ССЗ сопровождаются нарушением функций эндотелия [1] – плоского слоя клеток, выстилающего внутреннюю поверхность сосудов и служащего барьером между кровью и внутренними структурами сосуда. Эндотелий выполняет множество функций, включая гемостаз (равновесие между тромбозом и антикоагуляцией), регуляцию тонуса сосудов (баланс между вазоконстрикцией и вазодилатацией), а также ангиогенез (рост сосудов), заживление ран, пролиферацию гладкомышечных клеток. Эндотелиальная дисфункция, как правило, сопровождается окислительным стрессом и воспалительными реакциями. Все это делает чрезвычайно актуальной разработку новых лекарственных препаратов против избыточного воспаления и повреждающего действия окислительного стресса.
Транскрипционный фактор NRF2 – один из основных регуляторов гомеостаза, обеспечивает функционирование клетки в условиях стресса [2]. NRF2 контролирует экспрессию множества генов, участвующих в антиоксидантной защите, поддержании редокс-гомеостаза и детоксикации различных веществ. Известно, что снижение экспрессии NRF2 вызывает окислительный стресс и разбалансированный воспалительный ответ: гиперактивацию клеток врожденного иммунитета, высокую продукцию цитокинов и активных форм кислорода (АФК), которые не только уничтожают патогены, но и повреждают ткани организма-хозяина. Индукция NRF2 приводит к снижению окислительного стресса и восстановлению гомеостаза, что способствует уменьшению воспалительного ответа и разрешению воспаления.
В настоящем обзоре представлен всесторонний анализ данных о функциях NRF2 в сосудистом эндотелии. Рассмотрены механизмы и потенциальные мишени NRF2, влияющие на функцию эндотелиальных клеток, а также плюсы и минусы активации NRF2 для профилактики и лечения ССЗ.
NRF2: СТРУКТУРА, ФУНКЦИИ И РЕГУЛЯЦИЯ
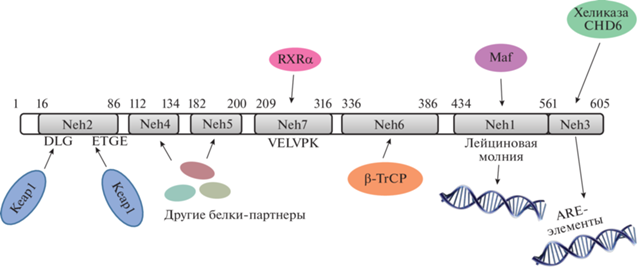
NRF2 (Nuclear factor erythroid 2-related factor 2) – фактор транскрипции, контролирующий экспрессию множества генов, отвечающих за антиоксидантную защиту клетки и метаболизм ксенобиотиков. NRF2 относится к белкам Cap’n’Collar (CNC), содержащим лейциновую молнию. NRF2 человека состоит из 605 а.о. и содержит семь высококонсервативных аминокислотных последовательностей, известных как Neh-домены (NRF2-ECH homology domain). Домен Neh1 (а.о. 434–561) представляет собой лейциновую молнию и отвечает за взаимодействие с малыми белками семейства Maf [3]. Во взаимодействии с другими белками-партнерами NRF2 участвуют домены Neh4 (а.о. 112–134) и Neh5 (а.о. 182–200) [4, 5]. В комплексе с белковыми партнерами NRF2 связывается с ARE-элементами – последовательностями, располагающимися в промоторах генов-мишеней NRF2. В активации транскрипции генов, содержащих ARE-последовательности, участвует домен Neh3 (а.о. 561–605), находящийся в C-концевой части NRF2. Домен Neh3 содержит VFLVPK-мотив, который выполняет роль связывающего звена между NRF2 и хеликазой CHD6. Домен Neh2 (а.о. 1–86) содержит две последовательности – DLG и ETGE, которые отвечают за связывание NRF2 со своим негативным регулятором KEAP1 (Kelch-like ECH-associated protein 1) [6]. В домене Neh6 (а.о. 336–386) находятся две последовательности, которые распознает E3-убиквитинлигаза β-TrCP [7, 8]. Домен Neh7 (а.о. 209–316) отвечает за связывание NRF2 с рецептором ретиноевой кислоты RXRα [9]. Схема расположения Neh-доменов и взаимодействующих с ними белковых партнеров приведена на рис. 1.
Рис. 1.
Доменная структура транскрипционного фактора NRF2. NRF2 содержит семь консервативных Neh-доменов. При помощи Neh1 NRF2 взаимодействует с малыми белками семейства Maf, Neh7 связывает рецептор ретиноевой кислоты RXRα, а Neh4 и Neh5 – другие коактиваторы и корепрессоры. Домен Neh2 содержит DLG и ETGE-мотивы, с которыми взаимодействует негативный регулятор NRF2 KEAP1. Домен Neh3, находящийся в C-концевой области, представляет собой связующее звено между NRF2 и хеликазой CDH6, благодаря чему NRF2 активирует транскрипцию своих генов-мишеней, промоторы которых содержат ARE-элементы. Домен Neh6 содержит два дегрона (последовательности, которые узнает Е3-убиквитинлигаза β-TrCP).
В обычных физиологических условиях концентрация NRF2 в цитоплазме и ядре составляет 149 и 273 нМ соответственно [10]. Относительно низкое количество NRF2 в отсутствие стресса поддерживается благодаря его постоянному протеолизу, который инициирует негативный регулятор NRF2 – KEAP1 [11]. KEAP1 играет роль адапторного белка для E3-убиквитинлигазного комплекса Cullin 3. Связывание NRF2 с KEAP1 приводит к убиквитинилированию семи остатков лизина в домене Neh2 с последующей протеасомной деградацией NRF2 [12].
KEAP1 выполняет роль сенсора окислителей и электрофилов благодаря остаткам цистеина, экспонированным в цитоплазму [12, 13]. В 2009 году была предложена концепция “цистеинового кода”, согласно которой разные классы соединений-индукторов NRF2 связываются с разными цистеиновыми остатками KEAP1, основные из которых Cys151 и Cys273 [14]. Так, различные комбинации модификаций остатков цистеина вызывают специфические биологические эффекты, характерные для каждого индуктора NRF2 [15]. Для нормального функционирования KEAP1 необходима связь этого белка с актиновым цитоскелетом [16]. KEAP1 функционирует в виде димера и связывается с NRF2 с помощью Kelch-доменов. Оба Kelch-домена димера KEAP1 связываются с Neh2-доменом NRF2: один – с DLG-мотивом с низкой аффинностью, другой – с ETGE-мотивом с высокой аффинностью [17, 18]. Благодаря тому, что взаимодействие двух белков KEAP1 с NRF2 происходит с разной аффинностью, была предложена модель “hinge and latch”, согласно которой KEAP1, связанный с ETGE-мотивом, выполняет роль “молнии”, обеспечивая сильное взаимодействие с NRF2, а KEAP1, связанный с DLG-мотивом, играет роль “застежки”, которая размыкается под воздействием окислителей или электрофилов [19].
Взаимодействие окислителей или электрофилов с цистеиновыми остатками KEAP1 приводит к нарушению его взаимодействия с Е3-убиквитинлигазой Cullin 3, в результате чего протеасомная деградация NRF2 останавливается [20]. Из-за нарушения взаимодействия с DLG-мотивом NRF2 KEAP1 остается связанным с уже существующим NRF2, в отсутствие “вакантных” негативных регуляторов новосинтезированный NRF2 накапливается в цитоплазме [21]. Перемещению NRF2 в ядро и активации транскрипции его генов-мишеней способствует сигнал ядерной локализации [20, 22]. При добавлении индуктора NRF2 диэтилмалеата концентрация NRF2 в цитоплазме и ядре возрастает до 619 и 2733 нМ соответственно [10]. После восстановления редокс-гомеостаза KEAP1 перемещается в ядро при участии α6-кариоферина (KPNA6), чтобы “облегчить” экспорт NRF2 из ядра и возобновить его постоянный протеолиз [23, 24]. Так как KEAP1 является основным регулятором активности NRF2, действие многих индукторов NRF2 связано именно с воздействием на KEAP1. Так, многие вещества растительного происхождения (например, сульфорафан, содержащийся в растениях семейства Капустные), являются электрофилами, способными связываться с цистеиновыми остатками KEAP1 и таким образом активировать NRF2.
Помимо редокс-зависимой регуляции посредством связывания с остатками цистеина KEAP1, активность NRF2 может регулироваться и другими способами, не зависящими от редокс-баланса. Например, в домене Neh6 NRF2 мыши расположена последовательность, содержащая остатки серинов, которые могут фосфорилироваться киназой GSK-3β [8]. При фосфорилировании эта последовательность превращается в фосфодегрон, который распознается E3-убиквитинлигазой β-TrCP, в результате чего NRF2 подвергается деградации в протеасомах. По-видимому, регуляция активности с помощью β-TrCP происходит в условиях значительного угнетения активности KEAP1 [25].
NRF2 активируется также при стрессе эндоплазматического ретикулума. Накопление в клетке неправильно свернутых белков вызывает стрессовый ответ (UPR, unfolded protein response). Во время UPR-ответа активируется киназа эндоплазматического ретикулума, подобная протеинкиназе R (PERK), которая останавливает трансляцию и клеточный цикл, а также активирует NRF2 [26–28]. Кроме PERK, в UPR-ответе участвуют белки IRE1 и XBP1 [29], под контролем которых находится промотор гена Hrd1, кодирующего одноименную E3-убиквитинлигазу [30]. Hrd1 связывается с Neh4- и Neh5-доменами NRF2, что приводит к протеасомной деградации последнего. С активностью Hrd1 связано снижение уровня NRF2 в печени при циррозе [30].
Помимо Е3-убиквитинлигаз, регулирующих уровень NRF2, направляя его на протеасомную деградацию, существует ряд белков, в аминокислотной последовательности которых присутствуют мотивы, идентичные или напоминающие ETGE-мотив NRF2 [31]. Эти белки могут конкурировать с NRF2 за связывание с KEAP1, стабилизируя NRF2 в цитоплазме и вызывая его последующее перемещение в ядро. Самым известным белком, активирующим NRF2 таким образом, является p62/SQSTM1, содержащий STGE-мотив. p62/SQSTM1 входит в состав аутофагосом и участвует в транспортировке белков, которые должны подвергнуться деградации в процессе аутофагии [32]. Нарушение аутофагии приводит к накоплению аутофагосом, а соответственно, и белка p62, который связывает KEAP1 и таким образом активирует NRF2 [33].
DJ-1 – один из важных регуляторов антиоксидантной системы клетки, также способен активировать NRF2, однако молекулярные механизмы этой активации до сих пор не выяснены. Предполагалось, что DJ-1 может дестабилизировать комплекс KEAP1-NRF2 [34], но этот механизм не был подтвержден последующими исследованиями [35].
Продукт гена-мишени NRF2, фермент гемоксигеназа-1 (Hmox1), катализирует превращение гема в биливердин с одновременным высвобождением иона железа и CO [36]. СО обладает противовоспалительным и антиапоптотическим действием, что делает гемоксигеназу-1 важной терапевтической мишенью при эндотелиальной дисфункции [37]. Генетический нокаут гемоксигеназы-1 связан с увеличением окислительного стресса, а также с нарушением коагуляционной функции эндотелия [38, 39].
Еще одна мишень NRF2 – это ген Nqo1, кодирующий фермент NADPH:хинон-оксидоредуктазу-1, известную также как диафораза-1. Диафораза-1 катализирует двухэлектронное восстановление хинона до гидрохинона [40], что позволяет избежать одноэлектронного восстановления хинона другими ферментами, например, редуктазой цитохрома P450, которое сопровождается образованием супероксид-аниона (${\text{O}}_{2}^{{\centerdot - }}$). Антиоксидантное действие диафоразы-1 заключается в предотвращении генерации АФК в образовании антиоксидантных форм кофермента Q и α-токоферола [41, 42].
Биосинтез и метаболизм глутатиона (GSH) также находятся под контролем NRF2. GSH обеспечивает поддержание редокс-баланса в клетке за счет как прямого захвата проникающих в клетку окислителей, так и участия в работе ферментов антиоксидантной защиты (например, глутатионпероксидазы) в качестве кофактора [43, 44]. Так, активация NRF2 приводит к повышению экспрессии гена GCLC, кодирующего каталитическую субъединицу глутамат-цистеин-лигазы – ключевого фермента биосинтеза GSH, а также гена GSR, кодирующего глутатионредуктазу [45, 46].
СОСУДИСТЫЙ ЭНДОТЕЛИЙ
Эндотелий – однослойный пласт клеток, выстилающий внутреннюю полость сосудов, обеспечивает ключевые процессы в сердечно-сосудистой системе, такие как проницаемость и тонус сосудов, свертывание крови и регуляцию воспалительных реакций.
В регуляции тонуса сосудов участвует оксид азота (II) (NO), продуцируемый эндотелиальными клетками [47]. NO образуется при окислении L-аргинина до L-цитруллина в активном центре эндотелиальной NO-синтазы (eNOS) [48].
На обращенной внутрь сосуда поверхности эндотелиальных клеток располагается гликокаликс, состоящий из гликопротеинов, глюкозаминогликанов и протеогликанов [49]. Гликокаликс участвует в противосвертывающей функции эндотелия, так как один из глюкозаминогликанов – гепарансульфат – служит кофактором антитромбина III [50].
Эндотелий выполняет роль барьера между циркулирующими клетками крови и тканевой жидкостью. Этот барьер пластичен и может менять свою проницаемость в зависимости от условий окружающей среды. В нормальных условиях лейкоциты мало контактируют с эндотелием. Проникновение патогенов или появление в кровотоке маркеров повреждения клеток вызывает воспалительный ответ эндотелия, заключающийся в усиленной продукции цитокинов и экспрессии на поверхности клеток таких молекул адгезии, отвечающих за взаимодействие с лейкоцитами, как P- и E-селектины, интегрины, ICAM-1 и VCAM-1 [51–54]. В результате лейкоциты распластываются по поверхности эндотелия и мигрируют сквозь стенку сосуда в очаг воспаления, что сопровождается повышением проницаемости эндотелиального барьера и облегчает миграцию лейкоцитов через стенку сосуда [55]. В регуляции потока лейкоцитов участвует гликокаликс эндотелиальных клеток: разрушение гликокаликса приводило к повышению проницаемости сосудов и усиленной адгезии лейкоцитов к поверхности эндотелия [56, 57].
Воспаление является естественной иммунной реакцией организма на инфекцию, которая должна прекратиться после уничтожения патогена. Но некоторые воспалительные состояния, такие как сепсис, вызывают генерализованный и гипертрофированный ответ эндотелия. Окислительный стресс, сопутствующий воспалительному ответу, способствует развитию эндотелиальной дисфункции, что выражается в невозможности осуществления гомеостатических функций, избыточной продукции АФК и провоспалительных медиаторов, снижении способности к ангиогенезу, протромботической активности, в снижении биодоступности NO, а также в ухудшении вазодилатации и вазоконстрикции. Эндотелиальная дисфункция связана с развитием цитокинового шторма и таких заболеваний, как гипертензия, сахарный диабет, атеросклероз, венозный тромбоз [58–62].
NRF2 И ОКИСЛИТЕЛЬНЫЙ СТРЕСС В ЭНДОТЕЛИИ
В результате активности дыхательной цепи митохондрий, а также специализированных ферментов, таких как NADPH-оксидаза, в клетках постоянно синтезируются АФК – чрезвычайно реакционноспособные молекулы, которые в небольших количествах участвуют в передаче различных сигналов в клетках. К АФК относятся пероксид водорода (H2O2), гидроксильный радикал (ОН•) и супероксид-анион (${\text{O}}_{2}^{{\centerdot - }}$). В физиологических условиях АФК эффективно элиминируются системами антиоксидантной защиты клетки. Однако при образовании слишком большого количества АФК или при нарушении работы систем, отвечающих за их уничтожение, может развиться окислительный стресс. Возникновение окислительного стресса в клетках эндотелия может приводить к развитию эндотелиальной дисфункции, которая, как уже упоминалось, вносит значительный вклад в развитие сахарного диабета, атеросклероза, а также хронической болезни почек или острого респираторного дистресс-синдрома [63]. В некоторых случаях повреждения нуклеиновых кислот, белков и липидов, возникающие в результате окислительного стресса, вызывают апоптоз клеток [64].
Активация транскрипционного фактора NRF2 – многообещающий подход к терапии заболеваний, связанных с окислительным стрессом и с развивающейся впоследствии эндотелиальной дисфункцией. В ряде работ показано, что активация NRF2 способна предотвращать повышение уровня АФК и гибель клеток под действием различных окислительных стимулов. Так, индукция NRF2-ответа повышала выживаемость клеток эндотелия аорты, коронарных артерий и пупочной вены человека при окислительном стрессе, вызванном H2O2 [51–54, 65–67]. Частично это обусловлено способностью NRF2 поднимать уровень GSH за счет увеличения экспрессии гена GCLC [68]. Активация NRF2 также восстанавливала индуцированное трет-бутилгидропероксидом снижение экспрессии VE-кадгерина – белка межклеточных контактов – и нарушение его внутриклеточного распределения [69].
Активность NRF2 также снижала уровень окислительного стресса в эндотелиальных клетках, вызванного воздействием уремической сыворотки [70], лептина [71], бензо[а]пирена [72] и доксорубицина [73].
Под действием АФК в клетках эндотелия легочных артерий увеличивается секреция трансформирующего фактора роста TGFβ1 [74]. TGFβ1, в свою очередь, способен индуцировать эндотелиально-мезенхимальный переход, который вносит вклад в развитие легочной артериальной гипертензии [75, 76]. Легочная гипертензия сопровождается обструкцией легочных путей и может приводить к правожелудочковой недостаточности. При эндотелиально-мезенхимальном переходе клетки эндотелия приобретают мезенхимальный фенотип, что выражается в снижении экспрессии эндотелиальных маркеров (CD31 и VE-кадгерина) и повышении экспрессии маркеров клеток фибробластного типа (FSP1, виментина, проколлагена I и гладкомышечного α-актина) [77]. Также АФК вызывают повышение уровня провоспалительных цитокинов, что дополнительно вносит вклад в эндотелиально-мезенхимальный переход [78]. Индукция NRF2 под действием сальвианоловой кислоты А снижала уровень АФК и экспрессию маркеров мезенхимального фенотипа [79].
Дисфункция эндотелия приводит к невозможности выполнения им гомеостатических функций, например, в результате снижения выработки NO. Один из регуляторов активности eNOS – ее кофактор тетрагидробиоптерин (BH4), действует как аллостерический регулятор, способствующий связыванию L-аргинина с активным центром eNOS, а также может напрямую связывать супероксид-анион [80]. Нарушение соотношения BH4 : eNOS приводит к “разобщению” eNOS, в результате которого фермент начинает продуцировать супероксид-анион вместо NO. АФК, образование которых связано с развитием различных ССЗ, способны окислять BH4 в клетках эндотелия и нарушать тем самым стехиометрическое соотношение BH4 : eNOS, что ведет к разобщению eNOS и продукции супероксид-аниона [81–83]. Супероксид-анион, помимо своего собственного вклада в развитие окислительного стресса, способен реагировать с NO с образованием пероксинитрита – активной формы азота [84]. Снижение концентрации BH4 ведет к активации NRF2, который способствует восстановлению стехиометрического равновесия BH4: eNOS путем снижения экспрессии eNOS [85]. Дополнительный вклад в регуляцию количества eNOS может вносить способность NRF2 увеличивать количество гемоксигеназы-1, которая снижает биодоступность гема, входящего в состав активного центра eNOS [85].
Тем не менее, NRF2 также может вносить вклад в развитие окислительного стресса. Например, при гипероксии NRF2 способен перемещаться в ядро и стимулировать экспрессию гена Nox4, кодирующего NADPH-оксидазу 4 и содержащего в промоторе ARE-элемент. Nox4, в свою очередь, продуцирует супероксид-анион, что вносит вклад в развитие окислительного стресса [86, 87].
NRF2 И ВОСПАЛИТЕЛЬНЫЕ РЕАКЦИИ В ЭНДОТЕЛИИ
Воспалительные реакции в эндотелии могут быть вызваны как провоспалительными цитокинами, так и молекулами патогенов, узнаваемыми соответствующими рецепторами. В результате воспалительной активации в эндотелии происходят значительные морфологические и функциональные изменения. Так, например, обработка клеток эндотелия липополисахаридом (LPS) вызывает повышение уровня молекул межклеточной адгезии (ICAM-1 и VCAM-1) и секреции цитокинов (фактора некроза опухоли TNF и IL-1ß) [88, 89]. LPS вызывают также нарушение функций митохондрий: снижение мембранного потенциала и уровня АТP, высвобождение цитохрома c в цитоплазму, снижение количества антиапоптотического белка Bcl2 и увеличение содержания проапоптотического белка BAK, активацию каспазы-3. В то же время индукция пути NRF2/MafF/ARE с помощью MitoQ предотвращала негативные последствия воздействия LPS [63]. Активация NRF2 снижала также адгезию лейкоцитов к поверхности эндотелия и его проницаемость [89–91].
Избыточное воспаление в эндотелии считается причиной многих острых и хронических заболеваний. Индукция NRF2-ответа снижает воспалительную активацию эндотелия, вызванную TNF, путем ингибирования транскрипционного фактора NF-kB [92], поднимает уровень GSH в клетках, снижает экспрессию MCP-1 и VCAM-1 и адгезию моноцитов к поверхности эндотелия [65].
NRF2 И ГЕМОДИНАМИЧЕСКИЙ СТРЕСС
Эндотелий кровеносных сосудов постоянно подвергается физическому воздействию потоков крови [93]. Ламинарное течение крови, без завихрений, характерно для протяженных участков сосудов в состоянии покоя. В этом случае имеет место слоистое движение крови: вблизи стенок сосудов скорость потока плазмы крови минимальна, а в центре достигает максимума. Трение слоев крови характеризуется так называемым напряжением сдвига (shear stress). Действие напряжения сдвига при ламинарном течении крови способствует защите от атеросклероза [94].
Завихрения потоков крови в местах изгибов и разветвления артерий приводят к тому, что поток становится турбулентным, т.е. кровь начинает двигаться не только параллельно стенкам сосудов, но и перпендикулярно. Именно эти участки сосудов чаще всего подвергаются атерогенезу [94]. Возникшие атеросклеротические бляшки дополнительно усиливают турбулентные потоки, негативно влияющие на функции эндотелия.
Эндотелиальные клетки активно реагируют на изменения потока крови, распознавая их с помощью многочисленных механочувствительных рецепторов [95]. В результате этого активируются сигнальные пути, влияющие на окислительно-восстановительный баланс клетки, а также на экспрессию цитокинов, генов антиоксидантной защиты и межклеточных контактов [96]. Ламинарное напряжение сдвига через индукцию таких факторов, как eNOS и тромбомодулин (ТМ), обеспечивает атеропротективный, антиоксидантный, антикоагулянтный и противовоспалительный фенотип [97]. При этом гемодинамический стресс, вызываемый турбулентными потоками, приводит к обратным эффектам и способствует развитию патофизиологических процессов в эндотелии. Важно отметить, что для здоровья сосудов опасно и слишком низкое напряжение сдвига.
Впервые индукция NRF2 в эндотелии под действием ламинарных потоков была обнаружена 20 лет назад [98]. С тех пор ключевая роль NRF2 в обеспечении противовоспалительных свойств ламинарных потоков была многократно подтверждена и изучена в деталях (см. обзор [99]).
Транскрипционный Krüppel-подобный фактор 2 (KLF2) активируется в эндотелии под действием ламинарного потока крови и ингибируется турбулентными потоками [100]. KLF2 существенно усиливает активность NRF2, способствуя его перемещению в ядро [101]. Таким образом, в нормальных условиях NRF2 обеспечивает антиоксидантный и противовоспалительный фенотип эндотелия. При гемодинамическом стрессе активность KLF2 и, следовательно, NRF2 уменьшается, что приводит к развитию окислительного стресса и воспаления. В эндотелиальных клетках мышей, обработанных малой интерферирующей РНК к NRF2, а также в клетках с нокаутом гена Nrf2 гемодинамический стресс значительно усиливает провоспалительный ответ [102].
Таким образом, путь KLF2–NRF2 играет важную роль в обеспечении сосудистого гомеостаза. Молекулярные механизмы активации NRF2 при гемодинамическом стрессе изучены не полностью и включают активацию других сигнальных путей, таких как фосфоинозитид-3-киназа (PI3K)-AKT [103]. В индукции NRF2-ответа принимают участие АФК, синтезируемые NADPH-оксидазой, митохондриальной ЭТЦ и ксантиноксидазой [104]. В этом случае применение таких антиоксидантов, как NAC, приводит к уменьшению активности NRF2, что потенциально опасно для сосудистого эндотелия. Возможно, именно эта активность антиоксидантов и стала причиной неудачных многочисленных попыток использования антиоксидантов в клинической практике для профилактики ССЗ.
При этом роль NRF2 при гемодинамическом стрессе не сводится исключительно к защите эндотелия. Так, в отдельных случаях острый гемодинамический стресс может приводить к активации NRF2 и повышению синтеза провоспалительного хемокина IL-8, привлекающего иммунные клетки [105].
NRF2 И АТЕРОСКЛЕРОЗ
Атеросклероз – хроническое заболевание, характеризующееся образованием отложений липопротеинов на внутренней стенке сосудов. Окислительный стресс и эндотелиальная дисфункция, часто являющаяся следствием стресса, вносят большой вклад в развитие атеросклероза [106]. Окислительный стресс, возникающий под действием различных стимулов, таких как свободные жирные кислоты или их окисленные производные, может приводить к дисфункции или гибели клеток эндотелия [107, 108]. Экспрессия молекул адгезии и секреция провоспалительных цитокинов усиливают адгезию моноцитов к поверхности эндотелия, а нарушение его барьерной функции – к накоплению в интиме сосудов макрофагов и липидов, формирующих атеросклеротические бляшки [109]. Кроме того, клетки поврежденного эндотелия продуцируют большое количество факторов роста, которые способствуют пролиферации клеток гладкой мускулатуры сосудов, секреции самими клетками внеклеточного матрикса и увеличению нестабильности атеросклеротических бляшек [110, 111].
PAPC (1-пальмитоил-2-арахидоноил-sn-глицеро-3-фосфохолин) – компонент клеточных мембран и липопротеинов. Окисленный PAPC (oxPAPC) входит в состав атеросклеротических бляшек, окисленных липопротеинов низкой плотности (oxLDL), а также мембран апоптотических клеток [112]. Воздействие oxPAPC на клетки эндотелия приводит к увеличению экспрессии провоспалительных генов и молекул адгезии [113]. Обнаружено, что курение (один из факторов риска атеросклероза) связано с увеличенным образованием oxPAPC и репрессией защитного пути NRF2/ARE [114, 115]. Инкубация клеток линии HUVEC с сывороткой крови курящих людей, в которой повышен уровень oxPAPC, приводит к развитию в них окислительного стресса, вызванного повышением уровня АФК с одновременным снижением уровня GSH [116] за счет снижения экспрессии NRF2 и его гена-мишени GCLC. При этом стоит отметить, что, хотя большие концентрации oxPAPC вызывали репрессию NRF2/ARE-пути, относительно небольшая концентрация oxPAPC, наоборот, приводила к активации NRF2/ARE и увеличению экспрессии Hmox1, GCLC и NQO1 в клетках HUVEC in vitro и в артериях мышей in vivo [116, 117]. Показано также, что и сам никотин вызывает увеличение количества АФК в клетках эндотелия и активацию NLRP3-инфламмасомы, приводя, в конечном счете, к гибели клеток путем пироптоза, что успешно предотвращается активацией NRF2 [118, 119].
Кроме oxPAPC, воспалительным действием обладают также свободные жирные кислоты, количество которых в кровотоке существенно повышается при ожирении [120]. Активация NRF2/ARE снижает также воспалительный ответ клеток на пальмитиновую кислоту [121, 122], наиболее распространенную циркулирующую жирную кислоту.
В развитие эндотелиальной дисфункции и атеросклероза вносит вклад еще один фактор – oxLD-L [123]. Активация NRF2 под действием изотиоцианатов (сульфорафан, бензилизотиоцианат и фенетилизотиоцианат) также частично предотвращала активацию NF-kB и повышение экспрессии ICAM-1, VCAM-1, E-селектина, вызванное воздействием oxLDL на клетки эндотелия пупочной вены человека [124].
Развитие атеросклероза тесно связано с ферроптозом – одним из видов клеточной смерти, при котором происходит железозависимое окисление липидов [125, 126]. Это связано с нарушением метаболизма железа и снижением активности систем антиоксидантной защиты клетки [116, 127, 128]. Так, воздействие oxLDL на клетки эндотелия приводило к снижению экспрессии субъединицы 2 пренилдифосфатсинтазы (Prenyldiphosphate synthase subunit 2, PDSS2) – фермента, участвующего в синтезе коэнзима Q10 (CoQ10), а снижение контролируемой ею экспрессии NRF2 было связано с усиленной гибелью клеток путем ферроптоза, что успешно предотвращалось сверхэкспрессией PDSS2 [129]. Гибель клеток мог предотвращать и таншинон IIA, обладающий способностью активировать NRF2 [130].
Активация NRF2 с помощью различных веществ оказывала атеропротективное действие in vivo. Так, на мышах с дефицитом аполипопротеина E и рецептора липопротеинов низкой плотности (LDLR), которых держали на высокожировой диете, показано, что индукторы NRF2 снижали уровень общего холестерина, триглицеридов и LDL в сыворотке крови [131–133]. Добавление в корм мышей индукторов NRF2 приводило к уменьшению площади поражения сосудов атеросклеротическими бляшками, увеличению стабильности бляшек, а также к снижению накопления липидов в печени. Кроме того, сверхэкспрессия гена гемоксигеназы-1 – одного из генов-мишеней NRF2, также вызывала уменьшение площади поражения атеросклеротическими бляшками в корне аорты и снижала уровень накопления железа в атеросклеротических бляшках [134]. В то же время, у мышей с дефицитом аполипопротеина Е нокаут Hmox1 приводил к более тяжелому атеросклерозу.
Несмотря на то, что множество работ посвящено протективному влиянию активации NRF2 на течение атеросклероза, опубликованы работы, описывающие обратный эффект. Так, на модели атеросклероза, индуцированного высокожировой диетой у мышей с дефицитом ApoE, показано [135, 136], что нокаут Nrf2 снижал площадь поражения сосудов атеросклеротическими бляшками. Похожие результаты получены и в работе [137], но различия в течении атеросклероза наблюдали только у самцов с нокаутом гена Nrf2.
NRF2 И ГИПЕРГЛИКЕМИЯ
Ключевую роль в нарушении функций внутренних органов (почек, сетчатки, нервной системы, а также сердца и сосудов) при сахарном диабете играет гипергликемия и связанная с ней эндотелиальная дисфункция. Гипергликемия приводит к истончению гликокаликса, необходимого для нормального функционирования эндотелия, и усиленной продукции таких АФК, как пероксид водорода и супероксид-анион [138, 139]. Под воздействием высокого уровня глюкозы в клетках эндотелия происходит нарушение межклеточных контактов, что приводит к увеличению проницаемости эндотелиального барьера, а также к апоптозу клеток эндотелия [140, 141], увеличению экспрессии молекул адгезии и усиленной адгезии лейкоцитов к поверхности эндотелия, к снижению биодоступности NO [142, 143]. Под воздействием высокого содержания глюкозы снижается соотношение GSH/GSSG в цитоплазме и митохондриях, что обусловлено снижением количества GSH с одновременным увеличением уровня глутатионилированных белков [144]. Кроме того, при высоком уровне глюкозы нарушается нормальное функционирование митохондрий, что выражается в снижении потенциала на внутренней мембране митохондрий и усиленной продукции АФК в митохондриях [145]. Нарушение функций митохондрий, в свою очередь, приводит к образованию актиновых стресс-фибрилл и усиленному апоптозу эндотелиальных клеток [145].
Выявлены негативные последствия воздействия гипергликемии не только на зрелые, но и на прогениторные клетки эндотелия. Показано, что эндотелиальные прогениторные клетки, выделенные из костного мозга мышей с диабетом, характеризовались сниженной способностью к миграции и пролиферации, повышенным уровнем окислительного стресса и сниженной активностью защитного пути NRF2/ARE [146].
Негативное воздействие гипергликемии на клетки эндотелия выражается также в снижении активности защитного NRF2/ARE-ответа. Так, в клетках эндотелия пупочной вены, полученных от пациентов с гестационным диабетом, выявлено снижение адаптивного NRF2-ответа на добавление 4-гидроксиноненаля (4-HNE) [147]. Вероятно, одной из причин этого может быть снижение количества белка DJ-1, способного активировать NRF2, в клетках эндотелия больных гестационным диабетом [147]. Кроме того, в гипергликемических условиях в клетках эндотелия снижается экспрессия SET8 – метилтрансферазы, которая отвечает за метилирование Lys20 в гистоне H4 [148]. Метилирование гистона H4 (H4K20me1), располагающегося в непосредственной близости от промотора гена KEAP1, приводит к “замалчиванию” его транскрипции. Таким образом, снижение экспрессии SET8 вносит вклад в снижение активации NRF2/ARE-ответа при гипергликемии.
Гипергликемия не только нарушает нормальное функционирование эндотелия, но и снижает адаптивный NRF2-ответ. При этом активация NRF2 с помощью сверхэкспрессии или малых молекул-индукторов может снижать эндотелиальную дисфункцию, вызванную высоким уровнем глюкозы. Так, активация NRF2 препятствует повышению уровня АФК, малонового диальдегида и экспрессии ICAM-1 и VCAM-1 при высоком содержании глюкозы, а также восстанавливает способность клеток эндотелия к миграции и ангиогенезу [149, 150]. Вызванная инсулином активация NRF2 через сигнальный путь PI3K/AKT/mTOR приводит к увеличению количества и активности GCLC, восстанавливает соотношение GSH/GSSG в цитоплазме и митохондриях и предотвращает апоптотическую гибель клеток [144]. Митохондриально-направленный антиоксидант MitoQ, способный активировать NRF2, восстанавливал мембранный потенциал митохондрий, снижал продукцию митохондриальных АФК и уменьшал количество актиновых стресс-фибрилл и адгезию лейкоцитов к поверхности эндотелия [145].
Активация NRF2 восстанавливает уровень NO и приводит к более успешному расслаблению аорты мышей с диабетом под действием ацетилхолина, снижая окислительный стресс и эндотелиальную дисфункцию [151, 152].
В мышиной модели сахарного диабета, индуцированного внутрибрюшинным введением стрептозотоцина, бутират натрия (NaB), обладающий способностью активировать NRF2, предотвращал повышение уровня ICAM-1, VCAM-1, 4-HNE и уровня экспрессии индуцибельной NO-синтазы iNOS в аорте, а также улучшал способность аорты к релаксации под действием ацетилхолина [149]. Активация NRF2 с помощью трет-бутилгидрохинона в прогениторных эндотелиальных клетках восстанавливает их способность к миграции, пролиферации и секреции факторов роста, а также снижает уровень окислительного стресса [146].
NRF2 И ТРОМБОЗ
Нарушения свертываемости крови могут приводить как к неконтролируемым кровотечениям, так и к образованию тромбов (тромбозам), которые представляют серьезную угрозу для жизни и здоровья человека. Однако до настоящего времени взаимосвязи тромбозов и активности NRF2 в эндотелии посвящено ограниченное число исследований. Вместе с тем известно, что гибель эндотелиальных клеток, вызванная сильными воспалительными реакциями организма, например, при сепсисе, может быть причиной возникновения тромбозов [153]. Реакции воспаления всегда сопровождаются сильным окислительным стрессом [154], а поскольку NRF2 способен снижать как окислительный стресс, так и воспалительные реакции, логично предположить, что этот фактор может предотвращать гибель эндотелиальных клеток и, следовательно, развитие тромбоза.
Получены данные, косвенно подтверждающие эту гипотезу. Во-первых, показано, что липоксин A4 предотвращает тромбообразование, вызванное окислительным стрессом, с помощью активации NRF2 [155]. Во-вторых, у 89% пациентов с тромбозом глубоких вен обнаружены мутации в сигнальном пути NRF2/KEAP1, приводящие к снижению экспрессии мРНК гена NRF2 [156]. Тем не менее, требуются дальнейшие исследования роли NRF2 при заболеваниях, связанных с нарушением свертываемости крови.
NRF2 И ВАЗОКОНСТРИКЦИЯ
Вазоконстрикция – сужение просвета сосудов, обусловленное сокращением мышечных стенок артерий и артериол, обеспечивает повышение давления крови, а также участвует в защите организма от кровопотерь, вызванных серьезными травмами. Вазодилатация – это процесс обратный вазоконстрикции, при котором мышечные стенки сосудов расслабляются и кровяное давление падает.
Сигнальная молекула NO(II) в низких концентрациях обеспечивает вазодилатацию путем активации растворимой гуанилатциклазы в гладкомышечных клетках сосудов. В высоких концентрациях NO вызывает апоптоз и некроз клеток, в том числе эндотелиальных [157]. Сигнальный путь NRF2 активируется в присутствии NO за счет S-нитрозилирования ингибиторного белка KEAP1 [158], которое включает защитные клеточные реакции для уменьшения NO-индуцированного повреждения.
Для уничтожения попавших в организм патогенов клетки иммунной системы продуцируют NO в высокой концентрации. По-видимому, активация NRF2 в этом случае может уменьшать окислительный стресс и способствовать выживанию патогенов, поэтому следует с осторожностью применять индукторы NRF2 при инфекционных заболеваниях.
NRF2 И СТАРЕНИЕ ЭНДОТЕЛИЯ
С окислительным стрессом тесно связано хроническое воспаление, сопутствующее старению (“inflammaging”) [159]. Большинство возрастных заболеваний, в том числе ССЗ, имеют в своей основе воспалительный компонент [160]. В стареющих сосудах увеличивается содержание маркеров воспаления и окислительного стресса [161, 162], а также повышается апоптоз [163]. Снижение системного воспаления и окислительного стресса в сосудах можно рассматривать как перспективный подход к замедлению процессов преждевременного старения.
Существует множество исследований, показывающих, что в эндотелии сосудов с возрастом уменьшается как количество NRF2, так и его активность, что может способствовать развитию эндотелиальной дисфункции и патологий сосудистой системы [164–166]. Дисфункция NRF2 может быть потенциальным механизмом, лежащим в основе возрастного нарушения ангиогенеза, при этом активация NRF2 восстанавливает ангиогенез [167].
Под действием ряда соединений, влияющих на клеточную ДНК, клетки эндотелия могут превращаться в так называемые сенесцентные, или старческие клетки, которые не способны к дальнейшему делению. Эти клетки экспрессируют типичные маркеры клеточного старения (бета-галактозидазу, p16, p21 и p53) и не выполняют большинство из своих функций. Стареющие клетки также приобретают секреторный фенотип, ассоциированный со старением (SASP, senescence-associated secretory phenotype), при котором повышена секреция провоспалительных цитокинов, хемокинов и матриксных металлопротеиназ [168]. В мозговых артериях мышей с нокаутом Nrf2 усиливаются возрастные изменения, которые сопровождаются повышением содержания маркеров воспаления [169]. Активация NRF2, наоборот, предотвращает появление сенесцентных клеток [170]. Индукция NRF2 с помощью трет-бутилгидрохинона препятствовала развитию SASP в прогениторных эндотелиальных клетках мышей со стрептозотоциновым диабетом [146]. Таким образом, активацию NRF2 можно рассматривать как многообещающее направление профилактики ССЗ при старении [171, 172].
NRF2 И ЭНДОТЕЛИЙ ОПУХОЛЕЙ
Согласно принятым в настоящее время представлениям, активация NRF2 защищает клетки от мутагенеза [173], что объясняется антиоксидантным действием ферментов, экспрессия которых находится под контролем NRF2: снижение окислительного стресса уменьшает вероятность потенциально мутагенных окислительных повреждений ДНК. Тем не менее, конститутивная активация NRF2 способствует развитию, прогрессированию и метастазированию опухолей [174]. В опухолевых тканях зачастую повышен уровень экспрессии NRF2, чья антиоксидантная активность способствует выживанию опухолевых клеток [175]. Кроме того, активность NRF2 влияет на ангиогенез, вызванный метаболическими изменениями в опухолевых клетках [176]. Показано, что нокдаун NRF2 ингибирует образование кровеносных сосудов путем уменьшения количества белка HIF-1α, что приводит к снижению уровней фактора роста эндотелия сосудов, тромбоцитарного фактора роста, ангиопоэтина и ангиогенина [177, 178]. Таким образом, активность NRF2 способствует как выживаемости опухолей, так и формированию новых сосудов. Это позволяет рассматривать NRF2 как перспективную мишень для терапии опухолей.
ЗАКЛЮЧЕНИЕ
Транскрипционный фактор NRF2 является главным регулятором антиоксидантного ответа клетки, который, активируя транскрипцию своих генов-мишеней, способствует снижению или предотвращению окислительного стресса и связанного с ним воспаления (рис. 2). К числу мишеней NRF2 относятся гены, продукты которых обладают антиоксидантными или противовоспалительными свойствами. Например, Hmox1 кодирует гемоксигеназу-1, продуктом гена NQO1 является диафораза, а GCLC кодирует каталитическую субъединицу глутамат-цистеин-лигазы – ключевого фермента биосинтеза GSH. NRF2 активируется под воздействием целого ряда стимулов: это могут быть окислители и электрофилы; воспалительные стимулы, такие как oxPAPC или ультрафиолетовое излучение; ксенобиотики; биоактивные соединения, продуцируемые самим организмом (NO, инсулин или мелатонин), а также физическое воздействие тока крови на клетки [72, 117, 119, 144, 179, 180].
Рис. 2.
Роль NRF2 в функциях эндотелия. К положительным эффектам NRF2 относятся снижение окислительного стресса и воспалительной активации эндотелия, восстановление нормальной функции митохондрий, увеличение биодоступности NO за счет предотвращения разобщения eNOS. Однако активация NRF2 может иметь и такие негативные последствия, как увеличение экспрессии гена Nox4, продукт которого, NADPH-оксидаза, продуцирует АФК. NRF2 играет двойную роль в образовании опухолей: механизмы, защищающие нормальные клетки от мутагенеза и опухолевой трансформации, способствуют выживанию опухолевых клеток. Кроме того, способность NRF2 стимулировать ангиогенез вносит дополнительный вклад в прогрессирование опухолей, способствуя их васкуляризации. Роль NRF2 в развитии атеросклероза остается изученной не до конца: имеются данные, указывающие как на защитное, так и на проатерогенное действие NRF2.
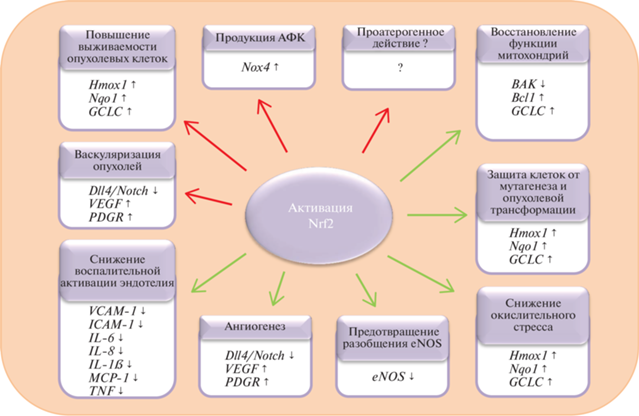
Эндотелий, выстилающий внутренние полости сосудов, участвует во многих процессах, необходимых для поддержания гомеостаза. Он обладает антитромботической активностью, контролирует миграцию лейкоцитов сквозь стенку сосуда в ткани, регулирует сосудистый тонус и ангиогенез, при этом в ангиогенезе участвует транскрипционный фактор NRF2, способный ингибировать сигнальный путь Dll4/Notch [181]. Эндотелиальная дисфункция, возникающая при окислительном стрессе клеток эндотелия, является спутником таких заболеваний, связанных с хроническим воспалением, как диабет и атеросклероз. Активация NRF2 способствует снижению окислительного стресса, предотвращению эндотелиальной дисфункции и ее последствий. Так, активация NRF2 снижает уровень выработки АФК и повышает выживаемость клеток под действием H2O2, трет-бутилгидропероксида, уремической сыворотки, лептина, бензо[а]пирена и доксорубицина [65–67, 69–73]. Индукция NRF2/ARE-ответа вызывала антиатерогенный эффект in vivo и in vitro, снижая адгезию лейкоцитов к поверхности эндотелия и уменьшая площадь поражения сосудов атеросклеротическими бляшками [121, 122, 124, 131–133]. Также активация NRF2 спасала эндотелиальные клетки от воспаления, вызываемого гипергликемией [144, 145, 149, 151, 152].
Несмотря на многочисленные работы, посвященные защитному действию NRF2, активация NRF2 может иметь и негативные последствия (рис. 2). Так, NRF2 может увеличивать экспрессию гена Nox4, продукт которого – NADPH-оксидаза – способен продуцировать АФК, внося тем самым дополнительный вклад в развитие окислительного стресса [86, 87]. Снижение площади поражения сосудов атеросклеротическими бляшками у мышей с дефицитом аполипопротеина Е и LDLR и с нокаутом Nrf2 указывает на возможную проатерогенную активность NRF2 [51–54, 135, 137]. В контексте развития опухолей NRF2 также играет двойную роль – активация NRF2 в нормальных клетках помогает предотвращать мутагенез и опухолевую трансформацию, при этом она же увеличивает выживаемость опухолевых клеток и способствует васкуляризации опухолей [173–178]. Положительные и отрицательные эффекты активации NRF2 в клетках эндотелия приведены на рис. 2.
Таким образом, активация транскрипционного фактора NRF2 представляется перспективным подходом к профилактике и лечению ССЗ, в том числе связанных со старением. Однако для выяснения побочного действия такой активации необходимо проведение дальнейших исследований.
Авторы благодарят Анастасию Приходько за конструктивную критику и помощь в редактировании текста.
Работа выполнена при поддержке гранта Российского научного фонда (23-14-00061).
Настоящая работа выполнена без привлечения людей или животных в качестве объектов исследований.
Авторы заявляют об отсутствии конфликта интересов.
Список литературы
Widmer R.J., Lerman A. (2014) Endothelial dysfunction and cardiovascular disease. Glob. Cardiol. Sci. Pract. 2014(3), 291–308. https://doi.org/10.5339/gcsp.2014.43
Kaspar J.W., Niture S.K., Jaiswal A.K. (2009) Nrf2:INrf2 (Keap1) signaling in oxidative stress. Free Radic. Biol. Med. 47(9), 1304–1309. https://doi.org/10.1016/j.freeradbiomed.2009.07.035
Motohashi H., Katsuoka F., Engel J.D., Yamamoto M. (2004) Small Maf proteins serve as transcriptional cofactors for keratinocyte differentiation in the Keap1–Nrf2 regulatory pathway. Proc. Natl. Acad. Sci. USA. 101(17), 6379–6384. https://doi.org/10.1073/pnas.0305902101
Nioi P., Nguyen T., Sherratt P.J., Pickett C.B. (2005) The carboxy-terminal Neh3 domain of Nrf2 is required for transcriptional activation. Mol. Cell Biol. 25(24), 10 895–10 906. https://doi.org/10.1128/MCB.25.24.10895-10906.2005
Katoh Y., Itoh K., Yoshida E., Miyagishi M., Fukamizu A., Yamamoto M. (2001) Two domains of Nrf2 cooperatively bind CBP, a CREB binding protein, and synergistically activate transcription. Genes Cells. 6(10), 857–868. https://doi.org/10.1046/j.1365-2443.2001.00469.x
Tong K.I., Katoh Y., Kusunoki H., Itoh K., Tanaka T., Yamamoto M. (2006) Keap1 recruits Neh2 through binding to ETGE and DLG motifs: characterization of the two-site molecular recognition model. Mol. Cell. Biol. 26(8), 2887–2900. https://doi.org/10.1128/MCB.26.8.2887-2900.2006
McMahon M., Thomas N., Itoh K., Yamamoto M., Hayes J.D. (2004) Redox-regulated turnover of Nrf2 is determined by at least two separate protein domains, the redox-sensitive Neh2 degron and the redox-insensitive Neh6 degron. J. Biol. Chem. 279(30), 31 556–31 567. https://doi.org/10.1074/jbc.M403061200
Rada P., Rojo A.I., Chowdhry S., McMahon M., Hayes J.D., Cuadrado A. (2011) SCF/{beta}-TrCP promotes glycogen synthase kinase 3-dependent degradation of the Nrf2 transcription factor in a Keap1-independent manner. Mol. Cell Biol. 31(6), 1121–1133. https://doi.org/10.1128/MCB.01204-10
Wang H., Liu K., Geng M., Gao P., Wu X., Hai Y., Li Y., Li Y., Luo L., Hayes J.D., Wang X.J., Tang X. (2013) RXRα inhibits the NRF2-ARE signaling pathway through a direct interaction with the Neh7 domain of NRF2. Cancer Res. 73(10), 3097–3108. https://doi.org/10.1158/0008-5472.CAN-12-3386
Iso T., Suzuki T., Baird L., Yamamoto M. (2016) Absolute amounts and status of the Nrf2-Keap1-Cul3 complex within cells. Mol. Cell. Biol. 36(24), 3100–3112. https://doi.org/10.1128/MCB.00389-16
Kobayashi A., Kang M.-I., Okawa H., Ohtsuji M., Zenke Y., Chiba T., Igarashi K., Yamamoto M. (2004) Oxidative stress sensor Keap1 functions as an adaptor for Cul3-based E3 ligase to regulate proteasomal degradation of Nrf2. Mol. Cell. Biol. 24(16), 7130–7139. https://doi.org/10.1128/MCB.24.16.7130-7139.2004
Zhang D.D., Lo S.-C., Cross J.V., Templeton D.J., Hannink M. (2004) Keap1 is a redox-regulated substrate adaptor protein for a Cul3-dependent ubiquitin ligase complex. Mol. Cell. Biol. 24(24), 10941–10953. https://doi.org/10.1128/MCB.24.24.10941-10953.2004
Dinkova-Kostova A.T., Holtzclaw W.D., Cole R.N., Itoh K., Wakabayashi N., Katoh Y., Yamamoto M., Talalay P. (2002) Direct evidence that sulfhydryl groups of Keap1 are the sensors regulating induction of phase 2 enzymes that protect against carcinogens and oxidants. Proc. Natl. Acad. Sci. USA. 99(18), 11 908–11 913. https://doi.org/10.1073/pnas.172398899
Kobayashi M., Li L., Iwamoto N., Nakajima-Takagi Y., Kaneko H., Nakayama Y., Eguchi M., Wada Y., Kumagai Y., Yamamoto M. (2009) The antioxidant defense system Keap1-Nrf2 comprises a multiple sensing mechanism for responding to a wide range of chemical compounds. Mol. Cell. Biol. 29(2), 493–502. https://doi.org/10.1128/MCB.01080-08
Suzuki T., Takahashi J., Yamamoto M. (2023) Molecular basis of the KEAP1-NRF2 signaling pathway. Mol. Cells. 46(3), 133–141. https://doi.org/10.14348/molcells.2023.0028
Kang M.-I., Kobayashi A., Wakabayashi N., Kim S.-G., Yamamoto M. (2004) Scaffolding of Keap1 to the actin cytoskeleton controls the function of Nrf2 as key regulator of cytoprotective phase 2 genes. Proc. Natl. Acad. Sci. USA. 101(7), 2046–2051. https://doi.org/10.1073/pnas.0308347100
McMahon M., Thomas N., Itoh K., Yamamoto M., Hayes J.D. (2006) Dimerization of substrate adaptors can facilitate cullin-mediated ubiquitylation of proteins by a “tethering” mechanism: a two-site interaction model for the Nrf2-Keap1 complex. J. Biol. Chem. 281(34), 24756–24768. https://doi.org/10.1074/jbc.M601119200
Tong K.I., Kobayashi A., Katsuoka F., Yamamoto M. (2006) Two-site substrate recognition model for the Keap1-Nrf2 system: a hinge and latch mechanism. Biol. Chem. 387(10–11), 1311–1320. https://doi.org/10.1515/BC.2006.164
Tong K.I., Padmanabhan B., Kobayashi A., Shang C., Hirotsu Y., Yokoyama S., Yamamoto M. (2007) Different electrostatic potentials define ETGE and DLG motifs as hinge and latch in oxidative stress response. Mol. Cell. Biol. 27(21), 7511–7521. https://doi.org/10.1128/MCB.00753-07
Kobayashi A., Kang M.-I., Watai Y., Tong K.I., Shibata T., Uchida K., Yamamoto M. (2006) Oxidative and electrophilic stresses activate Nrf2 through inhibition of ubiquitination activity of Keap1. Mol. Cell. Biol. 26(1), 221–229. https://doi.org/10.1128/MCB.26.1.221-229.2006
Baird L., Llères D., Swift S., Dinkova-Kostova A.T. (2013) Regulatory flexibility in the Nrf2-mediated stress response is conferred by conformational cycling of the Keap1-Nrf2 protein complex. Proc. Natl. Acad. Sci. USA. 110(38), 15259–15264. https://doi.org/10.1073/pnas.1305687110
Jain A.K., Bloom D.A., Jaiswal A.K. (2005) Nuclear import and export signals in control of Nrf2. J. Biol. Chem. 280(32), 29158–29168. https://doi.org/10.1074/jbc.M502083200
Sun Z., Wu T., Zhao F., Lau A., Birch C.M., Zhang D.D. (2011) KPNA6 (Importin {alpha}7)-mediated nuclear import of Keap1 represses the Nrf2-dependent antioxidant response. Mol. Cell. Biol. 31(9), 1800–1811. https://doi.org/10.1128/MCB.05036-11
Sun Z., Zhang S., Chan J.Y., Zhang D.D. (2007) Keap1 controls postinduction repression of the Nrf2-mediated antioxidant response by escorting nuclear export of Nrf2. Mol. Cell. Biol. 27(18), 6334–6349. https://doi.org/10.1128/MCB.00630-07
Kuga A., Tsuchida K., Panda H., Horiuchi M., Otsuk-i A., Taguchi K., Katsuoka F., Suzuki M., Yama-moto M. (2022) The β-TrCP-mediated pathway cooperates with the Keap1-mediated pathway in Nrf2 degradation in vivo. Mol. Cell. Biol. 42(7), e0056321. https://doi.org/10.1128/mcb.00563-21
Brewer J.W., Diehl J.A. (2000) PERK mediates cell-cycle exit during the mammalian unfolded protein response. Proc. Natl. Acad. Sci. USA. 97(23), 12 625–12 630. https://doi.org/10.1073/pnas.220247197
Harding H.P., Zhang Y., Ron D. (1999) Protein translation and folding are coupled by an endoplasmic-reticulum-resident kinase. Nature. 397(6716), 271–274. https://doi.org/10.1038/16729
Cullinan S.B., Zhang D., Hannink M., Arvisais E., Kaufman R.J., Diehl J.A. (2003) Nrf2 is a direct PERK substrate and effector of PERK-dependent cell survival. Mol. Cell. Biol. 23(20), 7198–7209. https://doi.org/10.1128/MCB.23.20.7198-7209.2003
Back S.H., Schröder M., Lee K., Zhang K., Kaufman R.J. (2005) ER stress signaling by regulated splicing: IRE1/HAC1/XBP1. Methods. 35(4), 395–416. https://doi.org/10.1016/j.ymeth.2005.03.001
Wu T., Zhao F., Gao B., Tan C., Yagishita N., Nakajima T., Wong P.K., Chapman E., Fang D., Zhang D.D. (2014) Hrd1 suppresses Nrf2-mediated cellular protection during liver cirrhosis. Genes Dev. 28(7), 708–722. https://doi.org/10.1101/gad.238246.114
Hast B.E., Goldfarb D., Mulvaney K.M., Hast M.A., Siesser P.F., Yan F., Hayes D.N., Major M.B. (2013) Proteomic analysis of ubiquitin ligase KEAP1 reveals associated proteins that inhibit NRF2 ubiquitination. Cancer Res. 73(7), 2199–2210. https://doi.org/10.1158/0008-5472.CAN-12-4400
Pankiv S., Clausen T.H., Lamark T., Brech A., Bruun J.-A., Outzen H., Øvervatn A., Bjørkøy G., Johansen T. (2007) p62/SQSTM1 binds directly to Atg8/LC3 to facilitate degradation of ubiquitinated protein aggregates by autophagy. J. Biol. Chem. 282(33), 24131–24145. https://doi.org/10.1074/jbc.M702824200
Lau A., Wang X.-J., Zhao F., Villeneuve N.F., Wu T., Jiang T., Sun Z., White E., Zhang D.D. (2010) A noncanonical mechanism of Nrf2 activation by autophagy deficiency: direct interaction between Keap1 and p62. Mol. Cell. Biol. 30(13), 3275–3285. https://doi.org/10.1128/MCB.00248-10
Clements C.M., McNally R.S., Conti B.J., Mak T.W., Ting J.P.-Y. (2006) DJ-1, a cancer- and Parkinson’s disease-associated protein, stabilizes the antioxidant transcriptional master regulator Nrf2. Proc. Natl. Acad. Sci. USA. 103(41), 15091–15096. https://doi.org/10.1073/pnas.0607260103
Gan L., Johnson D.A., Johnson J.A. (2010) Keap1-Nrf2 activation in the presence and absence of DJ-1. Eur. J. Neurosci. 31(6), 967–977. https://doi.org/10.1111/j.1460-9568.2010.07138.x
Tenhunen R., Marver H.S., Schmid R. (1968) The enzymatic conversion of heme to bilirubin by microsomal heme oxygenase. Proc. Natl. Acad. Sci. USA. 61(2), 748–755. https://doi.org/10.1073/pnas.61.2.748
Calay D., Mason J.C. (2014) The multifunctional role and therapeutic potential of HO-1 in the vascular endothelium. Antioxid. Redox Signal. 20(11), 1789–1809. https://doi.org/10.1089/ars.2013.5659
Yachie A., Niida Y., Wada T., Igarashi N., Kaneda H., Toma T., Ohta K., Kasahara Y., Koizumi S. (1999) Oxidative stress causes enhanced endothelial cell injury in human heme oxygenase-1 deficiency. J. Clin. Invest. 103 (1), 129–135. https://doi.org/10.1172/JCI4165
Radhakrishnan N., Yadav S.P., Sachdeva A., Pruthi P.K., Sawhney S., Piplani T., Wada T., Yachie A. (2011) Human heme oxygenase-1 deficiency presenting with hemolysis, nephritis, and asplenia. J. Pediatr. Hematol. Oncol. 33(1), 74–78. https://doi.org/10.1097/MPH.0b013e3181fd2aae
Ernster L. (1967) [56] DT diaphorase. In: Methods in Enzymology. Acad. Press. 10, 309–317. https://doi.org/10.1016/0076-6879(67)10059-1
Beyer R.E., Segura-Aguilar J., Di Bernardo S., Cavazzoni M., Fato R., Fiorentini D., Galli M.C., Setti M., Landi L., Lenaz, G. (1996) The role of DT-diaphorase in the maintenance of the reduced antioxidant form of coenzyme Q in membrane systems. Proc. Natl. Acad. Sci. USA. 93(6), 2528‒2532. https://doi.org/10.1073/pnas.93.6.2528
Siegel D., Bolton E.M., Burr J.A., Liebler D.C., Ross D. (1997) The reduction of α-tocopherolquinone by human NAD(P)H: quinone oxidoreductase: the role of α-tocopherolhydroquinone as a cellular antioxidant. Mol. Pharmacol. 52(2), 300–305. https://doi.org/10.1124/mol.52.2.300
Wu G., Fang Y.-Z., Yang S., Lupton J.R., Turner N.D. (2004) Glutathione metabolism and its implications for health. J. Nutr. 134(3), 489–492. https://doi.org/10.1093/jn/134.3.489
Han D., Hanawa N., Saberi B., Kaplowitz N. (2006) Mechanisms of liver injury. III. Role of glutathione redox status in liver injury. Am. J. Physiol. Gastrointest. Liver Physiol. 291(1), G1–G7. https://doi.org/10.1152/ajpgi.00001.2006
Harvey C.J., Thimmulappa R.K., Singh A., Blake D.J., Ling G., Wakabayashi N., Fujii J., Myers A., Biswal S. (2009) Nrf2-regulated glutathione recycling independent of biosynthesis is critical for cell survival during oxidative stress. Free Radic. Biol. Med. 46(4), 443–453. https://doi.org/10.1016/j.freeradbiomed.2008.10.040
Chan J.Y., Kwong M. (2000) Impaired expression of glutathione synthetic enzyme genes in mice with targeted deletion of the Nrf2 basic-leucine zipper protein. Biochim. Biophys. Acta. 1517(1), 19–26. https://doi.org/10.1016/s0167-4781(00)00238-4
Furchgott R.F., Zawadzki J.V. (1980) The obligatory role of endothelial cells in the relaxation of arterial smooth muscle by acetylcholine. Nature. 288(5789), 373–376. https://doi.org/10.1038/288373a0
Griffith O.W., Stuehr D.J. (1995) Nitric oxide synthases: properties and catalytic mechanism. Annu. Rev. Physiol. 57, 707–736. https://doi.org/10.1146/annurev.ph.57.030195.003423
Reitsma S., Slaaf D.W., Vink H., van Zandvoort M.A.M.J., oude Egbrink M.G.A. (2007) The endothelial glycocalyx: composition, functions, and visualization. Pflugers Arch. 454(3), 345–359. https://doi.org/10.1007/s00424-007-0212-8
Sugahara K., Mikami T., Uyama T., Mizuguchi S., Nomura K., Kitagawa H. (2003) Recent advances in the structural biology of chondroitin sulfate and dermatan sulfate. Curr. Opin. Struct. Biol. 13(5), 612–620. https://doi.org/10.1016/j.sbi.2003.09.011
McEver R.P., Moore K.L., Cummings R.D. (1995) Leukocyte trafficking mediated by selectin-carbohydrate interactions. J. Biol. Chem. 270(19), 11 025–11 028. https://doi.org/10.1074/jbc.270.19.11025
Dustin M.L., Rothlein R., Bhan A.K., Dinarello C.A., Springer T.A. (1986) Induction by IL 1 and interferon-gamma: tissue distribution, biochemistry, and function of a natural adherence molecule (ICAM-1). J. Immunol. 137(1), 245–254. https://doi.org/10.4049/jimmunol.137.1.245
Sans M., Panés J., Ardite E., Elizalde J.I., Arce Y., Elena M., Palacín A., Fernández-Checa J.C., Anderson D.C., Lobb R., Piqué J.M. (1999) VCAM-1 and ICAM-1 mediate leukocyte-endothelial cell adhesion in rat experimental colitis. Gastroenterology. 116(4), 874–883. https://doi.org/10.1016/s0016-5085(99)70070-3
Lampugnani M.G., Resnati M., Dejana E., Marchisio P.C. (1991) The role of integrins in the maintenance of endothelial monolayer integrity. J. Cell Biol. 112(3), 479–490. https://doi.org/10.1083/jcb.112.3.479
Gotsch U., Borges E., Bosse R., Böggemeyer E., Simon M., Mossmann H., Vestweber D. (1997) VE-cadherin antibody accelerates neutrophil recruitment in vivo. J. Cell Sci. 110(5), 583–588. https://doi.org/10.1242/jcs.110.5.583
Constantinescu A.A., Vink H., Spaan J.A.E. (2003) Endothelial cell glycocalyx modulates immobilization of leukocytes at the endothelial surface. Arterioscler. Thromb. Vasc. Biol. 23(9), 1541–1547. https://doi.org/10.1161/01.ATV.0000085630.24353.3D
Jacob M., Bruegger D., Rehm M., Welsch U., Conzen P., Becker B.F. (2006) Contrasting effects of colloid and crystalloid resuscitation fluids on cardiac vascular permeability. Anesthesiology. 104(6), 1223–1231. https://doi.org/10.1097/00000542-200606000-00018
Castro-Ferreira R., Cardoso R., Leite-Moreira A., Mansilha A. (2018) The role of endothelial dysfunction and inflammation in chronic venous disease. Ann. Vasc. Surg. 46, 380–393. https://doi.org/10.1016/j.avsg.2017.06.131
Weber C., Noels H. (2011) Atherosclerosis: current pathogenesis and therapeutic options. Nat. Med. 17(11), 1410–1422. https://doi.org/10.1038/nm.2538
Ng H.H., Leo C.H., Parry L.J., Ritchie R.H. (2018) Relaxin as a therapeutic target for the cardiovascular complications of diabetes. Front. Pharmacol. 9, 501. https://doi.org/10.3389/fphar.2018.00501
Baszczuk A., Kopczyński Z., Thielemann A. (2014) Endothelial dysfunction in patients with primary hypertension and hyperhomocysteinemia. Postepy Hig. Med. Dosw. 68, 91–100. https://doi.org/10.5604/17322693.1087521
De Lorenzo A., Escobar S., Tibiriçá E. (2020) Systemic endothelial dysfunction: a common pathway for COVID-19, cardiovascular and metabolic diseases. Nutr. Metab. Cardiovasc. Dis. 30(8), 1401–1402. https://doi.org/10.1016/j.numecd.2020.05.007
Cen M., Ouyang W., Zhang W., Yang L., Lin X., Dai M., Hu H., Tang H., Liu H., Xia J., Xu, F. (2021) MitoQ protects against hyperpermeability of endothelium barrier in acute lung injury via a Nrf2-dependent mechanism. Redox Biol. 41, 101936. https://doi.org/10.1016/j.redox.2021.101936
Grimsrud P.A., Xie H., Griffin T.J., Bernlohr D.A. (2008) Oxidative stress and covalent modification of protein with bioactive aldehydes. J. Biol. Chem. 283(32), 21837–21841. https://doi.org/10.1074/jbc.R700019200
Chen X.-L., Dodd G., Thomas S., Zhang X., Wasserman M.A., Rovin B.H., Kunsch C. (2006) Activation of Nrf2/ARE pathway protects endothelial cells from oxidant injury and inhibits inflammatory gene expression. Am. J. Physiol. Heart Circ. Physiol. 290(5), H1862–H1870. https://doi.org/10.1152/ajpheart.00651.2005
Donovan E.L., McCord J.M., Reuland D.J., Miller B.F., Hamilton K.L. (2012) Phytochemical activation of Nrf2 protects human coronary artery endothelial cells against an oxidative challenge. Oxid. Med. Cell. Longev. 2012, 132931. https://doi.org/10.1155/2012/132931
Chen M., Zhang M., Zhang X., Li J., Wang Y., Fan Y., Shi R. (2015) Limb ischemic preconditioning protects endothelium from oxidative stress by enhancing Nrf2 translocation and upregulating expression of antioxidases. PLoS One. 10, e0128455. https://doi.org/10.1371/journal.pone.0128455
Cortese M.M., Suschek C.V., Wetzel W., Kröncke K.-D., Kolb-Bachofen V. (2008) Zinc protects endothelial cells from hydrogen peroxide via Nrf2-dependent stimulation of glutathione biosynthesis. Free Radic. Biol. Med. 44(12), 2002–2012. https://doi.org/10.1016/j.freeradbiomed.2008.02.013
Li X., Zhang Q., Hou N., Li J., Liu M., Peng S., Zhang Y., Luo Y., Zhao B., Wang S., Zhang Y. (2019) Carnosol as a Nrf2 activator improves endothelial barrier function through antioxidative mechanisms. Int. J. Mol. Sci. 20(4), 800. https://doi.org/10.3390/ijms20040880
Chen Z.-W., Miu H.-F., Wang H.-P., Wu Z.-N., Wang W.-J., Ling Y.-J., Xu X.-H., Sun H.-J., Jiang X. (2018) Pterostilbene protects against uraemia serum-induced endothelial cell damage via activation of Keap1/Nrf2/HO-1 signaling. Int. Urol. Nephrol. 50(3), 559–570. https://doi.org/10.1007/s11255-017-1734-4
Teixeira T.M., da Costa D.C., Resende A.C., Soulage C.O., Bezerra F.F., Daleprane J.B. (2017) Activation of Nrf2-antioxidant signaling by 1,25-dihydroxycholecalciferol prevents leptin-induced oxidative stress and inflammation in human endothelial cells. J. Nutr. 147(4), 506–513. https://doi.org/10.3945/jn.116.239475
Rajendran P., Alzahrani A.M., Ahmed E.A., Veeraraghavan V.P. (2021) Kirenol inhibits B[a]P-induced oxidative stress and apoptosis in endothelial cells via modulation of the Nrf2 signaling pathway. Oxid. Med. Cell. Longev. 2021, 5585303. https://doi.org/10.1155/2021/5585303
Ismail M.B., Rajendran P., AbuZahra H.M., Veeraraghavan V.P. (2021) Mangiferin inhibits apoptosis in doxorubicin-induced vascular endothelial cells via the Nrf2 signaling pathway. Int. J. Mol. Sci. 22(8), 4259. https://doi.org/10.3390/ijms22084259
Montorfano I., Becerra A., Cerro R., Echeverría C., Sáez E., Morales M.G., Fernández R., Cabello-Verrugio C., Simon F. (2014) Oxidative stress mediates the conversion of endothelial cells into myofibroblasts via a TGF-β1 and TGF-β2-dependent pathway. Lab. Invest. 94(10), 1068–1082. https://doi.org/10.1038/labinvest.2014.100
Saito A. (2013) EMT and EndMT: regulated in similar ways? J. Biochem. 153(6), 493–495. https://doi.org/10.1093/jb/mvt032
Good R.B., Gilbane A.J., Trinder S.L., Denton C.P., Coghlan G., Abraham D.J., Holmes A.M. (2015) Endothelial to mesenchymal transition contributes to endothelial dysfunction in pulmonary arterial hypertension. Am. J. Pathol. 185(7), 1850–1858. https://doi.org/10.1016/j.ajpath.2015.03.019
Zeisberg E.M., Tarnavski O., Zeisberg M., Dorfman A.L., McMullen J.R., Gustafsson E., Chandraker A., Yuan X., Pu W.T., Roberts A.B., Neilson E.G. (2007) Endothelial-to-mesenchymal transition contributes to cardiac fibrosis. Nat. Med. 13(8), 952–961. https://doi.org/10.1038/nm1613
Rieder F., Kessler S.P., West G.A., Bhilocha S., de la Motte C., Sadler T.M., Gopalan B., Stylianou E., Fiocchi C. (2011) Inflammation-induced endothelial-to-mesenchymal transition: a novel mechanism of intestinal fibrosis. Am. J. Pathol. 179(5), 2660–2673. https://doi.org/10.1016/j.ajpath.2011.07.042
Chen Y., Yuan T., Zhang H., Yan Y., Wang D., Fang L., Lu Y., Du G. (2017) Activation of Nrf2 attenuates pulmonary vascular remodeling via inhibiting endothelial-to-mesenchymal transition: an insight from a plant polyphenol. Int. J. Biol. Sci. 13(8), 1067–1081. https://doi.org/10.7150/ijbs.20316
Vásquez-Vivar J., Kalyanaraman B., Martásek P., Hogg N., Masters B.S., Karoui H., Tordo P., Pritchard K.A. Jr. (1998) Superoxide generation by endothelial nitric oxide synthase: the influence of cofactors. Proc. Natl. Acad. Sci. USA. 95(16), 9220–9225. https://doi.org/10.1073/pnas.95.16.9220
Stuehr D., Pou S., Rosen G.M. (2001) Oxygen reduction by nitric-oxide synthases. J. Biol. Chem. 276(18), 14533–14536. https://doi.org/10.1074/jbc.R100011200
Alp N.J., Channon K.M. (2004) Regulation of endothelial nitric oxide synthase by tetrahydrobiopterin in vascular disease. Arterioscler. Thromb. Vasc. Biol. 24(3), 413–420. https://doi.org/10.1161/01.ATV.0000110785.96039.f6
Li H., Förstermann U. (2013) Uncoupling of endothelial NO synthase in atherosclerosis and vascular disease. Curr. Opin. Pharmacol. 13(2), 161–167. https://doi.org/10.1016/j.coph.2013.01.006
Beckman J.S., Koppenol W.H. (1996) Nitric oxide, superoxide, and peroxynitrite: the good, the bad, and ugly. Am. J. Physiol. 271(5), C1424–C1437. https://doi.org/10.1152/ajpcell.1996.271.5.C1424
Heiss E.H., Schachner D., Werner E.R., Dirsch V.M. (2009) Active NF-E2-related factor (Nrf2) contributes to keep endothelial NO synthase (eNOS) in the coupled state: role of reactive oxygen species (ROS), eNOS, and heme oxygenase (HO-1) levels. J. Biol. Chem. 284(46), 31579–31586. https://doi.org/10.1074/jbc.M109.009175
Pendyala S., Gorshkova I.A., Usatyuk P.V., He D., Pennathur A., Lambeth J.D., Thannickal V.J., Natarajan V. (2009) Role of Nox4 and Nox2 in hyperoxia-induced reactive oxygen species generation and migration of human lung endothelial cells. Antioxid. Redox Signal. 11(4), 747–764. https://doi.org/10.1089/ars.2008.2203
Pendyala S., Moitra J., Kalari S., Kleeberger S.R., Zhao Y., Reddy S.P., Garcia J.G.N., Natarajan V. (2011) Nrf2 regulates hyperoxia-induced Nox4 expression in human lung endothelium: identification of functional antioxidant response elements on the Nox4 promoter. Free Radic. Biol. Med. 50(12), 1749–1759. https://doi.org/10.1016/j.freeradbiomed.2011.03.022
Chen H., Xie K., Han H., Li Y., Liu L., Yang T., Yu Y. (2015) Molecular hydrogen protects mice against polymicrobial sepsis by ameliorating endothelial dysfunction via an Nrf2/HO-1 signaling pathway. Int. Immunopharmacol. 28(1), 643–654. https://doi.org/10.1016/j.intimp.2015.07.034
Lin Q., Qin X., Shi M., Qin Z., Meng Y., Qin Z., Guo S. (2017) Schisandrin B inhibits LPS-induced inflammatory response in human umbilical vein endothelial cells by activating Nrf2. Int. Immunopharmacol. 49, 142–147. https://doi.org/10.1016/j.intimp.2017.05.032
Gao F., Li J.-M., Xi C., Li H.-H., Liu Y.-L., Wang Y.-P., Xuan L.-J. (2019) Magnesium lithospermate B protects the endothelium from inflammation-induced dysfunction through activation of Nrf2 pathway. Acta Pharmacol. Sin. 40(7), 867–878. https://doi.org/10.1038/s41401-018-0189-1
Li C., Zhang W.-J., Frei B. (2016) Quercetin inhibits LPS-induced adhesion molecule expression and oxidant production in human aortic endothelial cells by p38-mediated Nrf2 activation and antioxidant enzyme induction. Redox Biol. 9, 104–113. https://doi.org/10.1016/j.redox.2016.06.006
Fratantonio D., Speciale A., Molonia M.S., Bashllari R., Palumbo M., Saija A., Cimino F., Monastra G., Virgili F. (2018) Alpha-lipoic acid, but not di-hydrolipoic acid, activates Nrf2 response in primary human umbilical-vein endothelial cells and protects against TNF-α induced endothelium dysfunction. Arch. Biochem. Biophys. 655, 18–25. https://doi.org/10.1016/j.abb.2018.08.003
Gimbrone M.A., Jr., García-Cardeña G. (2013) Vascular endothelium, hemodynamics, and the pathobiology of atherosclerosis. Cardiovasc. Pathol. 22(1), 9–15. https://doi.org/10.1016/j.carpath.2012.06.006
Davies P.F. (2009) Hemodynamic shear stress and the endothelium in cardiovascular pathophysiology. Nat. Clin. Pract. Cardiovasc. Med. 6(1), 16–26. https://doi.org/10.1038/ncpcardio1397
Fang Y., Wu D., Birukov K.G. (2019) Mechanosensing and mechanoregulation of endothelial cell functions. Compr. Physiol. 9(2), 873–904. https://doi.org/10.1002/cphy.c180020
Davies P.F., Civelek M., Fang Y., Fleming I. (2013) The atherosusceptible endothelium: endothelial phenotypes in complex haemodynamic shear stress regions in vivo. Cardiovasc. Res. 99(2), 315–327. https://doi.org/10.1093/cvr/cvt101
Nayak L., Lin Z., Jain M.K. (2011) “Go with the flow”: how Krüppel-like factor 2 regulates the vasoprotective effects of shear stress. Antioxid. Redox Signal. 15(5), 1449–1461. https://doi.org/10.1089/ars.2010.3647
Chen X.-L., Varner S.E., Rao A.S., Grey J.Y., Thomas S., Cook C.K., Wasserman M.A., Medford R.M., Jaiswal A.K., Kunsch C. (2003) Laminar flow induction of antioxidant response element-mediated genes in endothelial cells: a novel anti-inflammatory mechanism . J. Biol. Chem. 278(2), 703–711. https://doi.org/10.1074/jbc.M203161200
Ishii T., Warabi E., Mann G.E. (2021) Mechanisms underlying unidirectional laminar shear stress-mediated Nrf2 activation in endothelial cells: amplification of low shear stress signaling by primary cilia. Redox Biol. 46, 102103. https://doi.org/10.1016/j.redox.2021.102103
Dekker R.J., van Soest S., Fontijn R.D., Salamanca S., de Groot P.G., VanBavel E., Pannekoek H., Horrevoets A.J.G. (2002) Prolonged fluid shear stress induces a distinct set of endothelial cell genes, most specifically lung Krüppel-like factor (KLF2). Blood. 100(5), 1689–1698. https://doi.org/10.1182/blood-2002-01-0046
Fledderus J.O., Boon R.A., Volger O.L., Hurttila H., Ylä-Herttuala S., Pannekoek H., Levonen A.-L., Horrevoets A.J.G. (2008) KLF2 primes the antioxidant transcription factor Nrf2 for activation in endothelial cells. Arterioscler. Thromb. Vasc. Biol. 28(7), 1339–1346. https://doi.org/10.1161/ATVBAHA.108.165811
Takabe W., Warabi E., Noguchi N. (2011) Anti-atherogenic effect of laminar shear stress via Nrf2 activation. Antioxid. Redox Signal. 15(5), 1415–1426. https://doi.org/10.1089/ars.2010.3433
Dai G., Vaughn S., Zhang Y., Wang E.T., Garcia-Cardena G., Gimbrone M.A. Jr. (2007) Biomechanical forces in atherosclerosis-resistant vascular regions regulate endothelial redox balance via phosphoinositol 3-kinase/Akt-dependent activation of Nrf2. Circ. Res. 101(7), 723–733. https://doi.org/10.1161/CIRCRESAHA.107.152942
Warabi E., Takabe W., Minami T., Inoue K., Itoh K., Yamamoto M., Ishii T., Kodama T., Noguchi N. (2007) Shear stress stabilizes NF-E2-related factor 2 and induces antioxidant genes in endothelial cells: role of reactive oxygen/nitrogen species. Free Radic. Biol. Med. 42(2), 260–269. https://doi.org/10.1016/j.freeradbiomed.2006.10.043
Ward A.O., Sala-Newby G.B., Ladak S., Angelini G.D., Caputo M., Suleiman M.-S., Evans P.C., George S.J., Zakkar M. (2022) Nrf2-Keap-1 imbalance under acute shear stress induces inflammatory response in venous endothelial cells. Perfusion. 37(6), 582–589. https://doi.org/10.1177/02676591211012571
Kattoor A.J., Pothineni N.V.K., Palagiri D., Mehta J.L. (2017) Oxidative stress in atherosclerosis. Curr. Atheroscler. Rep. 19(11), 42. https://doi.org/10.1007/s11883-017-0678-6
Gimbrone M.A. Jr, García-Cardeña G. (2016) Endothelial cell dysfunction and the pathobiology of atherosclerosis. Circ. Res. 118(4), 620–636. https://doi.org/10.1161/CIRCRESAHA.115.306301
Xu Y.-J., Zheng L., Hu Y.-W., Wang Q. (2018) Pyroptosis and its relationship to atherosclerosis. Clin. Chim. Acta. 476, 28–37. https://doi.org/10.1016/j.cca.2017.11.005
Crea F., Libby P. (2017) Acute coronary syndromes: the way forward from mechanisms to precision treatment. Circulation. 136, 1155–1166. https://doi.org/10.1161/CIRCULATIONAHA.117.029870
Celletti F.L., Waugh J.M., Amabile P.G., Brendolan A., Hilfiker P.R., Dake M.D. (2001) Vascular endothelial growth factor enhances atherosclerotic plaque progression. Nat. Med. 7(4), 425–429. https://doi.org/10.1038/86490
Bennett M.R., Sinha S., Owens G.K. (2016) Vascular smooth muscle cells in atherosclerosis. Circ. Res. 118(4), 692‒702. https://doi.org/10.1161/CIRCRESAHA.115.306361
Fruhwirth G.O., Loidl A., Hermetter A. (2007) Oxidized phospholipids: from molecular properties to disease. Biochim. Biophys. Acta. 1772(7), 718–736. https://doi.org/10.1016/j.bbadis.2007.04.009
Bochkov V.N., Oskolkova O.V., Birukov K.G., Levonen A.-L., Binder C.J., Stöckl J. (2010) Generation and biological activities of oxidized phospholipids. Antioxid. Redox Signal. 12(8), 1009–1059. https://doi.org/10.1089/ars.2009.2597
Garbin U., Pasini A.F., Stranieri C., Cominacini M., Pasini A., Manfro S., Lugoboni F., Mozzini C., Gu-idi G.C., Faccini G., Cominacini L. (2009) Cigarette smoking blocks the protective expression of Nrf2/ARE pathway in peripheral mononuclear cells of young heavy smokers favouring inflammation. PLoS One. 4, e8225. https://doi.org/10.1371/journal.pone.0008225
Cui M., Cui R., Liu K., Dong J.-Y., Imano H., Hayama-Terada M., Muraki I., Kiyama M., Okada T., Kitamura A., Umesawa M., Yamagishi K., Ohira T., Iso H. (2018) Associations of tobacco smoking with impaired endothelial function: the circulatory risk in communities study (CIRCS). J. Atheroscler. Thromb. 25(9), 836–845. https://doi.org/10.5551/jat.42150
Fratta Pasini A., Albiero A., Stranieri C., Cominacini M., Pasini A., Mozzini C., Vallerio P., Cominacini L., Garbin U. (2012) Serum oxidative stress-induced repression of Nrf2 and GSH depletion: a mechanism potentially involved in endothelial dysfunction of young smokers. PLoS One. 7, e30291. https://doi.org/10.1371/journal.pone.0030291
Jyrkkänen H.-K., Kansanen E., Inkala M., Kivelä A.M., Hurttila H., Heinonen S.E., Goldsteins G., Jauhiainen S., Tiainen S., Makkonen H., Oskolkova O., Afonyushkin T., Koistinaho J., Yamamoto M., Bochkov V.N., Ylä-Herttuala S., Levonen A.-L. (2008) Nrf2 regulates antioxidant gene expression evoked by oxidized phospholipids in endothelial cells and murine arteries in vivo. Circ. Res. 103, e1–e9. https://doi.org/10.1161/CIRCRESAHA.108.176883
Wu X., Zhang H., Qi W., Zhang Y., Li J., Li Z., Lin Y., Bai X., Liu X., Chen X., Yang H., Xu C., Zhang Y., Yang B. (2018) Nicotine promotes atherosclerosis via ROS-NLRP3-mediated endothelial cell pyroptosis. Cell Death Dis. 9(2), 171. https://doi.org/10.1038/s41419-017-0257-3
Zhao Z., Wang X., Zhang R., Ma B., Niu S., Di X., Ni L., Liu C. (2021) Melatonin attenuates smoking-induced atherosclerosis by activating the Nrf2 pathway via NLRP3 inflammasomes in endothelial cells. Aging. 13(8), 11363–11380. https://doi.org/10.18632/aging.202829
Opie L.H., Walfish P.G. (1963) Plasma free fatty acid concentrations in obesity. N. Engl. J. Med. 268, 757–760. https://doi.org/10.1056/NEJM196304042681404
Fratantonio D., Speciale A., Ferrari D., Cristani M., Saija A., Cimino F. (2015) Palmitate-induced endothelial dysfunction is attenuated by cyanidin-3-O-glucoside through modulation of Nrf2/Bach1 and NF-κB pathways. Toxicol. Lett. 239(3), 152–160. https://doi.org/10.1016/j.toxlet.2015.09.020
Mahmoud A.M., Wilkinson F.L., Jones A.M., Wilkinson J.A., Romero M., Duarte J., Alexander M.Y. (2017) A novel role for small molecule glycomimetics in the protection against lipid-induced endothelial dysfunction: involvement of Akt/eNOS and Nrf2/ARE signaling. Biochim. Biophys. Acta Gen. Subj. 1861, 3311–3322. https://doi.org/10.1016/j.bbagen.2016.08.013
Gao S., Zhao D., Wang M., Zhao F., Han X., Qi Y., Liu J. (2017) Association between circulating oxidized LDL and atherosclerotic cardiovascular disease: a meta-analysis of observational studies. Can. J. Cardiol. 33, 1624–1632. https://doi.org/10.1016/j.cjca.2017.07.015
Huang C.-S., Lin A.-H., Liu C.-T., Tsai C.-W., Chang I.-S., Chen H.-W., Lii C.-K. (2013) Isothiocyanates protect against oxidized LDL-induced endothelial dysfunction by upregulating Nrf2-dependent antioxidation and suppressing NFκB activation. Mol. Nutr. Food Res. 57, 1918–1930. https://doi.org/10.1002/mnfr.201300063
Dixon S.J., Lemberg K.M., Lamprecht M.R., Skouta R., Zaitsev E.M., Gleason C.E., Patel D.N., Bauer A.J., Cantley A.M., Yang W.S., Morrison B., Stockwell B.R. (2012) Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 149(5), 1060–1072. https://doi.org/10.1016/j.cell.2012.03.042
Bai T., Li M., Liu Y., Qiao Z., Wang Z. (2020) Inhibition of ferroptosis alleviates atherosclerosis through attenuating lipid peroxidation and endothelial dysfunction in mouse aortic endothelial cell. Free Radic. Biol. Med. 160, 92–102. https://doi.org/10.1016/j.freeradbiomed.2020.07.026
Vinchi F., Porto G., Simmelbauer A., Altamura S., Passos S. T., Garbowski M., Silva A. M. N., Spaich S., Seide S.E., Sparla R., Hentze M.W., Muckenthaler M.U. (2020) Atherosclerosis is aggravated by iron overload and ameliorated by dietary and pharmacological iron restriction. Eur. Heart J. 41, 2681–2695. https://doi.org/10.1093/eurheartj/ehz112
Guo Z., Ran Q., Roberts L.J. 2nd, Zhou L., Richardson A., Sharan C., Wu D., Yang H. (2008) Suppression of atherogenesis by overexpression of glutathione peroxidase-4 in apolipoprotein E-deficient mice. Free Radic. Biol. Med. 44, 343–352. https://doi.org/10.1016/j.freeradbiomed.2007.09.009
Yang K., Song H., Yin D. (2021) PDSS2 inhibits the ferroptosis of vascular endothelial cells in atherosclerosis by activating Nrf2. J. Cardiovasc. Pharmacol. 77, 767–776. https://doi.org/10.1097/FJC.0000000000001030
He L., Liu Y.-Y., Wang K., Li C., Zhang W., Li Z.-Z., Huang X.-Z., Xiong Y. (2021) Tanshinone IIA protects human coronary artery endothelial cells from ferroptosis by activating the NRF2 pathway. Biochem. Biophys. Res. Commun. 575, 1–7. https://doi.org/10.1016/j.bbrc.2021.08.067
Meng N., Chen K., Wang Y., Hou J., Chu W., Xie S., Yang F., Sun C. (2022) Dihydrohomoplantagin and homoplantaginin, major flavonoid glycosides from Salvia plebeia R. Br. inhibit oxLDL-induced endothelial cell injury and restrict atherosclerosis via activating Nrf2 anti-oxidation signal pathway. Molecules. 27(6), 1990. https://doi.org/10.3390/molecules27061990
Zhang T., Hu Q., Shi L., Qin L., Zhang Q., Mi M. (2016) Equol attenuates atherosclerosis in apolipoprotein E-deficient mice by inhibiting endoplasmic reticulum stress via activation of Nrf2 in endothelial cells. PLoS One. 11(12), e0167020. https://doi.org/10.1371/journal.pone.0167020
Zhu Y., Zhang Y., Huang X., Xie Y., Qu Y., Long H., Gu N., Jiang W. (2019) Z-ligustilide protects vascular endothelial cells from oxidative stress and rescues high fat diet-induced atherosclerosis by activating multiple NRF2 downstream genes. Atherosclerosis. 284, 110–120. https://doi.org/10.1016/j.atherosclerosis.2019.02.010
Juan S.H., Lee T.S., Tseng K.W., Liou J.Y., Shyue S.K., Wu K.K., Chau L.Y. (2001) Adenovirus-mediated heme oxygenase-1 gene transfer inhibits the development of atherosclerosis in apolipoprotein E-deficient mice. Circulation. 104(13), 1519–1525. https://doi.org/10.1161/hc3801.095663
Sussan T.E., Jun J., Thimmulappa R., Bedja D., Antero M., Gabrielson K.L., Polotsky V.Y., Biswal S. (2008) Disruption of Nrf2, a key inducer of antioxidant defenses, attenuates ApoE-mediated atherosclerosis in mice. PLoS One. 3(11), e3791. https://doi.org/10.1371/journal.pone.0003791
Freigang S., Ampenberger F., Spohn G., Heer S., Shamshiev A.T., Kisielow J., Hersberger M., Yamamoto M., Bachmann M.F., Kopf M. (2011) Nrf2 is essential for cholesterol crystal-induced inflammasome activation and exacerbation of atherosclerosis. Eur. J. Immunol. 41(7), 2040–2051. https://doi.org/10.1002/eji.201041316
Barajas B., Che N., Yin F., Rowshanrad A., Orozco L.D., Gong K.W., Wang X., Castellani L.W., Reue K., Lusis A.J., Araujo J.A. (2011) NF-E2-related factor 2 promotes atherosclerosis by effects on plasma lipoproteins and cholesterol transport that overshadow antioxidant protection. Arterioscler. Thromb. Vasc. Biol. 31(1), 58–66. https://doi.org/10.1161/ATVBAHA.110.210906
Folli F., Corradi D., Fanti P., Davalli A., Paez A., Giaccari A., Perego C., Muscogiuri G. (2011) The role of oxidative stress in the pathogenesis of type 2 diabetes mellitus micro- and macrovascular complications: avenues for a mechanistic-based therapeutic approach. Curr. Diabetes Rev. 7(5), 313–324. https://doi.org/10.2174/157339911797415585
Nieuwdorp M., van Haeften T.W., Gouver-neur M.C.L.G., Mooij H.L., van Lieshout M.H.P., Levi M., Meijers J.C.M., Holleman F., Hoekstra J.B.L., Vink H., Kastelein J.J.P., Stroes E.S.G. (2006) Loss of endothelial glycocalyx during acute hyperglycemia coincides with endothelial dysfunction and coagulation activation in vivo. Diabetes. 55(2), 480–486. https://doi.org/10.2337/diabetes.55.02.06.db05-1103
Nobe K., Miyatake M., Sone T., Honda K. (2006) High-glucose-altered endothelial cell function involves both disruption of cell-to-cell connection and enhancement of force development. J. Pharmacol. Exp. Ther. 318(2), 530–539. https://doi.org/10.1124/jpet.106.105015
Baumgartner-Parzer S.M., Wagner L., Pettermann M., Grillari J., Gessl A., Waldhäusl W. (1995) High-glucose–triggered apoptosis in cultured endothelial cells. Diabetes. 44(11), 1323–1327. https://doi.org/10.2337/diab.44.11.1323
Du X.L., Edelstein D., Dimmeler S., Ju Q., Sui C., Brownlee M. (2001) Hyperglycemia inhibits endothelial nitric oxide synthase activity by posttranslational modification at the Akt site. J. Clin. Invest. 108(9), 1341–1348. https://doi.org/10.1172/JCI11235
Morigi M., Angioletti S., Imberti B., Donadelli R., Micheletti G., Figliuzzi M., Remuzzi A., Zoja C., Remuzzi G. (1998) Leukocyte-endothelial interaction is augmented by high glucose concentrations and hyperglycemia in a NF-kB-dependent fashion. J. Clin. Invest. 101(9), 1905–1915. https://doi.org/10.1172/JCI656
Okouchi M., Okayama N., Alexander J.S., Aw T.Y. (2006) NRF2-dependent glutamate-L-cysteine ligase catalytic subunit expression mediates insulin protection against hyperglycemia-induced brain endothelial cell apoptosis. Curr. Neurovasc. Res. 3(4), 249–261. https://doi.org/10.2174/156720206778792876
Yang M.-Y., Fan Z., Zhang Z., Fan J. (2021) MitoQ protects against high glucose-induced brain microvascular endothelial cells injury via the Nrf2/HO-1 pathway. J. Pharmacol. Sci. 145(1), 105–114. https://doi.org/10.1016/j.jphs.2020.10.007
Wang R.-Y. Liu L.-H., Liu H., Wu K.-F., An J., Wang Q., Liu E., Bai L.-J., Qi B.-M., Qi B.-L., Zhang L. (2018) Nrf2 protects against diabetic dysfunction of endothelial progenitor cells via regulating cell senescence. Int. J. Mol. Med. 42(3), 1327–1340. https://doi.org/10.3892/ijmm.2018.3727
Cheng X., Chapple S.J., Patel B., Puszyk W., Sugden D., Yin X., Mayr M., Siow R.C.M., Mann G.E. (2013) Gestational diabetes mellitus impairs Nrf2-mediated adaptive antioxidant defenses and redox signaling in fetal endothelial cells in utero. Diabetes. 62(12), 4088–4097. https://doi.org/10.2337/db13-0169
Chen X., Qi J., Wu Q., Jiang H., Wang J., Chen W., Mao A., Zhu M. (2020) High glucose inhibits vascular endothelial Keap1/Nrf2/ARE signal pathway via downregulation of monomethyltransferase SET8 expression. Acta Biochim. Biophys. Sin. 52(5), 506–516. https://doi.org/10.1093/abbs/gmaa023
Wu J., Jiang Z., Zhang H., Liang W., Huang W., Zhang H., Li Y., Wang Z., Wang J., Jia Y., Liu B., Wu H. (2018) Sodium butyrate attenuates diabetes-induced aortic endothelial dysfunction via P300-mediated transcriptional activation of Nrf2. Free Radic. Bio-l. Med. 124, 454–465. https://doi.org/10.1016/j.freeradbiomed.2018.06.034
Sun C.C., Lai Y.N., Wang W.H., Xu X.M., Li X.Q., Wang H., Zheng J.Y., Zheng J.Q. (2020) Metformin ameliorates gestational diabetes mellitus-induced endothelial dysfunction via downregulation of p65 and upregulation of Nrf2. Front. Pharmacol. 11, 575390. https://doi.org/10.3389/fphar.2020.575390
Wang F., Pu C., Zhou P., Wang P., Liang D., Wang Q., Hu Y., Li B., Hao X. (2015) Cinnamaldehyde prevents endothelial dysfunction induced by high glucose by activating Nrf2. Cell. Physiol. Biochem. 36(1), 315–324. https://doi.org/10.1159/000374074
Wang D., Hou J., Wan J., Yang Y., Liu S., Li X., Li W., Dai X., Zhou P., Liu W., Wang P. (2021) Dietary chlorogenic acid ameliorates oxidative stress and improves endothelial function in diabetic mice via Nrf2 activation. J. Int. Med. Res. 49(1), 300060520985363. https://doi.org/10.1177/0300060520985363
Verhamme P., Hoylaerts M.F. (2006) The pivotal role of the endothelium in haemostasis and thrombosis. Acta Clin. Belg. 61(5), 213–219. https://doi.org/10.1179/acb.2006.036
Lum H., Roebuck K.A. (2001) Oxidant stress and endothelial cell dysfunction. Am. J. Physiol. Cell Physiol. 280 (4), C719–C741. https://doi.org/10.1152/ajpcell.2001.280.4.C719
Yang S., Zheng Y., Hou X. (2019) Lipoxin A4 restores oxidative stress-induced vascular endothelial cell injury and thrombosis-related factor expression by its receptor-mediated activation of Nrf2-HO-1 axis. Cell. Signal. 60, 146–153. https://doi.org/10.1016/j.cellsig.2019.05.002
Akin-Bali D.F., Eroglu T., Ilk S., Egin Y., Kankilic T. (2020) Evaluation of the role of Nrf2/Keap1 pathway-associated novel mutations and gene expression on antioxidant status in patients with deep vein thrombosis. Exp. Ther. Med. 20(2), 868–881. https://doi.org/10.3892/etm.2020.8790
Li C.-Q., Wogan G.N. (2005) Nitric oxide as a modulator of apoptosis. Cancer Lett. 226(1), 1–15. https://doi.org/10.1016/j.canlet.2004.10.021
Um H.-C., Jang J.-H., Kim D.-H., Lee C., Surh Y.-J. (2011) Nitric oxide activates Nrf2 through S-nitrosylation of Keap1 in PC12 cells. Nitric Oxide. 25(2), 161–168. https://doi.org/10.1016/j.niox.2011.06.001
Franceschi C., Campisi J. (2014) Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J. Gerontol. A Biol. Sci. Med. Sci. 69(Suppl. 1), S4–S9. https://doi.org/10.1093/gerona/glu057
Guarner V., Rubio-Ruiz M.E. (2015) Low-grade systemic inflammation connects aging, metabolic syndrome and cardiovascular disease. Interdiscip. Top. Gerontol. 40, 99–106. https://doi.org/10.1159/000364934
Csiszar A., Ungvari Z., Edwards J.G., Kaminski P., Wolin M.S., Koller A., Kaley G. (2002) Aging-induced phenotypic changes and oxidative stress impair coronary arteriolar function. Circ. Res. 90(11), 1159–1166. https://doi.org/10.1161/01.res.0000020401.61826.ea
Ungvari Z., Tarantini S., Donato A.J., Galvan V., Csiszar A. (2018) Mechanisms of vascular aging. Circ. Res. 123(7), 849–867. https://doi.org/10.1161/CIRCRESAHA.118.311378
Csiszar A., Ungvari Z., Koller A., Edwards J.G., Kaley G. (2004) Proinflammatory phenotype of coronary arteries promotes endothelial apoptosis in aging. Physiol. Genomics. 17, 21–30. https://doi.org/10.1152/physiolgenomics.00136.2003
Ungvari Z., Bailey-Downs L., Sosnowska D., Gautam T., Koncz P., Losonczy G., Ballabh P., de Cabo R., Sonntag W.E., Csiszar A. (2011) Vascular oxidative stress in aging: a homeostatic failure due to dysregulation of NRF2-mediated antioxidant response. Am. J. Physiol. Heart Circ. Physiol. 301(2), H363–H372. https://doi.org/10.1152/ajpheart.01134.2010
Chapple S.J., Siow R.C.M., Mann G.E. (2012) Crosstalk between Nrf2 and the proteasome: therapeutic potential of Nrf2 inducers in vascular disease and aging. Int. J. Biochem. Cell Biol. 44(8), 1315–1320. https://doi.org/10.1016/j.biocel.2012.04.021
Kloska D., Kopacz A., Piechota-Polanczyk A., Nowak W.N., Dulak J., Jozkowicz A., Grochot-Przeczek A. (2019) Nrf2 in aging – Focus on the cardiovascular system. Vascul. Pharmacol. 112, 42–53. https://doi.org/10.1016/j.vph.2018.08.009
Valcarcel-Ares M.N., Gautam T., Warrington J.P., Bailey-Downs L., Sosnowska D., de Cabo R., Losonczy G., Sonntag W.E., Ungvari Z., Csiszar A. (2012) Disruption of Nrf2 signaling impairs angiogenic capacity of endothelial cells: implications for microvascular aging. J. Gerontol. A Biol. Sci. Med. Sci. 67(8), 821–829. https://doi.org/10.1093/gerona/glr229
van Deursen J.M. (2014) The role of senescent cells in ageing. Nature. 509, 439–446. https://doi.org/10.1038/nature13193
Fulop G.A., Kiss T., Tarantini S., Balasubramanian P., Yabluchanskiy A., Farkas E., Bari F., Ungvari Z., Csiszar A. (2018) Nrf2 deficiency in aged mice exacerbates cellular senescence promoting cerebrovascular inflammation. Geroscience. 40, 513–521. https://doi.org/10.1007/s11357-018-0047-6
Romero A., San Hipólito-Luengo Á., Villalobos L.A., Vallejo S., Valencia I., Michalska P., Pajuelo-Lozano N., Sánchez-Pérez I., León R., Bartha J.L., Sanz M.J., Erusalimsky J.D., Sánchez-Ferrer C.F., Romacho T., Peiró C. (2019) The angiotensin-(1-7)/Mas receptor axis protects from endothelial cell senescence via klotho and Nrf2 activation. Aging Cell. 18(3), e12913. https://doi.org/10.1111/acel.12913
Arefin S., Buchanan S., Hobson S., Steinmetz J., Alsalhi S., Shiels P.G., Kublickiene K., Stenvinkel P. (2020) Nrf2 in early vascular ageing: calcification, senescence and therapy. Clin. Chim. Acta. 505, 108–118. https://doi.org/10.1016/j.cca.2020.02.026
Zinovkin R.A., Kondratenko N.D., Zinovkina L.A. (2022) Does Nrf2 play a role of a master regulator of mammalian aging? Biochemistry. 87, 1465–1476. https://doi.org/10.1134/S0006297922120045
Pillai R., Hayashi M., Zavitsanou A.-M., Papagiannakopoulos T. (2022) NRF2: KEAPing tumors protected. Cancer Discov. 12(3), 625–643. https://doi.org/10.1158/2159-8290.CD-21-0922
Wu S., Lu H., Bai Y. (2019) Nrf2 in cancers: a double-edged sword. Cancer Med. 8(5), 2252–2267. https://doi.org/10.1002/cam4.2101
Rojo de la Vega M., Chapman E., Zhang D.D. (2018) NRF2 and the hallmarks of cancer. Cancer Cell. 34(1), 21–43. https://doi.org/10.1016/j.ccell.2018.03.022
Wang Y.-Y., Chen J., Liu X.-M., Zhao R., Zhe H. (2018) Nrf2-mediated metabolic reprogramming in cancer. Oxid. Med. Cell. Longev. 2018, 9304091. https://doi.org/10.1155/2018/9304091
Ji X., Wang H., Zhu J., Zhu L., Pan H., Li W., Zhou Y., Cong Z., Yan F., Chen S. (2014) Knockdown of Nrf2 suppresses glioblastoma angiogenesis by inhibiting hypoxia-induced activation of HIF-1α. Int. J. Cancer. 135(3), 574–584. https://doi.org/10.1002/ijc.28699
Toth R.K., Warfel N.A. (2017) Strange bedfellows: nuclear factor, erythroid 2-like 2 (Nrf2) and hypoxia-inducible factor 1 (HIF-1) in tumor hypoxia. Antioxidants (Basel). 6(2), 27. https://doi.org/10.3390/antiox6020027
Liu C., Vojnovic D., Kochevar I.E., Jurkunas U.V. (2016) UV-A irradiation activates Nrf2-regulated antioxidant defense and induces p53/caspase3-dependent apoptosis in corneal endothelial cells. Invest. Ophthalmol. Vis. Sci. 57, 2319–2327. https://doi.org/10.1167/iovs.16-19097
Chen X.-L., Varner, S.E., Rao, A.S., Grey, J.Y., Thomas, S., Cook, C.K., Wasserman M.A., Medford R.M., Jaiswal A.K., Kunsch C. (2003) Laminar flow induction of antioxidant response element-mediated genes in endothelial cells: a novel anti-inflammatory mechanism. J. Biol. Chem. 278(2), 703–711. https://doi.org/10.1074/jbc.M203161200
Wei Y., Gong J., Thimmulappa R.K., Kosmider B., Biswal S., Duh E.J. (2013) Nrf2 acts cell-autonomously in endothelium to regulate tip cell formation and vascular branching. Proc. Natl. Acad. Sci. USA. 110(41), E3910–E3918. https://doi.org/10.1073/pnas.1309276110
Дополнительные материалы отсутствуют.
Инструменты
Молекулярная биология