

Российские нанотехнологии, 2021, T. 16, № 2, стр. 232-243
ИССЛЕДОВАНИЕ ВОЗДЕЙСТВИЯ ВНЕШНИХ ФАКТОРОВ НА СТРУКТУРНО-ДИНАМИЧЕСКИЕ ПАРАМЕТРЫ МАТЕРИАЛОВ ПЛЕНОЧНОГО ТИПА НА ОСНОВЕ ПОЛИ-3-ГИДРОКСИБУТИРАТА И ПОЛИЛАКТИДА
С. Г. Карпова 1, *, А. А. Ольхов 1, 2, 3, А. А. Попов 1, 2, А. Л. Жулькина 3, Р. Ю. Косенко 3, А. Л. Иорданский 3
1 Институт биохимической физики им. Н.М. Эмануэля РАН
Москва, Россия
2 Российский экономический университет им. Г.В. Плеханова
Москва, Россия
3 Институт химической физики им. Н.Н. Семенова РАН
Москва, Россия
* E-mail: karpova@sky.chph.ras.ru
Поступила в редакцию 08.07.2020
После доработки 02.09.2020
Принята к публикации 08.09.2020
Аннотация
Исследована структура пленочных и волокнистых материалов на основе смесей поли-3-гидроксибутирата и полилактида. Ультратонкие волокна получены методом электроформования полимерного раствора, пленки изготовлены методом полива на стеклянную подложку. С помощью оптической микроскопии показано, что волокна в зависимости от состава имеют различный диаметр и геометрию. Зондовым методом электронного парамагнитного резонанса и методом дифференциальной сканирующей калориметрии исследованы особенности кристаллической и аморфной структуры композиций. Добавление 10, 30% полилактида в композицию приводит к замедлению молекулярной подвижности в пленочном и волокнистом материале, а также росту энтальпии плавления ультратонких волокон. При 30% полилактида в смеси наблюдается излом на зависимостях: энтальпия плавления, время корреляции, концентрация радикала. Отжиг при температуре 140°С и экспозиция в водной среде при 25 и 70°С в течение 3 ч оказывают сильный эффект на структуру полимеров: растет энтальпия плавления, молекулярная динамика, снижается концентрация радикала в ультратонких волокнах и пленочном материале. Озонолиз сложным образом оказывает влияние на структуру ультратонких волокон.
ВВЕДЕНИЕ
Проблема утилизации полимерных отходов остро стоит во всем мире [1]. Основным источником отходов на основе синтетических полимеров, практически не разрушающихся в условиях компостирования, является упаковочная индустрия. Чтобы заменить традиционные синтетические полимеры на биодеградируемые, в ведущих научных организациях мира создают новые биополимеры, разрабатывают смесевые и композиционные полимерные материалы на основе природных полимеров, дисперсных наполнителей [2, 3]. На основе таких материалов с применением различных технологических методов создаются современные полностью биоразлагаемые упаковочные материалы с требуемым уровнем газо- и паропроницаемости, антисептическими и другими функциональными свойствами [4]. Полигидроксиалканоаты, полилактид, их сополимеры – биополимеры синтетического и природного происхождения – являются одними из главных кандидатов в качестве основы для создания инновационных типов биополимерных упаковок и тары [5]. Эти материалы способны разрушаться в окружающей среде на экологически безопасные продукты от месяца до года. Важной особенностью этих полимеров, а также изделий из них является биодеградация их макромолекулярной цепи по механизму гидролиза или ферментативного гидролиза, где конечными продуктами распада выступают безопасные для организма продукты: углекислый газ и вода [6]. Для ускорения процессов диффузии, гидролитической деструкции и биодеградации упаковочных материалов пленочного типа необходимо увеличение удельной площади поверхности полимерной матрицы. С этой точки зрения пленочные системы уступают пористым матрицам или нетканым волокнистым материалам на основе нано- и ультратонких полимерных волокон.
Упаковочные материалы, одноразовые фильтры, скаффолды и другие продукты на основе нетканых волокнистых биополимерных материалов, получаемых методом электроформования, являются наиболее перспективными и изучаемыми объектами. Их эффективность обусловлена, с одной стороны, простой экономичностью технологии изготовления волокон, а с другой, возможностью варьирования диаметра волокна в широком диапазоне (от 100 нм до 12 мкм) [7].
На процессы биодеградации, окисления и гидролиза существенно влияют не только морфология поверхности пленочного или волокнистого материала, но и структура аморфной и кристаллической фаз [8]. Скорость этих процессов возрастает, если структура волокон является гетерогенной. Наиболее гетерогенной структурой обладают смеси полимеров, которые, как правило, являются термодинамически несовместимыми [9]. Полимеры в смесях оказывают взаимное влияние на процессы кристаллизации, стеклования, приводя к увеличению диффузионно-транспортных характеристик [10], ускорению кинетики биодеградации, окисления и гидролиза композитных пленок и волокон [11]. В [12, 13] представлены результаты предварительных исследований влияния воды, УФ-излучения и термического воздействия на динамические характеристики и кристаллическую структуру волокнистых материалов на основе смесей природный поли-3-гидроксибутират (ПГБ) и полилактид (ПЛА).
Поскольку различные технологические приемы формования образцов на основе полимерных растворов и характеристики исходных полимеров оказывают существенное влияние на комплекс структурно-динамических характеристик, в настоящей работе предпринята попытка провести сравнительный анализ изменения структуры пленочных и нетканых волокнистых материалов на основе смесей ПГБ и ПЛА при воздействии некоторых факторов окружающей среды: воды, температуры и озона.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
В работе использовали ПГБ серии 16F (продукт микробиологического синтеза компании “BIOMER®” (Германия)) со средневязкостной молекулярной массой 4.6 × 105 и ПЛА марки “NatureWorks® Ingeo™ 3801X Injection Grade PLA” (“SONGHAN Plastics Technology Co., Ltd.”) со средневязкостной молекулярной массой 1.9 × 105. Смесевые пленки готовили по методике полива раствора полимеров в хлороформе на стеклянную подложку. Полимерные растворы для формования пленок и волокон готовили на основе следующих соотношений ПГБ/ПЛА: 1/0, 1/9, 3/7, 5/5, 0/1. Общая концентрация полимеров в растворе составляла 7 мас. %. Для улучшения растворимости ПГБ полимеры спрессовывали в пленки при 180°С в течение 30 с, затем охлаждали и помещали в растворитель СНСl3. Приготовление смесевых растворов ПГБ/ПЛА в различных соотношениях для электроформования и отлива пленок проводили в растворителе СНСl3. Расчетное количество ПЛА (в зависимости от состава) растворяли и оставляли на сутки. Полученный раствор доводили до кипения и постепенно при интенсивном перемешивании вносили необходимое количество ПГБ до полного растворения. Часть раствора шла для электроформования, часть для отлива пленок. Раствор заливали в чашки Петри и оставляли при комнатной температуре до полного испарения растворителя. Перемешивание осуществляли с помощью автоматической магнитной мешалки при температуре 50°С.
Спектры электронного парамагнитного резонанса (ЭПР) (Х-диапазона) регистрировали на автоматизированном спектрометре ЭПР-В (Институт химической физики РАН). Значение микроволновой мощности в резонаторе не превышало 7 мВт, что позволяло избежать эффектов насыщения. При записи спектров амплитуда модуляции всегда была существенно меньше ширины резонансной линии и не превышала 0.5 Гс.
Зондом служил стабильный нитроксильный радикал ТЕМПО. Его вводили в волокна из паров при температуре 70°C. Концентрация радикала в полимере не превышала 10–3 моль/л. Экспериментальные спектры спинового зонда в области медленных движений (время корреляции вращения зонда τ > 10–9 с) анализировали в рамках модели изотропного броуновского вращения по программе, описанной в [14]. При моделировании спектров использовали следующие значения g-тензора и тензора сверхтонкого взаимодействия радикала: gxx = 2.0096, gyy = 2.0066, gzz = = 2.0025, Ахх = 7.0, Ayy = 5.0, Azz = 35.0 Гc. Отметим, что величину Azz определяли экспериментально из ЭПР-спектров нитроксильного радикала в полимере при 77 К; она не сильно отличалась от значения, приведенного в [15]. Времена корреляции вращения зонда τ в области быстрых вращений (5 × 10–11 < τ < 10–9 с) находили из спектров ЭПР по формуле [16]:
(1)
$\tau = \Delta {{Н}_{ + }}[{{({{I}_{ + }}{\text{/}}{{I}_{--}})}^{{0.5}}}--1]\,6.65{\text{ }} \times {{10}^{{--10}}},$Морфологию образцов нетканых материалов изучали на оптическом микроскопе “Micromed polar 3 ToupCam 5.1 MP” (микроскоп поляризационный проходящего и отраженного света, Россия). Микрофотографии обрабатывали с помощью стандартного пакета программ.
Исследование образцов методом дифференциальной сканирующей калориметрии (ДСК) проводили на приборе DSC Q-20 фирмы “TA Instruments” (США) в атмосфере азота при скорости нагревания 10 град/мин. Среднестатистическая ошибка измерения тепловых эффектов приблизительно составляла ±3%.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Геометрия волокна ПГБ–ПЛА. Как демонстрируют данные электронной микроскопии (рис. 1а–1в), нетканые волокнистые материалы ПГБ–ПЛА имеют гетерогенную структуру и состоят из волокон со множественными утолщениями. Гетерогенность растет с увеличением концентрации ПГБ в смесях. Диаметр волокон колеблется от 3 до 9 мкм. Утолщения имеют произвольную геометрию и протяженность от 10 до 50 мкм. В результате возникновения такой гетерогенной структуры рассматриваемые нетканые волокнистые материалы обладают отличной друг от друга пространственной упорядоченностью волокон и плотностью упаковки. Наличие в материалах волокон различного диаметра и геометрии является причиной их плотной укладки (упаковки) в процессе формирования нетканого материала.
Рис. 1.
Микрофотографии поверхности материалов ПГБ/ПЛА (%): волокнистые: а – 70/30, б – 50/50, в – 10/90; пленочные: г – 70/30, д – 50/50, е – 10/90.

На рис. 1 с ростом содержания ПЛА количество и размеры утолщений на волокнах уменьшаются. Вероятно, данный эффект обусловлен уменьшением вязкости полимерного раствора. При этом средний диаметр волокон практически не меняется. Отметим, что волокнистые материалы с преобладанием ПЛА имеют сравнительно небольшое количество узлов зацеплений и характеризуются относительно упорядоченной укладкой волокон. По-видимому, ПЛА имеет более сильное сродство с используемым растворителем хлороформом, чем ПГБ, что приводит к снижению скорости десорбции растворителя из образующихся волокон и, как следствие, возникновению локальных утолщений. В процессе формования визуально фиксировали, что из формовочного раствора ПЛА хлороформ испарялся медленнее, чем из раствора ПГБ. При рассмотрении поверхности пленочных материалов (рис. 1г–1е) можно заметить уменьшение шероховатости пленок с ростом концентрации ПЛА, что достаточно хорошо согласуется с морфологией смесевых нетканых волокнистых материалов.
Изменяющаяся гетерогенность рассматриваемых материалов, связанная с термодинамической несовместимостью смешиваемых полимеров, может привести к формированию кристаллических образований с различными геометрическими параметрами и морфологией. Далее подробно изучим кристаллическую и аморфную фазы волокнистых и пленочных материалов с применением структурно-динамических методов анализа.
Термограммы ДСК ультратонких волокон и пленок ПГБ–ПЛА. Взаимодействие макромолекул полимера с растворителем определяется его природой. Одной из основных причин структурных различий материалов, получаемых переработкой через раствор, на макро- и микроуровнях является различие в термодинамическом сродстве между растворителем и растворенным веществом [17, 18]. Важнейшими условиями совмещения полимеров можно назвать межмолекулярные взаимодействия между ними, а также между полимерами и молекулами растворителя, что определяет способность полимерных цепей к конформационным переходам. Это влечет за собой изменение комплекса свойств полимеров.
В “хорошем” растворителе, когда взаимодействие макромолекул полимера с молекулами растворителя преобладает над взаимодействием между макромолекулами полимеров, реализуется возможность макромолекул принять равновесное состояние. Влияние растворителя на эффект совместимости и диапазон температуры стеклования обусловлен двумя основными факторами – термодинамическим качеством растворителя и его летучестью. В [19] показано, что в хорошем растворителе имеют место высокая совместимость полимеров (сильный межмолекулярный контакт между макромолекулами) и, как результат, наиболее низкая молекулярная подвижность цепей полимеров. Разный способ получения полимерного материала влияет на его структуру и свойства. При формовании волокон летучесть растворителя интенсивнее, чем при получении пленок, поэтому этот фактор также может повлиять на структуру полимера, что и будет показано далее.
На основании работ последних лет показано, что если между полимерами имеет место образование водородных связей или электронодонорно-акцепторных комплексов, можно создавать термодинамически совместимые системы [20, 21]. Известно, что при смешении ПГБ и ПЛА образуются водородные связи, что дает основание полагать о совместимости данных полимеров. Наличие водородных связей между компонентами было обнаружено в образцах на основе систем ПГБ–полиамид [22] и ПГБ–полиуретан [23]. При смешении ПГБ и ПЛА наряду с изменением морфологии ультратонких волокон следует ожидать изменения их теплофизических и структурных характеристик.
Для изучения теплофизических характеристик волокон использовали метод ДСК. В табл. 1 представлены данные энтальпии плавления (∆Нэндо-, ∆Нэкзо-процессы), температуры эндо-, экзопроцессов, температуры стеклования. Получены также данные энтальпии и температуры кристаллизации пленочного и волокнистого материалов ПГБ/ПЛА.
Таблица 1.
Экспозиция в водной среде в течение 3 ч. Энтальпия плавления (∆Нэндо, ∆Нэкзо), температура плавления (Тэндо, Тэкзо), стеклования (Тст) волокон и пленок смесей ПГБ с полилактидом, исследованных методом ДСК
| ПЛА 100% |
ПЛА/ПГБ 90/10% |
ПЛА/ПГБ 70/30% |
ПЛА/ПГБ 50/50% |
ПЛА/ПГБ 30/70% |
ПЛА/ПГБ 10/90% |
ПЛА | ||
|---|---|---|---|---|---|---|---|---|
| Пленки исходные |
Тст | |||||||
| ∆Нэндо | 26.4 | 32.6 | 48.2 | 52.7 | 67.5 | 77 | 88.8 | |
| Тэндо | 170 | 170 | 170.4 | 175 | 179 | 178.5 | 178.6 | |
| ∆Нэкзо | 14 | |||||||
| Тэкзо | 85 | |||||||
| 25°С | Тст | |||||||
| ∆Нэндо | 28 | 34 | 49 | 53.7 | 70.3 | 80 | 90.7 | |
| Тэндо | 170.4 | 171 | 171 | 175.5 | 177.3 | 178 | 178 | |
| ∆Нэкзо | 4.4 | 7.8 | ||||||
| Тэкзо | 83.2 | 92 | ||||||
| 70°С | Тст | |||||||
| ∆Нэндо | 27 | 40.14 | 53 | 61,8 | 71.2 | 81 | 87.3 | |
| Тэндо | 170 | 169.7 | 170 | 177 | 177 | 177 | 177.6 | |
| ∆Нэкзо | ||||||||
| Тэкзо | ||||||||
| Волокна исходные | Тст | 64 | 62.5 | 62 | 58 | |||
| ∆Нэндо | 31.5 | 30.3 | 43.5 | 52.3 | 83.8 | 80.6 | 80 | |
| Тэндо | 168.4 | 167.9 | 168 | 174.5 | 177 | 175 | 175.7 | |
| ∆Нэкзо | 25 | 21.2 | 13.6 | 9 | ||||
| Тэкзо | 102 | 95.4 | 95.5 | 92 | ||||
| 25°С | Тст | 61.2 | ||||||
| ∆Нэндо | 33.6 | 34 | 34.8 | 56.2 | 84 | 78 | 80 | |
| Тэндо | 167.3 | 167 | 168 | 167.9 | 177 | 177.7 | 177.4 | |
| ∆Нэкзо | ||||||||
| Тэкзо | ||||||||
| 70°С | Тст | |||||||
| ∆Нэндо | 38.5 | 45 | 34.6 | 45.4 | 82.3 | 80 | 81.4 | |
| Тэндо | 167.6 | 168 | 168 | 169 | 177 | 177 | 176 | |
| ∆Нэкзо | ||||||||
| Тэкзо |
Рассмотрим изменения энтальпии плавления (ΔН) в волокнистом материале. Характерной особенностью для данного материала является очень низкое значение ΔН (9 Дж/г) в ПЛА и достаточно высокое в волокнах ПГБ (80 Дж/г). В ПЛА и композициях ПЛА/ПГБ 90/10, 70/30 и 50/50% термограмма плавления волокна является бимодальной с экзо- и эндопиками (табл. 1). В волокнах при добавлении ПЛА к ПГБ Тст уменьшается от 64 до 58°С и отсутствует в волокнах с 30, 10% ПЛА и в ПГБ, что свидетельствует о межмолекулярном взаимодействии между полимерами. На рис. 2а представлены изменения энтальпии плавления ΔН для смесевых композиций ультратонких волокон ПГБ/ПЛА. При добавлении ПЛА к ПГБ до 30% (включительно) ΔН незначительно возрастает, при более высокой концентрации ПЛА в смеси происходит резкое снижение энтальпии плавления. Особенностью изучаемых композиций является наличие экзопиков при Т = 92–101°С при плавлении волокнистого материала, что свидетельствует о процессах докристаллизации (и образовании паракристаллических структур) и, как следствие, о высокой степени выпрямленности цепей в ПЛА. Так как значения температуры плавления ПГБ и ПЛА близки, приведена суммарная энтальпия плавления системы.
Рис. 2.
Зависимость энтальпии плавления ∆Н для волокнистого (а) и пленочного материала (б) от его состава: 1 – исходные образцы, 2 – после экспозиции в течение 3 ч при Т = 25°С, 3 – при Т = 70°С.
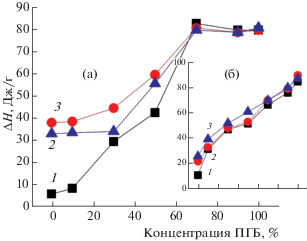
Причина кристаллизации системы при нагревании – конформационный критерий k* = h/L (где h – среднее расстояние между концами цепей, а L – контурная длина цепи), определяющий тенденцию макромолекулы к распрямлению и доориентации или к сворачиванию в клубок [24]. Для каждого полимера своя критическая величина k*. Цепи, у которых k > k*, стремятся к доориентации, макромолекулы, у которых k < k*, стремятся принять конформацию клубка. В процессе получения волокон формируется определенная доля выпрямленных макромолекул. Нагрев полимера облегчает процесс распрямления цепей, у которых k > k*.
Зависимость температуры плавления системы от содержания компонент также претерпевает излом при содержании ПЛА 30 и 70% в смеси (табл. 1). Наблюдаемые закономерности могут быть объяснены следующими факторами. Можно полагать, что цепь ПЛА (большая ее часть) находится в достаточно выпрямленной конформации, так как при первом сканировании определенная ее часть кристаллизуется при Т = 92–102°С (возможно, образуются паракристаллические структуры) (табл. 1). При добавлении невысокой доли ПЛА к ПГБ, при наличии достаточно выпрямленной конформации и сильного межмолекулярного взаимодействия с цепями ПГБ образуются паракристаллические структуры, что способствует повышению энтальпии смеси. При более высоком содержании ПЛА в смеси (более 30%), по-видимому, имеют место изолированные структуры ПЛА и ПГБ, причем ПЛА находится в стеклообразном состоянии с низкой энтальпией плавления. Наблюдается практически линейное уменьшение ΔН (рис. 2а).
Рассмотрим изменение энтальпии плавления в пленках ПЛА/ПГБ. На рис. 2б представлены данные изменения ΔН в смесевых композициях пленок (бимодальный характер плавления с экзо- и эндопиками имеет место только в пленках ПЛА), эта зависимость имеет нелинейный характер и при содержании 30, 50 и 10% ПЛА имеют место изломы. Сопоставляя данные энтальпии плавления для пленочного материала и волокон, видим, что практически для всех составов (исключая состав 30/70% ПЛА/ПГБ в волокнах) ΔН имеет более высокие значения. Структура пленочного материала находится в высокоэластическом состоянии для всех изучаемых составов. При первом сканировании в противоположность волокнистому материалу дополнительной кристаллизации не происходит (за исключением ПЛА). Можно полагать, что структура пленочного материала находится в достаточно равновесном состоянии по сравнению с волокнистым вследствие медленного испарения растворителя с высокой долей выпрямленных цепей и, как результат, ПЛА находится в высокоэластическом состоянии с более высокой энтальпией плавления.
В области 30/70% ПЛА/ПГБ в пленках, так же как и в волокнах, наблюдается излом на зависимостях Тпл и ΔН от состава полимера (табл. 1). Отметим, что как в пленочном, так и в волокнистом материале ПЛА и смеси ПЛА/ПГБ 90/10 и 80/20% в цикле охлаждения полимера отсутствует пик кристаллизации, что свидетельствует о высокой вязкости систем из-за высокой доли выпрямленных цепей.
Характерной особенностью изучаемых систем являются наличие стеклования и достаточно низкая энтальпия плавления при втором сканировании для всех изучаемых полимеров (как пленок, так и волокон, за исключением ПГБ), что указывает на определяющую роль не только растворителя, но и метода получения полимера на его структуру. Так, если в пленке ПЛА до плавления ΔН составляла 12.3 Дж/г, то при втором сканировании эта величина составляет только 3 Дж/г, в ПЛА/ПГБ такие изменения происходят от 32 до 2.5 Дж/г.
Абсорбция воды в волокнах ПЛА/ПГБ представляет самостоятельный интерес как при рассмотрении кинетики гидролитических реакций, где основным реагентом является вода, так и при описании физико-химических явлений, наблюдаемых на поверхности и в объеме волокна и пленки. Поскольку биологические объекты, как правило, контактируют с водной средой при комнатной и повышенных значениях температуры, исследовали влияние продолжительности экспозиции в дистиллированной водной среде при 25 и 70°С на кристалличность ультратонких волокон и пленочного материала ПЛА/ПГБ.
Волокна ПГБ имеют неравновесную структуру, кристаллиты и аморфные области в них в общем случае не отвечают минимуму свободной энергии, что приводит к незавершенности процесса кристаллизации. Стремление аморфно-кристаллической системы к минимуму свободной энергии реализуется, когда макромолекулы получают достаточную подвижность.
По классификации [25] ПГБ относится к классу полимеров с умеренной гидрофобностью, т.е. его растворимость в воде близка к растворимости гидрофобных пластиков, таких как синтетические полиэфиры. Полилактид относится к классу полимеров с высокой гидрофильностью.
Продолжительность экспозиции в дистиллированной водной среде при 25 и 70°С в течение 3 ч оказывает значительное влияние на кристалличность ультратонких волокон и пленочного материала ПЛА/ПГБ.
Рассмотрим зависимость изменения ΔН при экспозиции в водной среде ультратонких волокон от состава смеси. Характерно, что термограмма плавления всех образцов имеет только эндопик, стеклование полимера отсутствует (за исключением ПЛА при экспозиции 25°С). На рис. 2а представлены зависимости ΔН от состава смеси при экспозиции в водной среде в течение 3 ч при Т = 25 и 70°С. В композициях с концентрацией ПЛА до 30% включительно энтальпия плавления после водного воздействия возрастает незначительно, в то время как в смесях с высоким содержанием ПЛА эти изменения значительно больше. После экспозиции в воде при 70°С все смесевые композиции и исходные полимеры находятся в высокоэластическом состоянии. Можно полагать, что диффузия воды, снижая межмолекулярное взаимодействие в ПЛА и смесевых композициях ПЛА/ПГБ, обусловливает распрямление макромолекул и поэтому увеличивается доля паракристаллических структур в полимере. Из кинетической теории кристаллизации известно, что размер кристаллитов L при изотермическом отжиге зависит от времени отжига по закону L ∼ Вlogt (t – время отжига, В – коэффициент, характеризующий температуру отжига). Отметим, что наиболее сильные изменения ΔН от температуры отжига в водной среде имеют место в ПЛА (гидрофильный полимер).
Таким образом, экспозиция изучаемых систем в воде пластифицирует структуру, способствует их переходу в равновесное высокоэластическое состояние с ростом доли кристаллических и паракристаллических структур.
Рассмотрим влияние экспозиции в воде в течение 3 ч на пленочный материал. Из данных табл. 1 и рис. 2б видно, что отжиг в воде при 25°С приводит к небольшому росту энтальпии плавления, оставляя характер зависимости ΔН от состава смеси неизменным. Экспозиция в воде пленочного материала при 70°С обусловливает практически линейную зависимость энтальпии плавления от состава композиции. Наиболее значительные изменения ΔН, так же как и в волокнах, после водного воздействия при 25 и 70°С наблюдаются в ПЛА. Для всех изучаемых систем как в исходном состоянии, так и после экспозиции в водной среде в процессе охлаждения (цикл нагрев–охлаждение) отсутствуют пики кристаллизации в образцах ПЛА, 90/10, 80/20 ПЛА/ПГБ, что свидетельствует о высокой вязкости системы вследствие большой доли выпрямленности цепей. При втором сканировании на термограммах всех образцов (исключая ПГБ) имеет место стеклование полимера.
Результаты исследований особенностей геометрической структуры волокон и их термофизического поведения позволяют полагать, что при смешении ПГБ и ПЛА возникает сложная гетерогенная структура. Волокна ПГБ и ПЛА имеют неравновесную структуру, кристаллиты и аморфные области в них в общем случае не отвечают минимуму свободной энергии, что приводит к незавершенности процесса кристаллизации. Стремление аморфно-кристаллической системы к минимуму свободной энергии реализуется, когда макромолекулы получают достаточную подвижность. Видно, что термическая обработка в водной среде переводит кристаллическую фазу ультратонких волокон и пленочного материала в стабильное состояние с ростом энтальпии плавления и переходом полимера в высокоэластическое состояние. Смещение Тст в волокнах с уменьшением содержания ПЛА (до 50% включительно) и отсутствие стеклования при более высоком содержании ПГБ в системе, а также после экспозиции в водной среде для всех изучаемых композиций указывают на достаточно сильное пластифицирующее действие воды и взаимодействие между ПГБ и ПЛА. Как результат, растет доля выпрямленных цепей в полимере.
Важно отметить, что данные по энтальпии плавления, полученные методом ДСК, дают информацию как о доле кристаллической фазы, так и о паракристаллических структурах (структуры из выпрямленных цепей с двумерным порядком) в волокне и пленке. Ранее были получены данные о влиянии экспозиции в водной среде ультратонких волокон ПГБ, содержащих комплекс марганец–хлор тетрафенилпорфирин, тетрафенилпорфирин [26, 27].
Динамические характеристики аморфной фазы волокон и пленок ПЛА/ПГБ. Структура аморфных областей в значительной степени определяется долей кристаллических и паракристаллических образований. При добавлении к ПГБ полилактида растет доля кристаллитов и паракристаллических структур, что приводит к изменению структурного и динамического состояния аморфных областей. Молекулярную динамику этих областей удобно исследовать методом ЭПР с использованием стабильных радикалов. ЭПР-спектры радикала ТЕМПО в матрицах ультратонких волокон ПЛА и ПГБ имеют сложный вид и представляют собой суперпозицию двух спектров, соответствующих двум популяциям радикалов с временами корреляции τ1 и τ2, характеризующих молекулярную подвижность в плотных и менее плотных аморфных областях соответственно (рис. 3). В работе выполнен расчет времени корреляции τ2 по спектрам ЭПР (5 × 10–11 < τ2 < 10–9 с). С использованием специальной программы NLSL аналогично процедуре, описанной в [14, 15], рассчитаны времена корреляции в плотных областях путем математической обработки спектров.
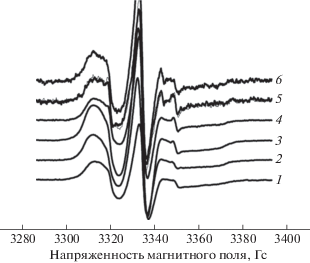
Рис. 3.
Экспериментальные спектры ЭПР нитроксильного радикала ТЕМПО пленочного материала: 1 – 0, 2 – 10/90, 3 – 30/70, 4 – 50/50, 5 – 70/30, 6 – 10/90, 7 – 100% ПЛА/ПГБ.
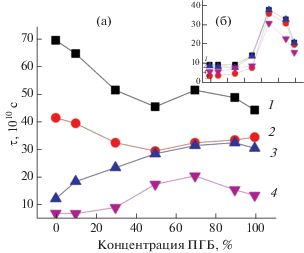
Рассмотрим динамику молекулярных движений, рассчитанную по спектрам ЭПР, в ультратонких волокнах ПЛА/ПГБ. Зависимость τ2 от состава смесевой композиции является нелинейной (рис. 4а). Отметим, что радикал в полимер вводили при Т ≈ 70°С, затем резко охлаждали (Тст ≈ 64°С). Так, если τ2 в ПЛА составляет около 73 × 10–10 с, то в ПГБ – 44 × 10–10, что свидетельствует о более высокой подвижности цепей в ПГБ. На рис. 4а видно, что добавление ПГБ вплоть до 50% в смесевую композицию приводит к снижению τ2, что подтверждает рост молекулярной подвижности. Добавление ПЛА до 30% включительно к ПГБ обусловливает рост времени корреляции. Именно в этом диапазоне концентраций ПЛА в смеси, как отмечалось ранее, наблюдается рост энтальпии плавления этих композиций. Как следствие, жесткость цепей возрастает, замедляется их подвижность. При концентрации ПЛА больше 50% τ2 растет, так как жесткость цепей ПЛА выше, чем в ПГБ.
Рис. 4.
Зависимость времени корреляции от состава волокнистого (а) и пленочного материала (б): 1 – исходные образцы, 2 – после экспозиции в течение 3 ч при Т = 25°С, 3 – при Т = 70°С, 4 – после отжига при 140°С.
Рассчитана равновесная концентрация радикала, адсорбированного в одинаковых по массе образцах волокон ПЛА/ПГБ, с использованием программного обеспечения фирмы “Bruker–winer and simfonia” (рис. 5). Видно, что концентрация радикала наиболее высока в волокнах ПГБ, она слабо уменьшается при добавлении ПЛА вплоть до 30% и при увеличении содержании этого полимера резко уменьшается, несмотря на сильное снижение энтальпии плавления полимера. Для данного полимера наблюдаемая закономерность может объясняться стеклообразным состоянием ПЛА, однако дальнейшее исследование пленочного материала, находящегося в высокоэластическом состоянии, показало, что основным фактором, влияющим на концентрацию радикала, является плотность упаковки цепей в полимере, о чем свидетельствует докристаллизация в процессе нагревания полимера (первый скан).
Рис. 5.
Изменения концентрации радикала в волокнистом (а) и пленочном материале (б) от его состава: 1 – исходные образцы, 2 – после экспозиции в течение 3 ч при Т = 25°С, 3 – при Т = 70°С, 4 – после отжига при 140°С.

Рассмотрим влияние экспозиции в воде ультратонких волокон при 25 и 70°С в течение 3 ч на молекулярную подвижность. Из рис. 4а видно, что отжиг в воде приводит к небольшому снижению τ2 относительно исходных полимеров с низкой концентрацией ПЛА в системе (до 50%). С ростом доли ПЛА в волокнах эти различия возрастают и наиболее сильные различия наблюдаются в волокнах 100%-ного ПЛА. Причиной уменьшения τ2 после экспозиции в водной среде для всех составов волокнистого материала является достаточно низкая концентрация радикала в плотных областях, доля и плотность которых возрастают с ростом температуры водного воздействия, о чем свидетельствуют данные метода ДСК (рис. 2а).
Концентрация радикала для всех составов волокон уменьшается после экспозиции в водной среде при 25 и 70°С, так как в плотных областях полимера его сорбция снижается (рис. 5а), а доля плотных структур, согласно ДСК, возрастает. Характерной особенностью изучаемого материала являются сильные изменения энтальпии плавления и времени корреляции именно в ПЛА и смесевой композиции 90/10% ПЛА/ПГБ.
Получены данные по изменению времени корреляции с ростом содержания ПГБ в пленочном материале ПГБ/ПЛА (рис. 4б). Видно, что добавление ПЛА к ПГБ до 30% включительно приводит к значительному росту τ и только при содержании ПЛА в смеси от 30 до 50% наблюдается резкое снижение этого параметра, дальнейший рост доли ПЛА в смеси к значительным изменениям τ не приводит. Можно полагать, что, как и в волокнистом материале, цепи ПЛА при невысокой их концентрации в пленке находятся в выпрямленной конформации, взаимодействуя с макромолекулами ПГБ, в результате определенная доля цепей ПГБ увеличивает степень выпрямленности. Этот эффект проявляется в пленочном материале ярче, чем в ультратонких волокнах, по причине более высокой доли выпрямленных цепей в пленочном материале (данные ДСК). В итоге интенсивнее, чем в волокнах, увеличивается время корреляции. При концентрации ПЛА от 30 до 50% имеет место инверсия фаз, при содержании ПЛА более 50% непрерывной фазой является ПЛА. Так как плотность ПЛА достаточно высока, радикал, не проникая в эти структуры, дает информацию о рыхлых областях с высокой молекулярной подвижностью.
Для пленочного материала получена зависимость концентрации радикала от состава смеси (рис. 5б). Аналогично изменениям в ультратонких волокнах концентрация радикала уменьшается для всех составов пленочного полимера. Такие изменения в составах с большой концентрацией ПЛА проявляются не столь значительно вследствие высокой плотности полимера. При сравнении зависимостей концентрации радикала в смесевых композициях пленочного и волокнистого материалов видно, что в последнем она выше вследствие достаточно низкой доли кристаллических и паракристаллических областей.
С использованием специальной программы NLSL аналогично процедуре, описанной в [14, 15], рассчитаны времена корреляции в плотных областях волокнистого и пленочного материалов путем математической обработки спектров. Получены данные по изменению молекулярной подвижности в плотных аморфных областях (рис. 6). В волокнах при добавлении ПГБ до 50% в смесевую композицию τ меняется слабо и при дальнейшем росте содержания ПЛА в системе подвижность значительно возрастает. Экспозиция в водной среде при 70°С в течение 3 ч приводит к снижению τ при добавлении ПЛА до 50% и при более высоком его содержании наблюдается значительное снижение этого параметра, что свидетельствует о росте молекулярной подвижности в аморфной фазе полимера. При сравнении данных времени корреляции для быстрой (рис. 4а) и медленной (рис. 6а) составляющих видно, что только в ПЛА и смесевых композициях с ПЛА выше 50% имеют место рост τ в исходных полимерах и его снижение после экспозиции в водной среде.
Рис. 6.
Зависимость τ от состава композиции в волокнистом (а) и пленочном (б) материале, рассчитанная по программе NLSL: 1 – исходный материал, 2 – после экспозиции в водной среде в течение 3 ч при 70°С.
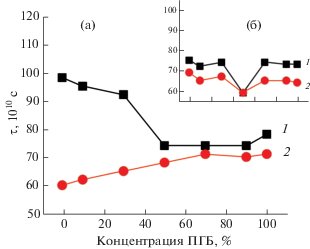
В пленочном материале, так же как в волокнистом, τ слабо меняется при добавлении ПЛА к ПГБ до 30% (включительно) и только при содержании ПЛА до 50% в смеси наблюдается снижение τ (рис. 6б). Добавление ПЛА выше 50% приводит к росту времени корреляции. Если время корреляции в рыхлых областях меняется слабо, то в плотных – возрастает. Аналогичные закономерности изменения τ в плотных аморфных областях получены в пленочном материале после экспозиции в водной среде при 70°С в течение 3 ч с той лишь разницей, что времена корреляции для всех систем характеризовались достаточно низкими значениями, что указывает на повышение молекулярной подвижности после отжига вследствие более низкой проницаемости радикала в плотные слои аморфной фазы.
Для перевода структуры полимеров в равновесное состояние образцы подвергали отжигу при 140°С. Из рис. 4 видно, что времена корреляции для всех составов волокнистого и пленочного материала характеризуются достаточно низкими значениями. Такие изменения обусловлены уплотнением материала при отжиге, когда радикал теряет способность проникать в плотные структуры и дает информацию о рыхлых областях полимера. Уплотнение материала при отжиге происходит вследствие роста доли кристаллитов. Из кинетической теории кристаллизации известно, что размер кристаллитов L при изотермическом отжиге зависит от времени отжига по закону L ∼ Вlogt. Поэтому как в пленочном, так и волокнистом материале кристалличность возрастает при отжиге 140°С [26, 28, 29], как следствие, концентрация радикала в плотных областях резко снижается и информация по времени корреляции поступает в основном о рыхлых областях с достаточно низкими временами корреляции. Именно такими изменениями структуры аморфной фазы объясняются наблюдаемые закономерности уменьшения τ после отжига в течение 3 ч при 140°С для всех изучаемых композиций ПЛА/ПГБ.
Интересно сопоставить времена корреляции образцов, полученных в результате комбинированного воздействия воды и температуры, и образцов, подвергнутых только температурной обработке. На рис. 4 видно, что воздействие воды и отжига при 140°С на волокна и пленки различного состава приводит к одному и тому же результату – снижению времени корреляции радикала (наиболее резко в ПЛА) и, следовательно, росту молекулярной подвижности в аморфных областях, доля плотных областей при этом возрастает (увеличивается энтальпия плавления образцов по данным [26, 28–30]).
Таким образом, с ростом температуры выдержки полимера в водной среде радикал все в меньшей концентрации проникает в плотные аморфные области волокон и дает информацию о наиболее рыхлой аморфной фазе.
Пластифицирующее действие воды, интенсивно реализующееся в аморфных областях пленок и волокон, в комбинации с повышенной температурой создает благоприятные условия для завершения дополнительной кристаллизации полимера, приводит к упорядоченному состоянию межкристаллитных областей и перераспределению соотношения между “рыхлой” и плотной фазами полимера в пользу последней. Ранее были проведены исследования воздействия водной среды на структуру полимеров [26–28, 30].
Воздействие озонолиза на динамику вращения радикала в ультратонких волокнах композиции ПЛА/ПГБ. Одновременно с механическим на изделия оказывает влияние экспозиция в атмосфере озона. Во-первых, он образуется при работе мощных электрических приборов, во-вторых, в некоторых случаях озон используется при стерилизации изделия. Несмотря на достаточно распространенную ситуацию воздействия озона на материал, его влияние на структурные и динамические характеристики остается мало исследованным. Изучено влияние озонолиза на ультратонкие волокна композиций ПЛА/ПГБ. В образцы предварительно вводили радикал, который затем окислялся. На рис. 7 видно, что на начальном этапе озонолиза (до 30 мин) для всех составов волокон имеет место рост времени корреляции зонда, что свидетельствует о замедлении молекулярной подвижности цепей в аморфных областях. При более длительном озонолизе прослеживается снижение τ, что указывает на рост молекулярной подвижности. При окислении протекают одновременно в основном два процесса: накопление полярных кислородсодержащих групп, продуктов озонолиза, которые приводят к усилению межмолекулярного взаимодействия между цепями; разрыв цепей при окислении, причем в первую очередь рвутся самые перенапряженные связи, в частности петли на поверхности кристаллитов [18]. На начальном этапе преобладает первый процесс, что приводит к росту жесткости цепей и степени их выпрямленности, а следовательно, к росту τ. В предыдущем разделе была представлена формула – конформационный критерий k* = $\sqrt 2 $/k = h/L (где h – среднее расстояние между концами цепей, L – контурная длина цепи, k – число сегментов в молекуле), что и определяет тенденцию макромолекулы к распрямлению и доориентации или к сворачиванию в клубок [24]. Для каждого полимера своя критическая величина k*. Цепи, у которых k > k*, стремятся распрямиться, макромолекулы, у которых k < k*, стремятся принять конформацию клубка. С увеличением жесткости цепи уменьшается число сегментов и возрастает k, растет число макромолекул с высокой степенью выпрямленности. В процессе получения волокон формируется определенная доля выпрямленных макромолекул. Именно в образцах ПГБ с 30% ПЛА максимальная энтальпия плавления и, следовательно, доля выпрямленных цепей. И именно в этих волокнах эффект сшивки макромолекул и их деструкции соизмерим, τ меняется незначительно. При достаточно глубоких степенях окисления процессы деструкции начинают играть значительную роль, что влечет уменьшение τ. Такие результаты получены при окислении смесевых композиций бутиленнитрильного каучука с хлорированным полиизопреном, ПГБ/Zn, ПГБ/Fe [19, 29, 30, 33–35]. Было показано, что чем выше доля выпрямленных цепей в полимере, тем сильнее возрастает τ. В данном случае τ возрастает в полимерах до 30 мин озонолиза, что обусловлено усилением межмолекулярного взаимодействия между цепями и замедлением молекулярной подвижности цепей, дальнейшее окисление приводит к снижению τ, а следовательно, к росту молекулярной подвижности макромолекул. Наибольший эффект уменьшения τ после озонного окисления при воздействии в течение 120 и 220 м проявляется в волокнах ПЛА и смесях ПЛА с 10, 30 и 50% ПГБ. Именно в этих полимерах доля плотных областей с высокой степенью выпрямленности невелика, поэтому имеет место интенсивная деструкция цепей с ростом молекулярной подвижности. В ПГБ при окислении в течение 120 и 220 м время корреляции заметно снижается и растет молекулярная подвижность.
Рис. 7.
Зависимость τ от состава композиции в волокнистом материале после озонолиза: 1 – исходный, 2 – 30, 3 – 120, 4 – 220 мин.
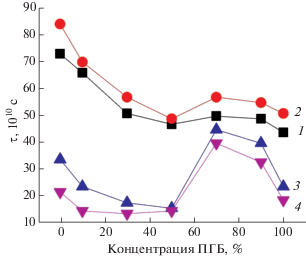
Таким образом, особенностью полученных зависимостей молекулярной подвижности от времени озонного воздействия на смесевые композиции ПЛА/ПГБ является рост τ при окислении до 30 мин и его снижении при увеличении времени озонолиза. Сильный эффект изменения τ после окисления проявляется в волокнах ПЛА и смесях ПЛА с 10, 30 и 50% ПГБ вследствие роста молекулярной подвижности по причине интенсивной деструкции макромолекул. По-видимому, данный эффект обусловлен достаточно рыхлой упаковкой цепей в данном полимере, когда молекулы озона достаточно легко сорбируются в структуре. Структура ультратонких волокон, сформированная волокнами ПГБ с 10, 30% ПЛА, имеет достаточно плотную упаковку с высоким содержанием выпрямленных цепей, и процессы окисления затрагивают малый объем образца, скорость деструкции цепей не столь значительно превалирует над скоростью сшивки. Эффект сшивки и деструкции соизмерим, τ меняется в течение 220 мин незначительно.
ЗАКЛЮЧЕНИЕ
Исследование ультратонких волокон и пленок смесевых композиций ПГБ–ПЛА показало, что способ приготовления полимера, его состав обусловливают значительные изменения в их надмолекулярной и молекулярной структуре. Эти изменения проявляются в динамике вращения зонда в аморфных областях и в энтальпии плавления полимеров. На основе структурно-динамических исследований при сочетании методов ЭПР и ДСК показано, что смешение ПГБ и полилактида влечет резкие изменения молекулярной динамики макромолекул в аморфной фазе и энтальпии плавления полимеров. Установлено, что надмолекулярная структура полимера находится в неравновесном состоянии. Отжиг при 140°С, экспозиция в водной среде при 25 и 70°С, озонолиз оказывают наибольший эффект (рост ∆Н и снижение τ) в ультратонких волокнах полилактида.
Взаимное влияние кристаллических и аморфных областей в биоразлагаемых полимерах и их композициях остается достаточно сложной и малоизученной проблемой современного материаловедения полимеров. Исследования состояния полимерной матрицы впервые позволили интерпретировать на молекулярном уровне воздействие ряда факторов (таких как температура и экспозиция в водной среде) на структурно-динамические характеристики пленочного и волокнистого материала ПГБ–ПЛА. Такие материалы могут быть использованы при конструировании терапевтических систем для программируемого транспорта лекарственных веществ.
В работе использовали приборы ЦКП Института биохимической физики РАН “Новые материалы и технологии”. В ИХФ РАН в рамках государственного задания Министерства высшего образования и науки РФ были проведены спектральные измерения (№ АААА-А18-118020890097-1) и калориметрические исследования (№ АААА-А17-117040610309-0).
Список литературы
Okan M., Aydin H.M., Barsbay M. // J. Chem. Technol. Biotechnol. 2019. V. 94. № 1. P. 8. https://doi.org/10.1002/jctb.5778
Jana S., Maiti S., Jana S. Biopolymer-Based Composites: Drug Delivery and Biomedical Application / Eds. Jana S. et al. Kidlington: Woodhead Publ. Elsevier Ltd., 2017. 408 p.
Zykova A.K., Pantyukhov P.V., Popov A.A. et al. // J. Polym. Environ. 2018. V. 26. № 4. P. 1343.
Sangroniz A., Zhu J.-B., Tang X. et al. // Nat. Commun. 2019. V. 10. Art. 3559. https://doi.org/10.1038/s41467-019-11525-x
Siracusa V., Karpova S., Olkhov A. et al. // Polymers. 2020. V. 12 (3). P. 647. https://doi.org/10.3390/polym12030647
Muniyasamy S., Seydibeyoğlu Ö., Thulasinathan B., Arun A. // Sustainable Biotechnology Enzymatic Resources of Renewable Energy / Eds. Singh O., Chandel A. Springer, Cham, 2018. https://doi.org/10.1007/978-3-319-95480-6_15
Cao K., Liu Y., Olkhov A.A. et al. // Drug Delivery Translational Res. 2018. V. 8 (1). P. 291.
Iordanskii A., Karpova S., Olkhov A. et al. // Eur. Polym. J. 2019. V. 117. P. 208.
Nawaz M., Baloch M.K., Rehman W. // J. Chil. Chem. Soc. 2010. V. 55. № 1. P. 90. https://doi.org/10.4067/S0717-97072010000100021
Ng C.W.W., So P.S., Lau S.Y. et al. // Eng. Geol. 2020. V. 272. P. 105631. https://doi.org/10.1016/j.enggeo.2020.105631
Chinaglia S., Tosin M., Degli-Innocenti F. // Polymer Degradation Stability. 2018. V. 147. P. 237. https://doi.org/10.1016/j.polymdegradstab.2017.12.011
Карпова С.Г., Ольхов А.А., Попов А.А. и др. // Высокомол. соединения. Сер. А. 2019. Т. 61. № 4. С. 335.
Ольхов А.А., Карпова С.Г., Тюбаева П.М. и др. // Перспективные материалы. 2020. № 3. С. 28.
Budil D.E., Lee S., Saxena S., Freed J.H. // J. Magn. Res. A. 1996. V. 120. P. 155.
Тимофеев В.П., Мишарин А.Ю., Ткачев Я.В. // Биофизика. 2011. Т. 56. № 3. С. 420.
Бучаченко А.Л., Вассерман А.М. Стабильные радикалы. М.: Химия, 1973.
Небратенко М.Ю., Наумова Ю.А., Филатов Ю.Н. // Вестн. МИТХТ. 2008. Т. 3. № 3. С. 31.
Карпова С.Г., Наумова Ю.А., Луканина Ю.К. и др. // Хим. физика. 2014. Т. 33. № 5. С. 89.
Karpova S.G., Naumova Yu.A., Lyusova L.R. et al. // Polym. Sci. A. 2019. V. 61. № 1. P. 9. https://doi.org/10.1134/S0965545X19010152
Зуев А.А., Люсова Л.Р., Борейко Н.П. // Промышленное производство и использование эластомеров. 2017. № 2. С. 30.
Котова С.В., Люсова Л.Р., Глаголев В.А. и др. // Вестн. МИТХТ. 2007. Т. 2. № 3. С. 38.
Ольхов А.А., Панкова Ю.Н., Гольдштрах М.А. и др. // Материаловедение. 2015. № 10. С. 20.
Olkhov A.A., Markin V.S., Kosenko R.Yu. et al. // Russ. J. Appl. Chem. 2015. V. 88. № 2. P. 308.
Озерин А.Н. Дисс. … канд. хим. наук. М.: Научно-исследовательский физико-химический ин-т им. Л.Я. Карпова, 1977.
Рейтлингер С.А. Проницаемость полимерных материалов. М.: Химия, 1974.
Karpova S.G., Ol’khov A.A., Lobanov A.V. et al. // Nanotechnologies in Russia. 2019. V. 14. № 3–4. P. 132. https://doi.org/10.1134/S1995078019020083
Karpova S.G., Ol’khov A.A., Chvalun S.N. et al. // Nanotechnologies in Russia. 2019. V. 14. № 7–8. P. 367. https://doi.org/10.1134/S1995078019040086
Karpova S.G., Ol’khov A.A., Tyubaeva P.M. et al. // Russian J. Phys. Chem. B. 2019. V. 13. № 2. P. 313. https://doi.org/10.1134/S1990793119020039
Karpova S.G., Ol’khov A.A., Krivandin A.V. et al. // Polym. Sci. A. 2019. V. 61. № 1. P. 70. https://doi.org/10.1134/S0965545X19010140
Karpova S.G., Olkhov A.A., Bakirov A.V. et al. // Russian J. Phys. Chem. B. 2018. V. 12. № 1. P. 142. https://doi.org/10.1134/S1990793118010190
Попов А.А., Рапопорт Н.Я., Заиков Г.Е. Окисление ориентированных и напряженных полимеров. М.: Химия, 1987.
Karpova S.G., Olkhov A.A., Popov A.A. et al. // Polym. Sci. A. 2019. V. 61. № 4. P. 480. https://doi.org/10.1134/S0965545X19040059
Karpova S.G., Ol’khov A.A., Iordanskii A.L. et al. // Polym. Sci. A. 2016. V. 58. № 1. P. 76.
Karpova S.G., Ol’khov A.A., Iordanskii A.L. et al. // Russian J. Phys. Chem. B. 2016. V. 10. № 4. P. 687. https://doi.org/10.1134/S1990793116040230
Ольхов А.А., Лобанов А.В., Карпова С.Г. и др. // Журн. прикл. химии. 2019. Т. 92. Вып. 4. С. 465.
Дополнительные материалы отсутствуют.
Инструменты
Российские нанотехнологии