

Российские нанотехнологии, 2022, T. 17, № 1, стр. 57-64
НАНОВОЛОКНИСТЫЙ МАТЕРИАЛ С ГИДРОФОБНО-ГИДРОФИЛЬНЫМИ СВОЙСТВАМИ НА ОСНОВЕ ПОЛИ(3-ГИДРОКСИБУТИРАТА) И ПОЛИ(2-ГИДРОКСИЭТИЛМЕТАКРИЛАТА)
С. Н. Холуйская 1, А. А. Ольхов 1, 2, *, С. Г. Карпова 3, М. А. Градова 1, Ю. Н. Зернова 1, А. Л. Иорданский 1
1 ФИЦ Институт химической физики им. Н.Н. Семенова РАН
Москва, Россия
2 Российский экономический университет им. Г.В. Плеханова
Москва, Россия
3 Институт биохимической физики им. Н.М. Эмануэля РАН
Москва, Россия
* E-mail: aolkhov72@yandex.ru
Поступила в редакцию 30.11.2020
После доработки 30.11.2020
Принята к публикации 20.12.2020
Аннотация
Получен новый материал с гидрофобно-гидрофильными свойствами на основе ультратонких волокон поли(3-гидроксибутирата) (ПГБ), модифицированных поли(2-гидроксиэтилметакрилатом). Надмолекулярная структура образцов исследована структурно динамическими методами: сканирующая электронная микроскопия, дифференциальная сканирующая калориметрия, электронный парамагнитный резонанс. Определены равновесно-кинетические характеристики сорбции воды материалом. Методом 1Н ЯМР получены доказательства химической модификации поверхности ПГБ вследствие участия его терминальных групп в реакции переэтерификации 2-гидроксиэтилметакрилата.
ВВЕДЕНИЕ
Полиэфиры 3-гидроксикарбоновых кислот, поли(3-гидроксиалканоаты) (ПГА) – важнейший объект медицинского материаловедения. Благодаря своей биосовместимости и биоразлагаемости они нашли широкое применение в хирургии, регенеративной инженерии мягких и скелетных тканей организма, в производстве систем контролируемой доставки лекарственных средств, матриксов, подложек, мембран и т.д. Основное препятствие в широком использовании ПГА для биологических и медицинских целей обусловлено их плохой смачиваемостью водой вследствие гидрофобности. Преодолеть эту проблему позволяет создание привитых и блочных сополимеров ПГА, содержащих такие функциональные группы, как гидроксил (–OH), карбоксильная (–COOH) и амино- (–NH2) группы [1]. Из мономеров с гидроксильными группами сополимеризацию часто проводят с полиэтиленгликолями (ПЭГ) [2–4] и гидроксилсодержащими (мет)акрилатами.
Представитель класса гидроксилсодержащих (мет)акрилатов, 2-гидроксиэтилметакрилат (ГЭМА), давно и широко применяется в медицине в силу своей гидрофильности, нетоксичности и биосовместимости [5]. Материалы на основе ГЭМА используют для производства контактных линз, медицинских клеев, искусственных имплантатов, систем доставки лекарств. Существенное ограничение использования поли(2-гидроксиэтилметакрилата) (ПГЭМА) связано с отсутствием биодеградации, поскольку продукты его разложения имеют высокую молекулярную массу, которая, однако, может быть снижена в результате сополимеризаци с биоразлагаемыми добавками.
Наибольший интерес с точки зрения улучшения эксплуатационных свойств ПГА представляет получение амфифильных сополимеров. Особое развитие в последние годы получило применение амфифильных сополимеров для контролируемой доставки лекарств, так как эти макромолекулы проявляют свойства самосборки с образованием мицелл в водных средах. Полимерные мицеллы состоят из гидрофильной оболочки и гидрофобного ядра. Таким образом, они могут повысить растворимость плохо растворимых лекарств и увеличить их биодоступность. В литературе имеются многочисленные примеры синтеза амфифильных полимеров на основе гидроксилсодержащих (мет)акрилатов и биоразлагаемых сложных полиэфиров: ПГЭМА и L-лактида [6–8], ПГЭМА и ε-капролактона [9–11], 2-полигидроксиэтилакрилата и L-лактида [12, 13].
Взаимопроникающие полимерные сетки – еще один тип биоматериалов с высоким потенциалом использования в тканевой инженерии и контролируемом высвобождении лекарств. В [14, 15] представлены данные о взаимопроникающих сетках из полилактида и ПГЭМА, причем в [15] на базе ГЭМА, D- и L-лактидов и ряда метакрилатов осуществлен синтез “умного” рН- и термочувствительного амфифильного гидрогеля.
Одним из первых полимеров из группы ПГА был получен поли(3-гидроксибутират) (ПГБ), который в настоящий момент является наиболее распространенным и широко изученным представителем этого класса полимеров. ПГБ обладает рядом ценных свойств: термопластичностью, биоразлагаемостью, нетоксичностью по отношению к живым системам, биосовместимостью с тканями организма и др. [16]. В то же время этот полимер имеет низкие термостабильность и механические характеристики, а именно, повышенную хрупкость и низкое относительное удлинение [1]. Стратегия улучшения свойств ПГБ включает в себя несколько направлений. Например, существенное улучшение механо-термических свойств ПГБ достигается введением наночастиц оксидов титана [17] и цинка [18], крахмала [19], целлюлозы [20], графена [21]. Другой эффективный подход состоит в сополимеризации ПГБ с различными мономерами. Тестирование более 150 биополимеров показало, что наилучшими характеристиками обладает поли(3-гидроксибутират-3-гидроксивалерат) (ПГБ-ГВ). Однако гидрофобный характер поверхности обоих полимеров ПГБ-ГВ и ПГБ снижает их ценность для медицины, поскольку смачиваемость материала является ключевым фактором для пролиферации клеток и регенерации тканей. Результативным подходом к решению этой проблемы стала модификация поверхности полимеров физическими (обработка УФ-излучением, плазмой) и химическими методами.
В литературе описано несколько примеров химической модификации поверхности ПГБ-ГВ и ПГБ путем прививки ГЭМА в результате радикальной полимеризации, инициированной γ-облучением [22], перекисью бензоила [23], УФ-облучением в присутствии фотоинициаторов [[24]. В последнем случае удалось получить полностью биоразлагаемый полимер ПГБ-ГВ с ПГЭМА при условии, что содержание ПГЭМА составляло менее 10%.
Повышенными гидрофильными свойствами помимо привитых сополимеров обладают амфифильные сополимеры ПГБ с полипропиленгликолем, ПЭГ, поли(2-диметиламиноэтилметакрилатом), поли(N-изопропилакриламидом), крахмалом, целлюлозой и другими мономерами и олигомерами [16]. Повысить гидрофильность ПГБ пленок или волокон можно также путем приготовления смесей ПГБ с более гидрофильными полимерами или дисперсными наполнителями: полиамидом [25], поливиниловым спиртом [26], хитозаном [27], металлокомплексами тетрафенилпорфирина [28], диоксидами титана и кремния [29] и др. Однако вводимые в ПГБ полимеры и дисперсные наполнители оказывают влияние на формирование надмолекулярной структуры полимера, что влечет за собой изменения комплекса физико-химических свойств.
С целью сохранения основных свойств ПГБ и увеличения гидрофильности материалов на его основе можно было бы использовать метод нанесения гидрофильного мономера на поверхность изделий из ПГБ с последующей полимеризацией. Данные о применении ГЭМА для этих целей в литературе отсутствуют. В то же время известно о получении взаимопроникающих полимерных сеток на основе ПГБ-ГВ и ПГЭМА методом фотополимеризации [30].
Цель настоящей работы – получить и охарактеризовать гидрофобно-гидрофильный композит на основе нетканого материала из ультратонких волокон ПГБ, модифицированного ПГЭМА.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Полигидроксибутират серии 16F, синтезированный микробиологическим путем компанией BIOMER® (Германия), со следующими характеристиками: размер частиц 5–20 мкм, Mη = 2.06 × × 105 г/моль, плотность d = 1.248 г/см3, Tпл = = 177°С, степень кристалличности α = 75%, использован для получения ультратонких волокон методом электроформования [31] из формовочного раствора с концентрацией ПГБ 7 мас. % в хлороформе. Формовочные растворы ПГБ готовили при 50°С с помощью автоматической магнитной мешалки с подогревом и ультразвуковой ванны.
Мономер ГЭМА (“ФГУП НИИ химии и технологии полимеров им. В.А. Каргина” г. Дзержинск) очищали от примесей дистилляцией в вакууме, отделяя фракцию с температурой кипения 67°С при давлении 3.5 мм рт.ст. Катализатор VO(DMSO)5(ClO4)2 синтезировали по методу [32].
Модификацию нетканого волокнистого материала из ПГБ проводили путем его пропитки ГЭМА, содержащим катализатор в концентрации 4.0 мМ, и прогреванием при 80°C на воздухе в течение двух часов под грузом с постоянным давлением 17.24 г/см2. Приготовлены материалы при следующих массовых соотношениях ПГБ/ГЭМА = = 1.6/1 и 1/2.2. Характеристики образцов получали после предварительного отмывания растворимой золь-фракции несшитого ПГЭМА и остаточного мономера. Для этого выдерживали образцы в дистиллированной воде до постоянной массы, затем высушивали в вакууме также до постоянной массы.
Исследование химической модификации проводили с исходным мелкодисперсным ПГБ: смешивали 0.2 г ПГБ с 0.4 мл ГЭМА, содержащего катализатор в концентрации 4.0 мМ, прогревали 2 ч при 80°С. Размолотый на мельнице продукт помещали на 18 ч в CD3OD, осадок отфильтровывали, раствор анализировали методом ЯМР. Высушенный от метанола осадок помещали в CDCl3, выдерживали несколько суток, анализировали жидкую фракцию, а также раствор исходного мелкодисперсного ПГБ в CDCl3 методом 1Н ЯМР. Спектры 1Н ЯМР регистрировали на спектрометре Brucker Avance III-500 MHz.
Сорбцию паров воды образцами ПГБ и ПГБ/ПГЭМА с соотношением ПГБ/ПГЭМА = = 1/2.2 определяли на весах Mак-Бена с кварцевой спиралью по методике, описанной в [33], в интервалах давления паров воды 0–9, 9–14.5, 14.5–18.5 мм рт.ст. Методом интервальной сорбции построена изотерма сорбции паров воды в координатах активность паров воды (а)–мас. % воды в образце. Активность паров воды определяется как отношение а = Pi/Pнас, где Pi – давление паров воды в колонне с образцом, Pнас – давление насыщенных паров воды.
Электронные микрофотографии поверхности образцов были получены при помощи сканирующего электронного микроскопа (СЭМ) JEOL JSM-T330А (Япония) при ускоряющем напряжении 10–15 кВ. Предварительная подготовка образцов включала в себя напыление тонким слоем золота (~10 нм) на установке вакуумного напыления JEOL JFC-1500. Регистрацию изображения проводили по синхронизованной схеме с использованием камеры Canon PowerShot А590 IS, управляемой с пульта электронного микроскопа.
Морфологию нетканых материалов исследовали методом оптической микроскопии в отраженном свете с помощью поляризационного микроскопа Микромед Полар-3 (Россия).
Молекулярную подвижность изучали методом спинового зонда на автоматизированном ЭПР-спектрометре ЭПР-В (Институт биохимической физики РАН им. Н.М. Эмануэля, Москва). В качестве зонда использовали стабильный нитроксильный радикал 2,2,6,6-тетраметилпиперидин-1-оксил. Радикал вводили в пленки из паров при температуре 60°С до концентрации, не превышающей 10–3 моль/л. Регистрацию спектров электронного парамагнитного резонанса (ЭПР) проводили в отсутствие насыщения, что проверяли по зависимости интенсивности сигнала от мощности микроволнового поля. Значения времени корреляции вращения зонда τ рассчитывали из спектров ЭПР по формуле
(1)
$\tau = \Delta {{H}_{ + }}[{{({{I}_{ + }}{\text{/}}{{I}_{ - }})}^{{0.5}}} - {\text{ }}1]6.65 \times {{10}^{{ - 10}}},$Исследование образцов методом дифференциальной сканирующей калориметрии (ДСК) было проведено на приборе DSC 204 F1 фирмы Netzsch в инертной среде аргона cо скоростью нагрева 10°C/мин. Среднестатистическая ошибка измерения тепловых эффектов составляла ±3%.
РЕЗУЛЬТАТЫ ИССЛЕДОВАНИЯ И ИХ ОБСУЖДЕНИЕ
Гидрофобные-гидрофильные свойства ПГБ/ПГЭМА и ПГБ/ГЭМА
На рис. 1 представлены микрофотографии исходных волокнистых материалов ПГБ и ПГБ с нанесенным ПГЭМА. Как видно на рис. 1а, 1б, исходные волокна ПГБ и ПГБ/ПГЭМА различаются по морфологии. Волокна в обоих нетканых материалах имеют близкие значения средних диаметров 8–16 мкм. При этом в структуре волокна присутствуют отдельные утолщения в форме веретена или капли протяженностью в продольном направлении до 150 мкм и поперечном – до 50–70 мкм. Происхождение этих структур связано с неоптимальной электропроводностью полимерного формовочного раствора [34]. Волокнистый материал ПГБ/ПГЭМА по сравнению с ПГБ характеризуется наличием одиночных пленочных элементов (спаек), скрепляющих волокна, протяженностью 100–200 мкм и средней толщиной 0.51 мкм. Четко видимая фактура волокнистого материала и наличие тонких спаек в межволоконном пространстве позволяют сделать вывод о практически равномерном покрытии ПГЭМА поверхности ПГБ-волокон. По данным микроскопического анализа средняя толщина слоя ПГЭМА на волокнах варьируется в диапазоне 0.5–1 мкм.
Рис. 1.
Образцы нетканых волокнистых материалов: ПГБ (а1 – оптическая микроскопия, а2, а3 – СЭМ) и композиционного материала ПГБ/ПГЭМА = 1/2.2 (б1 – оптическая микроскопия, б2, б3 – СЭМ).

При более детальном рассмотрении поверхности волокон с помощью СЭМ (рис. 1а2, 1а3) видно, что вся поверхность ПГБ-волокон имеет сложный рельеф, состоящий из раковин округлой формы со средним диаметром 0.05–0.1 мкм. Волокна ПГБ/ПГЭМА (рис. 1б2, 1б3) имеют сглаженный рельеф, который образуется в результате смачивания волокна мономером ГЭМА с последующей полимеризацией и образованием непрерывного тонкого слоя на поверхности волокна и пленочных структур (спаек) в межволоконном пространстве.
Для оценки степени адгезии ПГЭМА к ПГБ полученные образцы материалов выдерживали 1 ч в дистиллированной воде при комнатной температуре с последующей сушкой и изучением морфологии. На рис. 2 представлена микрофотография волокнистого материала ПГБ/ПГЭМА.
Рис. 2.
СЭМ-изображение нетканого волокнистого материала ПГБ/ПГЭМА = 1/2.2 после выдержки в воде. На вставке представлена увеличенная область с указанием размеров волокон.

Анализ микрофотографий материала позволяет судить об отсутствии морфологических изменений по сравнению с необработанными материалами. Данный факт свидетельствует о высокой адгезии слоя ПГЭМА к поверхности волокон ПГБ. На высокую степень полимеризации ГЭМА на поверхности волокон ПГБ указывает незначительная, около 5%, потеря массы образцов в ходе набухания в воде, что может быть обусловлено присутствием растворимой золь-фракции несшитого ПГЭМА. Отметим, что в результате поглощения воды материал не расслаивается и не разрушается.
Обращает на себя внимание характер распределения ПГЭМА на волокнах ПГБ: на рисунке видно, что метакрилат преимущественно располагается по поверхности волокон, происходит своего рода их инкапсуляция. Количество участков, где ПГЭМА образует пленку между волокнами, малó и снижается с увеличением соотношения ПГБ/ГЭМА. Данная картина может быть проявлением химического взаимодействия ГЭМА с подложкой.
По-видимому, в условиях приготовления образцов реакция переэтерификации ПГБ с разрывом цепи не является реалистичной. Однако, как известно, ПГБ содержит реакционноспособные концевые карбоксильные группы. Действительно, в [35] получен сополимер ПГБ путем этерификации его терминальных групп целлюлозой.
Проверку гипотезы о химической модификации ПГБ в результате взаимодействия с ГЭМА осуществляли методом спектроскопии ЯМР. Полимеризацию ГЭМА проводили в условиях наполнения мелкодисперсным ПГБ с целью увеличения площади контактной поверхности. Образец ПГБ-ПГЭМА после полимеризации обработали последовательно двумя дейтерированными растворителями – метанолом CD3OD для извлечения низкомолекулярных продуктов и хлороформом CDCl3, чтобы выявить признаки трансформации ПГБ, растворимого в хлороформе. В 1Н-спектре раствора CD3OD помимо сигналов остаточного мономера ГЭМА с характерными сдвигами обнаружены сигналы протонов гидроксильных групп (–ОН) с химическим сдвигом δ = 3.03 ppm и метиленовых протонов (–СН2) c δ = 3.61 ppm, относящихся к этиленгликолю. Контрольный эксперимент с добавкой этиленгликоля показал возрастание интенсивности указанных сигналов. Кроме того, наблюдается увеличение сигнала протонов воды по сравнению с водой исходного CD3OD. В растворе дейтерохлороформа спектр ЯМР представляет собой суперпозицию спектров ГЭМА и ПГБ, причем мономер ГЭМА находится в следовых количествах, и спектр ПГБ полностью совпадает со спектром исходного чистого ПГБ, за исключением неидентифицированной примеси с δ = 3.02 ppm.
Катализатор, используемый для полимеризации ГЭМА, представляет собой сильную кислоту Льюиса [36] и вполне может также эффективно катализировать реакцию переэтерификации ГЭМА с участием концевых карбоксильных групп ПГБ по схеме 1 :
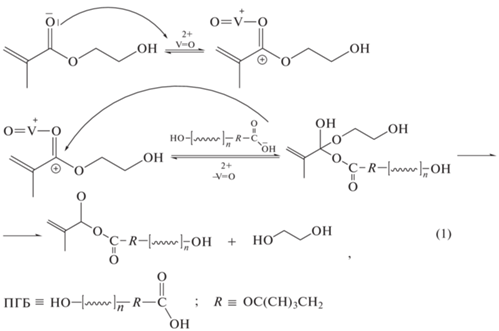
и реакцию этерификации карбоксильных терминальных групп ПГБ по схеме 2 :
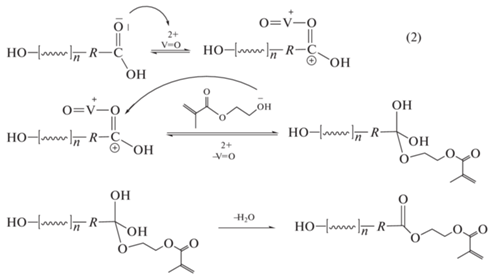
Из представленных схем следует, что основными низкомолекулярными продуктами этих реакций являются вода и этиленгликоль, сигналы которых и были обнаружены в спектрах 1Н. Таким образом, методом ЯМР получены доказательства химического взаимодействия ПГБ и ГЭМА.
Для сравнительной оценки гидрофильности волокнистого материала на основе ПГБ/ПГЭМА были получены изотермы сорбции паров воды (рис. 3) с помощью высокочувствительного метода изотермической гравиметрии [33]. Как показал анализ изотерм, гомополимер ПГБ и композиционный материал ПГБ/ГЭМА сильно различаются по степени гидрофильности и, соответственно, по сорбционной емкости (примерно в 7 раз). Существенный рост сорбционной емкости композитных волокон обусловлен гидрофильным покрытием ПГЭМА. Отметим, что диффузионные характеристики полимеров напрямую зависят от надмолекулярной структуры. Жесткоцепной ПГБ имеет высокую степень кристалличности (65–75%), что имеет следствием низкие значения коэффициента диффузии воды. Напротив, ПГЭМА относится к аморфным полимерам с химическими межмолекулярными сшивками, для которых характерна высокая проницаемость паров воды.
Рис. 3.
Зависимость массы образца нетканого волокнистого материала ПГБ (1) и композиционного материала ПГБ/ПГЭМА (2) от активности паров воды (изотермы сорбции) при температуре 23°С (ПГБ/ПГЭМА = 1/2.2).
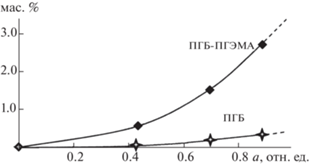
Таким образом, основная цель настоящей работы достигнута: ПГЭМА сильно увеличивает гидрофильность поверхности материала. Вместе с тем сорбция паров образцами в целом ниже (примерно в 5 раз), чем наблюдавшаяся в [37], где исследованы образцы блочного ПГЭМА, полученного в отсутствие и с добавками сшивающего агента. В [37] показано, что введение сшивающего агента и увеличение плотности сетки блочного ПГЭМА приводят к уменьшению сорбции водяных паров. В настоящей работе на стадии нанесения ПГЭМА использовали относительно высокие концентрации катализатора, при которых существенную роль начинают играть реакции сшивки макромолекул непосредственно на каталитическом центре, что приводит, по данным [38], к сильному увеличению плотности полимерной сетки и, соответственно, уменьшению влагопоглощения. В то же время именно благодаря высокой концентрации катализатора более эффективно проходит модификация поверхности с образованием химических связей между ПГБ и ПГЭМА, что в конечном итоге положительно влияет на прочность материала. Дальнейшей задачей исследований является оптимизация соотношения катализатор/ мономер ГЭМА для достижения достаточной прочности композиционного материала наряду с высоким водопоглощением.
Химическая модификация ПГБ – не единственная причина отсутствия гидролитический деструкции полученного композита. Хороший адгезионный контакт между ПГБ и ПГЭМА также обеспечивают полярные функциональные группы, присутствующие в основной цепи обоих полимеров. Формирование межмолекулярных физических связей между полимерами происходит исключительно на поверхности волокон и вносит изменение в молекулярную динамику полимеров лишь в прилегающих к поверхности слоях. Поэтому существенных изменений в структуре аморфных и кристаллических областей ПГБ волокон ожидать не следует. Тем не менее надмолекулярную структуру волокнистых материалов исследовали с использованием структурно чувствительных методов ДСК и ЭПР.
На рис. 4 представлены термограммы нагревания ДСК и спектры ЭПР волокнистых материалов ПГБ и с покрытием ПГЭМА. Как показал анализ термограмм нагревания (рис. 4а), температура и теплота плавления волокон ПГБ практически не меняются в результате формирования адгезионного покрытия ПГЭМА. Данный факт говорит о свободном протекании кристаллизационных процессов фазы ПГБ без каких-либо стерических затруднений.
Рис. 4.
Термограммы нагревания ДСК (а) и спектры ЭПР (б) образцов нетканых волокнистых материалов ПГБ/ПГЭМА.

Структуру и молекулярную динамику межкристаллических аморфных областей можно эффективно исследовать методом ЭПР с использованием стабильных нитроксильных радикалов. На рис. 4б представлены типичные спектры вращения стабильного радикала ТЕМПО в исследуемых образцах. Видно, что спектры представляют собой суперпозицию двух спектров, характеризующих вращение радикала в аморфных областях полимерного волокна различной степени упорядоченности и плотности. При анализе спектров вращения радикала ТЕМПО (рис. 4б) были вычислены средние времена корреляции (вращения). Величины времен корреляции (τ × 10–10, с) составили: 47 (ПГБ), 36 (ПГБ/ПГЭМА) и 45 (ПГБ/ПГЭМА после эксперимента сорбции паров воды и высушивания до постоянной массы в условиях вакуума). Видно, что в композитных волокнах время корреляции снизилось примерно на 10% относительно ПГБ. Снижение времени корреляции, которое указывает на увеличение свободного межмолекулярного объема, можно объяснить влиянием более “рыхлого” слоя ПГЭМА на поверхности волокон ПГБ. В этом слое концентрация радикала, по-видимому, более высокая, поэтому среднее время корреляции уменьшается. При исследовании образца нетканого материала ПГБ/ПГЭМА после сорбции и десорбции воды время корреляции увеличивается до значения, близкого к волокнам ПГБ. Увеличение времени корреляции можно объяснить дополнительным снижением молекулярной динамики в слое сшитого ПГЭМА за счет образования межмолекулярных водородных связей и водородных связей с молекулами воды [39]. Согласно результатам ДСК и ЭПР присутствие слоя ПГЭМА не изменяет существенным образом молекулярную динамику ПГБ.
ЗАКЛЮЧЕНИЕ
Методом полимеризационного наполнения получен новый биосовместимый композиционный материал на основе ультратонких волокон поли(3-гидроксибутирата) и 2-гидроксиэтилметакрилата. Композит характеризуется гидролитической устойчивостью, повышенным по сравнению с поли(3-гидроксибутиратом) влагопоглощением за счет гидрофильных свойств 2-гидроксиэтилметакрилата. Присутствие поли(2-гидроксиэтилметакрилата) не вносит существенных изменений в молекулярную динамику поли(3-гидроксибутирата) (ДСК, ЭПР). По данным СЭМ формирование слоя поли(2-гидроксиэтилметакрилата) происходит преимущественно на поверхности волокон поли(3-гидроксибутирата), его средняя толщина варьируется в диапазоне 0.5–1 мкм. Высокий адгезионный контакт компонентов материала является, в том числе, следствием их химического взаимодействия. Методом ЯМР установлено, что химическая модификация поверхности поли(3-гидроксибутирата) происходит вследствие участия его терминальных групп в реакции переэтерификации 2-гидроксиэтилметакрилата.
Список литературы
Albertsson A.C., Varma I.K. // Biomacromolecules. 2003. V. 4. № 6. P. 1466. https://doi.org/10.1021/bm034247a
Xiao R.Z., Zeng Z.W., Zhou G.L. et al. // Int. J. Nanomedicine. 2010. № 5. P. 1057. https://doi.org/10.2147/IJN.S14912
Shah M., Ullah N., Choi M.H. et al. // Eur. J. Pharm. Biopharm. 2012. V. 80. № 3. P. 518. https://doi.org/10.1016/j.ejpb.2011.11.014
Loh X.J., Goh S.H., Li J. // J. Phys. Chem. B. 2009. V. 113. № 35. P. 11822. https://doi.org/10.1021/jp903984r
Лавров Н.А. // Полимеры на основе 2-гидроксиэтилметакрилата. СПб.: ЦОП “Профессия”, 2017. С. 162.
Zhao C.S., Wu D.X., Huang N., Zhao N.H. // J. Polym. Sci. B: Polym. Phys. 2008. V. 46. № 6. P. 589. https://doi.org/10.1002/polb.21394
Cretu A., Kipping M., Adler H.J., Kuckling D. // Polym. Int. 2008. V. 57. № 7. P. 905. https://doi.org/10.1002/pi.2423
Zhu L., Liu F., Yu X., Xue L. // ACS Appl. Mater. Interfaces. 2015. V. 7. № 32. P. 17748. https://doi.org/10.1021/acsami.5b03951
Gatti S., Agostini A., Ferrari R., Moscatelli D // Polymers. 2017. № 9. P. 389. https://doi.org/10.3390/polym9090389
Cretu R., Gattin L., Brachais D., Barbier-Baudry. // Polym. Degrad. Stab. 2004. V. 83. № 3. P. 399. https://doi.org/10.1016/j.polymdegradstab.2003.09.001
Clément B., Trimaille T., Alluin O. et al. // Biomacromolecules. 2009. V. 10. № 6. P. 1436. https://doi.org/10.1021/bm900003f
Clément B., Decherchi P., Féron F. et al. // Macromol. Biosci. 2011. № 11. P. 1175. https://doi.org/10.1002/mabi.201100067
Yildrim I., Sungur P., Crecelius-Vitz A.C. et al. // Polym. Chem. 2017. V. 8. № 39. P. 6086. https://doi.org/10.1039/C7PY01176H
Passos M.F., Fernández-Gutiérrez M., Vázquez-Lasa B. et al. // Eur. Polym. J. 2016. V. 85. P. 150. https://doi.org/10.1016/j.eurpolymj.2016.10.023
Wu J., Shi X., Wang Z. et al. // Polymers. 2019. V. 11. № 12. P. 1940. https://doi.org/10.3390/polym11121940
Li Z., Yang J., Loh X.J. // NPG Asia Mater. 2016. V. 8. P. 265. https://doi.org/10.1038/am.2016.48
Valle Iulianelli G.C., dos S. David G., dos Santos T.N. et al. // Polym. Test. 2018. V. 65. P. 156. https://doi.org/10.1016/j.polymertesting.2017.11.018
Diez-Pascua A.M., Diez-Vicente A.L // Int. J. Mol. Sci. 2014. V. 15. № 6. P. 10950. https://doi.org/10.3390/ijms150610950
Zhang L., Deng X., Zhao S., Huang Z. // Polym. Int. 1997. V. 44. № 1. P. 104. https://doi.org/10.1002/(SICI)1097-0126(199709)44:1<104::AID-PI812>3.0.CO;2-%23
El-Shafee E., Saad G.R., Fahmy S.M. // Eur. Polym. J. 2001. V. 37. № 10. P. 2091. https://doi.org/10.1016/S0014-3057(01)00097-0
Hasheminejad K., Montazeri A. // Appl. Surf. Sci. 2020. V. 502. P. 144150. https://doi.org/10.1016/j.apsusc.2019.144150
Mitomo H., Enjôji T., Watanabe Y. et al. // J. Macromol. Sci. A. 1995. V. 32. № 3. P. 429. https://doi.org/10.1080/10601329508013674
Lao H.-K., Renard E., Linossier I. et al. // Biomacromolecules. 2007. V. 8. № 2. P. 416. https://doi.org/10.1021/bm0609700
Lao H.-K., Renard E., Langlois V. et al. // J. Appl. Polym. Sci. 2010. V. 116. № 1. P. 288. https://doi.org/10.1002/app.31507
Ольхов А.А., Маркин В.С., Косенко Р.Ю. и др. // Заводская лаборатория. Диагностика материалов. 2016. Т. 82. № 6. С. 33.
Ольхов А.А., Иорданский А.Л., Заиков Г.Е. и др. // Вестн. Казан. технол. ун-та. 2014. Т. 17. № 6. С. 169.
Karpova S.G., Ol’khov A.A., Iordanskii A.L. et al. // Polym. Sci. A. 2016. V. 58. № 1. P. 76.
Карпова С.Г., Ольхов А.А., Чвалун С.Н. и др. // Российские нанотехнологии. 2019. Т. 14. № 7–8. С. 71.
Ольхов А.А., Иорданский А.Л., Староверова О.В. и др. // Химические волокна. 2015. № 5. С. 8.
Gursel I., Balcik C., Arica Y. et al. // Biomaterials. 1998. V. 19. № 13. P. 1137. https://doi.org/10.1016/S0142-9612(98)00009-X
Филатов Ю.Н. Электроформование волокнистых материалов (ЭФВ-процесс). М.: Нефть и Газ, 1997. 297 с.
Selbin J., Holmes L.H. // J. Inorg. Nucl. Chem. 1962. V. 24. № 9. P. 1111. https://doi.org/10.1016/0022-1902(62)80256-5
Малкин А.Я., Чалых А.Е. Диффузия и вязкость полимеров. Методы измерения. М.: Химия, 1979. 301 с.
Ольхов А.А., Староверова О.В., Гольдштрах М.А. и др. // Химическая физика. 2016. Т. 35. № 10. С. 53.
Yalpani M., Marchessault R.H., Morin F.G., Monasterios C.J. // Macromolecules. 1991. V. 24. № 22. P. 6046. https://doi.org/10.1021/ma00022a024
Nikitin A.V., Kholuiskaja S.N., Rubailo V.L. // J. Chem. Res. (S). 1994. № 9. P. 358.
Sung Y.K., Jhon M.S., Gregonis D.E., Andrade J.D. // Polymer (Korea). 1984. V. 8. № 2. P. 123.
Холуйская С.Н., Филатова А.Г., Дубровский С.А. // Поверхность. Рентген., синхротр. и нейтр. исследования. 2014. № 9. С. 27.
Morita S. // Front. Chem. 2014. № 2. P. 1. https://doi.org/10.3389/fchem.2014.00010
Дополнительные материалы отсутствуют.
Инструменты
Российские нанотехнологии