

Российские нанотехнологии, 2022, T. 17, № 3, стр. 405-414
Влияние ультразвуковой обработки на функциональные группы и латеральный размер чешуек оксида графена
Ю. А. Грошкова 1, *, Д. Ю. Корнилов 1, С. В. Ткачев 1, С. П. Губин 2
1 ООО “НПО “Графеника”
Москва, Россия
2 Институт общей и неорганической химии им. Н.С. Курнакова РАН
Москва, Россия
* E-mail: JuliaGroshkova@yandex.ru
Поступила в редакцию 19.11.2021
После доработки 28.01.2022
Принята к публикации 28.01.2022
Аннотация
Исследовано влияние ультразвуковой обработки в течение разного времени при комнатной температуре на состав и структуру оксида графена, полученного по модифицированному методу Хаммерса. Отмечено, что при увеличении времени воздействия ультразвука на дисперсию оксида графена одновременно уменьшается латеральный размер чешуек и происходят количественные изменения функциональных кислородсодержащих групп, таких как гидроксильных, карбонильных и эпоксидных. Полученные материалы охарактеризованы различными физико-химическими методами (рентгенофазовый анализ, ИК-спектроскопия и спектроскопия комбинационного рассеяния, рентгеновская фотоэлектронная спектроскопия, сканирующая и просвечивающая электронная микроскопия, С,H,N-анализ и титрование по методу Боэма).
ВВЕДЕНИЕ
В настоящее время к графену и его производным, в частности оксиду графена (ОГ) [1–4], обращено пристальное внимание специалистов в различных областях. Данный наноматериал является достаточно перспективным, имеет и будет иметь множество применений: в электронике [5], в качестве катализатора [6], в датчиках [7], в энергетике [8], а также в электрохимии, особенно в химических источниках тока [9, 10].
ОГ – уникальное соединение, содержащее графеновые чешуйки, связанные ковалентными связями с кислородсодержащими функциональными группами, такими как гидроксильные (–OH), карбоксильные (–COOH), эпоксидные (–O–), карбонильные (С=O). Находятся обсуждаемые группы как в плоскости, так и на краях поверхности ОГ [11].
С помощью кислородсодержащих групп ОГ создает устойчивые дисперсии не только в гидрофильных [12], но и в полярных органических растворителях [13, 14]. Растворимость ОГ зависит от рН водных растворов и увеличивается при повышении водородного показателя. Происходит это благодаря разрыву водородных связей между карбоксигруппами расположенных на поверхности различных чешуек. В [15] показано, что при низких рН ОГ протонирован, образует агломераты, а при высоких значениях рН депротонированные карбоксигруппы образуют отдельные гидрофильные чешуйки ОГ и тем самым формируют коллоидные водные растворы.
В [16] показано, что при комнатной температуре содержание кислорода в ОГ постепенно снижается. На основании расчетов было доказано, что данное изменение напрямую связано с наличием водорода в структуре, благодаря которому происходит восстановление эпоксигрупп, что повышает концентрацию ОН-групп и приводит к образованию молекул воды.
Предполагается, что механические, сорбционные, электрохимические и электронные свойства будут зависеть не только от латерального размера чешуек ОГ, но и от их функционализации, а также от способа получения и очистки [17–21]. В [22] при увеличении времени окисления OH-группы переходят в эпоксидные, а при длительном нагревании до 200°С количество кислорода уменьшается. Также после отжига при температуре 200°С ~15% функциональных кислородсодержащих групп сохраняется на поверхности чешуек ОГ [23]. Предполагается, что варьирование условий воздействия ультразвука (УЗ) как одного из параметров для получения ОГ будет влиять на состав и структуру обсуждаемого продукта.
В связи с этим цель настоящей работы – изучение влияния УЗ-обработки в течение разного времени при комнатной температуре на распределение частиц по размерам, состав функциональных групп и их соотношение.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Исходные материалы для синтеза ОГ: природный китайский графит (Qingdao Guyu Graphite Co., Ltd) c размером частиц 150 мкм (99.9% чистоты), персульфат аммония (NH4)2S2O8, оксид фосфора(V) P2O5 “ч”, концентрированная серная кислота H2SO4 “ос.ч”, перманганат калия KMnO4 “чда”, пероксид водорода H2O2 (35–37%), соляная кислота НСl “хч” и дистиллированная вода H2O. Все химические реагенты использовали без дополнительной очистки.
Получение оксида графена. ОГ был получен по модифицированному методу Хаммерса [24] путем последовательного окисления чешуек природного графита (NH4)2S2O8, P2O5 и H2SO4 с последующей промывкой H2O до рН 7. Затем в H2SO4 на водяной бане с постоянным охлаждением добавляли полученный порошок интеркалированного соединения графена и порционно добавляли порошок KMnO4 в течение 1 ч, выдерживая температуру реакционной смеси <10°С. Далее по каплям добавляли H2O в течение 2 ч при температуре <35°С. Реакционную смесь выдерживали 2.5 ч, затем добавляли дополнительно дистиллированную воду и H2O2, наблюдая за изменением цвета суспензии с темно-коричневого на желто-зеленый. После седиментации, длящейся 18 ч, прозрачный супернатант декантировали, а осадок темно-коричневого цвета промывали водным раствором, содержащим 5 мас. % H2SO4, 10 мас. % НСl и 0.5 мас. % H2O2, и далее H2O до нейтрального рН. Таким образом был получен оксид графита с межплоскостным расстоянием 11–12 Å. Далее суспензию обрабатывали при помощи УЗ-установки в течение 20 мин. Небольшие количества частично окисленного исходного природного графита легко отделялись от конечной дисперсии центрифугированием при 6000 об./мин (исходная дисперсия ОГ – HGО-0).
Полученную водную дисперсию ОГ темно-коричневого цвета с концентрацией 3 мг/мл разделили на пять частей для дальнейших исследований, используя УЗ в течение 1, 2, 4 и 8 ч, с обозначением соответственно HGО-1, HGО-2, HGО-4 и HGО-8. Зависимость концентрации, массового содержания и рН от времени УЗ-обработки представлена в табл. 1.
Таблица 1.
Характеристики полученных образцов оксида графена
| Образцы ОГ | рН | с, мг/мл | ω, мас. % |
|---|---|---|---|
| HGO-0 | 2.53 | 3.00 | 1.34 |
| HGO-1 | 2.79 | 3.13 | 1.37 |
| HGO-2 | 2.56 | 3.19 | 1.51 |
| HGO-4 | 2.43 | 3.24 | 1.78 |
| HGO-8 | 2.05 | 3.41 | 1.9 |
Для изучения структуры, состава и свойств частиц ОГ все полученные образцы были исследованы методами физико-химического анализа: рентгенофазовый анализ (РФА) (спектрометр Bruker D8 Advance, излучение СuKα), ИК-спектроскопия (спектрометр VERTEX 70 v BRUKER методом нарушенного полного внутреннего отражения с использованием приставки GladyATR), спектроскопия комбинационного рассеяния (КР) (спектрометр RenishawInVia), рентгеновская фотоэлектронная спектроскопия (РФЭС) (электронный спектрометр для химического анализа Specs PHOIBOS 150 MCD, излучение MgKα), сканирующая электронная микроскопия (СЭМ) (микроскоп CarlZeissSupra 40-30-87), просвечивающая электронная микроскопия (ПЭМ) (установка JEOL JEM 1011 при ускоряющем напряжении 80–100 кВ), С,H,N-анализ (анализатор EA1108 CarloEbraInstruments) и измерение показателя активности ионов водорода (pH-150МИ).
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
УЗ-обработку ОГ проводили при следующих параметрах: удельная мощность 0.1–1 Вт/см3, частота 20.4 кГц. УЗ-обработка суспензии сопровождалась постоянным перемешиванием на магнитной мешалке. Отметим, что перемешивание способствовало гомогенизации дисперсии, а также заметно увеличивало количество образуемых пузырьков газа при кавитации. Образование данных пузырьков зависит от температуры, частоты обработки, свойств жидкости, поверхностного натяжения, вязкости используемого растворителя и силы сцепления. Известно, что внутри молекулы действуют высокие силы сцепления, препятствующие кавитации, а высокая температура, в свою очередь, способствует уменьшению когезионных сил (ван-дер-ваальсово и дипольное взаимодействие). Именно при высокой температуре происходит парообразная кавитация, снижающая при этом эффективность воздействия УЗ. Для того чтобы не допустить частичное восстановление ОГ при действии УЗ, обрабатывали ОГ на водяной бане при постоянном охлаждении.
Структурные характеристики
РФА. Согласно данным РФА природный графит успешно окисляется и функционализируется кислородсодержащими группами [25] (рис. 1). На рентгенограммах во всех образцах присутствует пик оксида графита в области 2θ = 12°, что соответствует межслойному d-расстоянию ~8 Å, вычисленному по уравнению Брэгга [26]. Анализ полученных данных показывает, что при увеличении воздействия УЗ в водных дисперсиях ОГ происходит снижение интенсивности рефлексов 001, характеризующих уменьшение количества слоев в структуре ОГ.
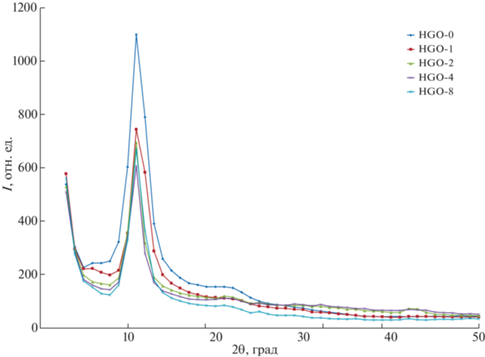
Титрование по методу Боэма [27]. Метод основан на определении концентрации кислородосодержащих групп на поверхности ОГ, таких как фенольных, лактонных и карбоксильных, за счет нейтрализации их основаниями различной силы – NaOH, Na2CO3, NaHCO3. Выявлено, что после УЗ-обработки (табл. 2) в течение 1 ч исходной дисперсии ОГ происходит увеличение количества фенольных групп и уменьшение карбоксильных групп более чем в 2 раза. Отметим, что при дальнейшем увеличении длительности УЗ-обработки дисперсий ОГ количество –ОН- и (‒(С=О)–О–)-групп уменьшается, а карбокси-групп – увеличивается. Исследование функциональных кислородсодержащих групп ОГ методом Боэма показало, что длительность воздействия УЗ влияет на изменение соотношения кислородсодержащих групп. Предполагается, что такой факт возможен благодаря взаимному превращению одних групп в другие и их частичного преобразования.
Таблица 2.
Содержание функциональных групп полученных образцов оксида графена
| Образцы ОГ | Общее количество групп, ммоль/г | Количество фенольных групп (–OH), ммоль/г | Количество карбоксильных групп (–СООН), ммоль/г | Количество лактонных групп (–(С=О)–О–), ммоль/г |
|---|---|---|---|---|
| HGО-0 | 7.46 | 2.57 | 1.18 | 3.71 |
| HGО-1 | 10.40 | 6.06 | 0.65 | 3.69 |
| HGО-2 | 9.12 | 4.40 | 1.10 | 3.62 |
| HGО-4 | 8.36 | 2.96 | 1.93 | 3.47 |
| HGО-8 | 7.13 | 1.26 | 2.67 | 3.20 |
С,H,N-анализ. По данным элементного анализа выявлено, что после дополнительных часов УЗ-обработки в химическом составе исследуемых образцов ОГ существенных изменений в массовом содержании элементов углерода, водорода и азота (табл. 3) не происходит. Полученные значения варьируются в пределах допустимой погрешности прибора. Высокое содержание кислорода в образцах свидетельствует об успешном окислении природного графита, а также о большом содержании кислородсодержащих групп. Атомное отношение С/О оксида графена во всех образцах составило ~0.9.
Таблица 3.
Элементный состав полученных образцов оксида графена
| Образцы ОГ | C, % | H, % | N, % | O, % (по разности) |
|---|---|---|---|---|
| HGО-0 | 47.01 | 2.79 | 0.00 | 50.20 |
| HGО-1 | 46.03 | 2.83 | 0.00 | 51.15 |
| HGО-2 | 46.98 | 2.75 | 0.00 | 50.28 |
| HGО-4 | 47.66 | 2.85 | 0.00 | 49.50 |
| HGО-8 | 47.96 | 2.93 | 0.00 | 49.11 |
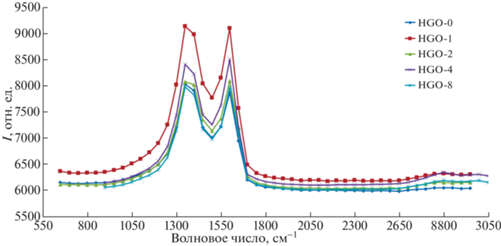
ИК-фурье-спектроскопия. Наличие определенных функциональных групп в структуре ОГ возможно определить при помощи еще одного основного исследования – ИК-фурье-спектроскопии. В высокочастотном диапазоне присутствуют главным образом сигналы валентных колебаний OH-групп (основания, фенолы и гидроксиды) – пик при 3121–3150 см–1 (рис. 2) [28]. При УЗ-обработке в течение 1 ч интенсивность резко возросла более чем в 2.5 раза, а пик в свою очередь сместился до 3150 см–1, но при дальнейшей обработке происходит возврат к первоначальному значению 3125 см–1. К валентным колебаниям карбонильных групп относится пик около 1725 см–1, это могут быть карбоксилы, кетоны или альдегиды [29]. Сигнал при 1620 см–1, предположительно, является составным: его соотносят с деформационными колебаниями воды, которая прочно связана со структурой ОГ, также в эту полосу вклад вносят колебания двойной связи С=С [30]. Валентные колебания ароматического кольца соответствуют поглощению при 1590–1575 см–1. Поглощение в области 1300 см–1 отвечает за искажения углеродного каркаса, взаимосвязанные с деформацией асимметричного строения эпоксидных групп. Схожа природа и в области 1200 см–1, соответствующей также искажению углеродного каркаса, но инициирующей колебания эпоксидных групп, которые были присоединены к графеновому скелету [31]. Полоса 1170 см–1 относится к колебаниям связей C–OH [32], а поглощения при 1046 см–1 – к колебаниям C–O-связей [33]. Наконец в области 977 см–1 могут лежать деформационные колебания эпоксидных групп.

При сравнении пиков образцов ОГ видно, что колебания функциональных кислородсодержащих групп и молекул воды проявляются не только в характерной, но и в высокочастотной области спектра. С увеличением количества часов УЗ-обработки дисперсии часть пиков, имеющих изначально слабо выраженный отклик, исчезает. Следовательно, такие группы, как лактонные, пропадают, а увеличение интенсивности образца HGО-1 указывает на повышение содержания –ОН-групп.
КР-спектроскопия является инструментальным методом для исследования графена и его производных [34], позволяющим оценить топологию сформированной графеновой структуры (дефекты структуры, присутствие химических примесей, а также количество слоев в чешуйке ОГ). Типичный КР-спектр листов ОГ состоит из D-, G-линий. Считается, что спектральная линия D (1330–1340 см–1) определяет деформации и дефекты в кристаллической структуре полученного ОГ, а G-линия (1580–1600 см–1) (графитоподобная зона) характеризует колебания системы sp2-углеродных связей [35, 36]. Полученные образцы содержат широкие, характерные для ОГ D- и G-полосы (рис. 3).
Сначала происходит потеря адсорбированной воды и лабильных функциональных групп, затем создаются новые домены OГ с меньшим латеральным размером, чем в НGO-0, более многочисленнные по количеству [37], что ведет к небольшому увеличению интенсивности.
Отношение интенсивностей D- и G-линий (ID/IG) отражает среднее расстояние между дефектами в структуре ОГ (табл. 4). Данное отношение полос пропорционально количественному отношению углерода со связями sp2- и sp3-гибридизации и количественному отношению атомов кислорода и атомов углерода (О/С) [38].
Рост интенсивности и ширины D-линии в образцах свидетельствует о повышении концентрации дефектов в структуре ОГ. Таким образом, чешуйки разбиваются и уменьшаются пропорционально в размерах и количестве слоев (увеличивается область обертонов выше 2500 см–1).
Методом РФЭС исследовали влияние длительного времени УЗ-обработки на химический состав поверхности ОГ. На рис. 4 представлены рентгеновские спектры, полученные с помощью обратной свертки, области C1s, записанные для каждого полученного образца OГ, которые состоят из четырех типов углеродных связей: C−C/C=C, C−O, C=O и O−C=O [43].
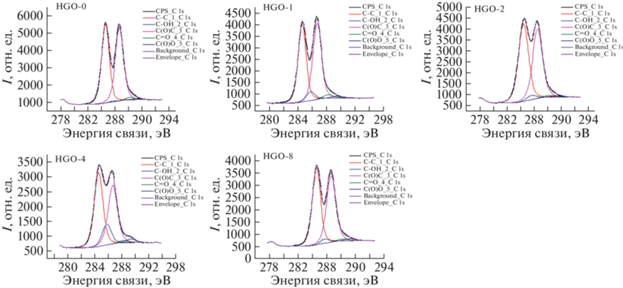
Наличие интенсивного пика при 284 эВ соответствует sp2-гибридизированным атомам углерода. Разложение C1s-спектра показывает наличие компонента C–OН при 287 эВ, связанных атомов углерода с гидроксильной группой и тремя высокоэнергетическими компонентами C=O при 288, CОO при 299 эВ и С–О при 287 эВ [44].
В образце НGO-0 наблюдается пик низкой интенсивности, относящийся к отклику гидроксильных и карбоксильных групп. При дальнейшей УЗ-обработке отмечается небольшое увеличение концентрации –СООН-группы. Возможно, наличие воды в дисперсии и присутствие воздуха в ОГ приводят к образованию некоторых новых кислородных групп (карбоксильных). Спад интенсивности эпоксидных групп свидетельствует о разрушении чешуек на более мелкие по латеральному размеру.
Морфологические характеристики
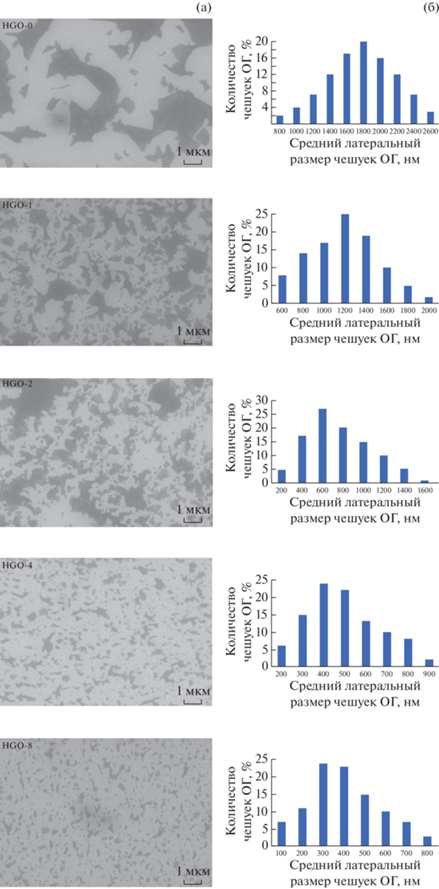
СЭМ-исследование морфологии поверхности образцов ОГ показало типичную слоистую структуру ОГ, состоящую из отдельных чешуек, латеральные размеры которых достигают нескольких микрон. Из рис. 5 видно, что в выбранных условиях диспергирования образцы ОГ имеют разное распределение частиц по размерам. Графеновые листы во всех образцах имеют неровные края и дефекты. По гистограмме распределения по размерам чешуйки HGО-0 составляют в среднем 1600–2000 нм, при увеличении времени УЗ-обработки средний размер чешуек полученного ОГ постепенно уменьшается, в среднем до 300 нм, а некоторые чешуйки достигают размера меньше 100 нм.
Рис. 5.
СЭМ-изображения (а) и соответствующие гистограммы распределений по размерам полученных образцов оксида графена (б). Гистограммы распределений ОГ по размерам получены путем подсчета более 200 чешуек для каждого образца.
ПЭМ-исследование слоистой структуры ОГ показало (рис. 6), что слои ОГ расслаиваются, но некоторые остаются в виде отдельных нанолистов, тесно связанных друг с другом сильными π–π-взаимодействиями. Наблюдаются также небольшие смятия, произошедшие из-за отслаивания листов, и сгибания. По гистограмме распределения по размерам отчетливо видно влияние времени обработки образцов ОГ на средний размер получаемых чешуек, который уменьшается пропорционально увеличению времени воздействия УЗ. На образце HGО-8 имеются графеновые листочки, характеризующиеся латеральным размером всего 100 нм.

ЗАКЛЮЧЕНИЕ
Изучено влияние длительности УЗ-обработки на характеристики ОГ различными физико-химическими методами анализа.
Методами СЭМ и ПЭМ показано, что при ультразвуковой обработке слоистого наноматериала – оксида графена – происходит изменение распределения частиц по размерам. Средний латеральный размер чешуек ОГ и их агломератов уменьшается с увеличением времени УЗ-обработки (с 2.6 мкм до 100 нм). Полученные дисперсии стабильны благодаря ван-дер-ваальсову взаимодействию между чешуйками ОГ и водородным связям 2D-кластеров воды в межслоевом пространстве.
Показано, что длительная УЗ-обработка согласно данным элементного анализа, КР-, ИК-спектроскопии, РФА и РФЭС не приводит к фазовым изменениям структуры ОГ и сопровождается несущественными изменениями концентрации определяемых элементов. Однако согласно методу Боэма имеет место изменение соотношения концентраций функциональных кислородсодержащих групп в структуре ОГ (особенно –ОН и –СООН).
Таким образом, изменяя длительность УЗ-воздействия на дисперсии ОГ, можно контролировать распределение чешуек ОГ по размерам и соотношение функциональных кислородсодержащих группировок в структуре ОГ.
Работа выполнена в рамках государственного задания ИОНХ РАН в области фундаментальных научных исследований.
Список литературы
Novoselov K.S., Geim A.K., Morozov S.V. et al. // Science. 2004. V. 306. P. 666. https://doi.org/10.1126/science.1102896
Kornilov D.Y., Gubin S.P. // Russ. J. Inorg. Chem. 2020. V. 65. P. 1965. https://doi.org/10.1134/S0036023620130021
Bai H., Li C., Shi G. // Adv. Mater. 2011. V. 23. P. 1089.
Иони Ю.В., Грошкова Ю.А., Губин С.П. и др. // Российские нанотехнологии. 2020. Т. 15. № 2. С. 181.
Dua V., Surwade S.P., Ammu S. et al. // Chem. Int. Ed. 2010. V. 49. P. 2154.
Scheuermann G.M., Rumi L., Steurer P. et al. // J. Am. Chem. Soc. 2009. V. 131. P. 8262.
Liu Y., Dong X., Chen P. // Chem. Soc. Rev. 2012. V. 41. P. 2283. https://doi.org/10.1039/C1CS15270J
Sun Y., Wu Q., Shi G. // Energy Environ. Sci. 2011. V. 4. P. 1113. https://doi.org/10.1039/C0EE00683A
Kornilov D.Y., Gubin S.P., Chuprov P.N. // Russ. J. Electrochem. 2017. V. 53. № 6. P. 622. https://doi.org/10.1134/s102319351706012x
Kornilov D.Yu., Gubin S.P. // Радиоэлектроника. Наносистемы. Информационные технологии. 2016. Т. 8. № 1. С. 39. https://doi.org/10.17725/rensit.2016.08.039
Singh V., Joung D., Zhai L. et al. // Prog. Mater. Sci. 2011. V. 56. P. 1178.
Boehm H.P., Clauss A., Fischer G.O. et al. // Proceedings of the 5th Conference on Carbon. Oxford: Pergamon Press, 1962. P. 73.
Park S., An J., Jung I. et al. // Nano Lett. 2009. V. 9. P. 1593.
Zhu Y., Stoller M.D., Cai W. et al. // ACS Nano. 2007. V. 4. № 2. P. 1227. https://doi.org/10.1021/nn901689k
Shih C.J., Lin S., Sharma R. et al. // Langmuir. 2012. V. 28. P. 235. https://doi.org/10.1021/la203607w
Jeong H.K., Lee Y.P., Jin M.H. et al. // Chem. Phys. Lett. 2009. V. 470. № 4. P. 255.
Chernysheva M.N., Rychagov A.Y., Kornilov D.Y. et al. // J. Electroanal. Chem. 2020. V. 858. P. 113774. https://doi.org/10.1016/j.jelechem.2019.113774
Kornilov D.Y., Kasharina L.A. // Inorgan. Mater.: Appl. Res. 2019. V. 10. № 5. P. 1072. https://doi.org/10.1134/S2075113319050125
Babaev A.A., Zobov M.E., Kornilov D.Y. et al. // Opt. Spectrosc. 2018. V. 125. № 6. P. 1014. https://doi.org/10.1134/S0030400X18120032
Rychagov A.Y., Gubin S.P., Chuprov P.N. et al. // Russ. J. Electrochem. 2017. V. 53. № 7. P. 721. https://doi.org/10.1134/s1023193517070102
Groshkova Y.A., Buslaeva E.Y., Gubin S.P. // Russ. Chem. Bull. 2019. V. 68. № 11. P. 2028. https://doi.org/10.1007/s11172-019-2662-5
Jeong H.K., Lee Y.P., Lahaye R.J. et al. // J. Am. Chem. Soc. 2008. V. 130. № 4. P. 1362.
Jeong H.K., Lee Y.P., Jin M.H. et al. // Chem. Phys. Lett. 2009. V. 470. № 4. P. 255.
Hummers W.S., Offeman R.E. // J. Am. Chem. Soc. 1958. V. 80. P. 1339.
Moussa S., Atkinson G., SamyEl-Shall M. et al. // J. Mater. Chem. 2011. V. 21. P. 9608. https://doi.org/10.1039/c1jm11228g
Zaaba N.I., Foo K.L., Hashima U. et al. // Procedia Engineering. 2017. V. 184. P. 469. https://doi.org/10.1016/j.proeng.2017.04.118
Боэм X.П. Химическая идентификация поверхностных групп. М.: Мир, 1968. 186 с.
Hantel M.M., Kaspar T., Nesper R. et al. // Chem. Eur. J. 2012. V. 18. P. 9125. https://doi.org/10.1002/chem.201200702
Seredych M., Rossin J.A., Bandosz T.J. // Carbon. 2011. V. 49. P. 4392.
Karthika P. // Soft Nanosci. Lett. 2012. V. 2. P. 59.
Hontoria-Lucas C., Lpez-Peinado A.J., Lopez-Gonzlez J. de D. et al. // Carbon. 1995. V. 33. № 11. P. 1585. https://doi.org/10.1016/0008-6223(95)00120-3
Galande C., Mohite A.D., Naumov A.V. et al. // Sci. Rep. 2011. V. 1. P. 85. https://doi.org/10.1038/srep00085
Fu M., Jiao Q., Zhao Y. et al. // J. Mater. Chem. A. 2014. V. 2. P. 735.
Saito R., Hofmann M., Dresselhaus G. et al. // Adv. Phys. 2011. V. 60. № 3. P. 413. https://doi.org/10.1080/00018732.2011.582251
Александров Г.Н., Смагулова С.А., Капитонов А.Н. и др. // Российские нанотехнологии. 2014. Т. 9. С. 18.
Kudin K.N., Ozbas B., Schniepp H.C. et al. // NanoLett. 2008. V. 8. P. 36. https://doi.org/10.1021/nl071822y
Stankovich S., Dikin D.A., Piner R.D. et al. // Carbon. 2007. V. 45. P. 1558. https://doi.org/10.1016/j.carbon.2007.02.034
Lucchese M.M., Stavale F., Ferreira E.H.M. et al. // Carbon. 2010. V. 48. P. 1592. https://doi.org/10.1016/j.carbon.2009.12.057
Matsumoto Y., Koinuma M., Kim S.Y. et al. // Appl. Mater. Interfaces. 2010. V. 2. № 12. P. 3461. https://doi.org/10.1021/am100900q
Stankovich S., Dikin D.A., Piner R.D. et al. // Carbon. 2007. V. 45. P. 1558. https://doi.org/10.1016/j.carbon.2007.02.034
Wang G., Yang J., Park J. et al. // J. Phys. Chem. C. 2008. V. 112. № 22. P. 8192.
Hong S., Jung S., Kang S. et al. // J. Nanosci. Nanotechnol. 2008. V. 8. № 1. P. 424.
Chen J., Yao B., Li C. et al. // Carbon. 2013. V. 64. P. 225. https://doi.org/10.1016/j.carbon.2013.07.055
Hunt A., Dikin D.A., Kurmaev E.Z. et al. // Adv. Funct. Mater. 2012. V. 22. P. 3950. https://doi.org/10.1002/adfm.201200529
Дополнительные материалы отсутствуют.
Инструменты
Российские нанотехнологии