

Нефтехимия, 2019, T. 59, № 2, стр. 194-199
Влияние содержания фосфора в носителе четырехкомпонентных NiMoW/P–Al2O3-катализаторов гидроочистки на их гидродесульфуризующую и гидрирующую активности
П. С. Солманов 1, *, Н. М. Максимов 1, В. В. Тимошкина 1, Н. Н. Томина 1, А. А. Пимерзин 1
1 Самарский государственный технический университет
Самара, Россия
* E-mail: spase07@yandex.ru
Поступила в редакцию 12.04.2018
После доработки 15.10.2018
Принята к публикации 07.07.2018
Аннотация
Синтезирована серия NiMoW/P–Al2O3-катализаторов с различным содержанием P2O5 в носителе (0; 0.5; 1.0; 2.0; 5.0 мас. % P2O5, соотношение Mo : W = 1 : 2). Предшественниками MoО3 и WО3 являлись гетерополикислоты H3PMo12O40 и H3PW12O40 соответственно, NiO – цитрат никеля. Для образцов проведено исследование морфологии и состава поверхности сульфидной фазы методами просвечивающей электронной микроскопии высокого разрешения (ПЭМ ВР) и рентгеновской фотоэлектронной спектроскопии (РФЭС), определена их каталитическая активность в реакции гидродесульфуризации дибензотиофена и гидрирования нафталина.
Введение в состав катализаторов гидроочистки неорганических модификаторов – один из основных способов повышения их активности. В качестве модифицирующих добавок предложено множество разнообразных соединений различных элементов. Модифицирование носителя катализаторов гидроочистки способствует изменению кислотно-основных свойств каталитической системы. Увеличение или уменьшение общей, бренстедовской или льюисовской кислотности ведет к изменению числа гидроксильных групп [1], а также к изменению силы взаимодействия предшественников активной фазы с поверхностью носителя [1–3], однородности их распределения [4], изменению морфологии сульфидной фазы [1, 5–9].
Наиболее широко для модифицирования носителя оксида алюминия используется фосфор [6–10]. Реже применялись и поэтому в меньшей степени исследованы добавки бора [11, 12], фтора [13, 14] и других модификаторов. Введение фосфора увеличивает активность катализаторов в отношении реакций гидродеазотирования (ГДА) [15–17] снижает скорость коксообразования [18], существенно увеличивает гидродесульфуризующую [3, 18, 19] и гидрирующую [5, 16, 18] активности катализатора в гидроочистке нефтяных фракций [20].
Один из наиболее изученных и часто предлагаемых модификаторов – фосфор (до 10 мас.% P2O5) [6–10]. Причины повышения активности катализаторов гидроочистки при введении оксида фосфора объясняются по-разному. Согласно одной точке зрения, оксид фосфора промотирует активность Ni–Mo/Al2O3-катализаторов в реакциях ГДА, способствуя образованию сильных и средних кислотных центров и повышая дисперсность активной фазы [21], что положительным образом влияет на каталитическую активность в реакциях ГДА и гидрирования (ГИД) [8, 9]. Напротив, авторы [22] считают, что введение оксида фосфора понижает дисперсность активной фазы. Таким образом, нет единой точки зрения на причины повышения активности при введении фосфора в катализаторы гидроочистки.
Несмотря на это, применение катализаторов на основе оксида алюминия, модифицированного фосфором, остается наиболее эффективным, поскольку такие катализаторы обладают повышенной устойчивостью к дезактивации и высокой гидрирующей активностью, как в отношении ароматических углеводородов (УВ), так и в отношении гетероорганических соединений, что обуславливает их значительный потенциал в процессах нефтепереработки, нефтехимии и водородной энергетики. [7, 10].
Модифицирование поверхности носителей катализаторов гидроочистки вышеперечисленными соединениями является одним из способов увеличения их каталитической активности, использующимся уже достаточно долгое время [23]. При этом следует отметить, что в большинстве случаев использование модифицирующих добавок сочетается с наиболее известными и часто используемыми соединениями-прекурсорами Mo(W)S2 и Co(Ni)S – а именно полимолибдатом (поливольфраматом) аммония и азотнокислыми солями Co и Ni. Работы, в которых одновременно применено модифицирование носителя и использованы гетерополикислоты (ГПК) Мо(W) как предшественники активной фазы, единичны [24, 25]. В связи с этим исследования одновременного модифицирования и использования ГПК в качестве предшественника активной фазы на сегодняшний день актуальны.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Cинтезирована серия NiMoW/P–Al2O3-катализаторов с различным содержанием P2O5 в носителе (0; 0.5; 1.0; 2.0; 5.0 мас. % P2O5, соотношение Mo : W = 1 : 2). Носитель γ-Al2O3 был модифицирован фосфором путем пропитки раствором ортофосфорной кислоты (х. ч.) по влагоемкости с последующими сушкой при температурах 60, 80, 110°C по 2 ч и прокаливанием при 550°C в течение 2 ч. С использованием синтезированных носителей была приготовлена серия образцов катализаторов – содержание P2O5 до 5 мас. % в расчете на Al2O3 (табл. 1). Катализаторы готовили методом пропитки носителя по влагоемкости совместным раствором соединений – предшественников активных компонентов (Мо и Ni): фосфорномолибденовой ГПК Н3РМо12О40 (х. ч.), фосфорновольфрамовой ГПК Н3РW12О40 (х. ч.), карбоната никеля (ч. д. а.); комплексообразователь – лимонная кислота. Сушку приготовленных катализаторов проводили при температурах 60, 80, 110°C в течение 2 ч.
Таблица 1.
Состав синтезированных NiMoW/P–Al2O3-катализаторов
| Катализатор | Содержание в катализаторе, мас. % | |||
|---|---|---|---|---|
| MoO3 | WO3 | NiO | P2O5 | |
| NiMoW/P(0)–Al2O3 | 3.5 | 11.3 | 2.7 | 0.0 |
| NiMoW/P(0.5)–Al2O3 | 3.5 | 11.3 | 2.7 | 0.5 |
| NiMoW/P(1.0)–Al2O3 | 3.5 | 11.3 | 2.7 | 1.0 |
| NiMoW/P(2.0)–Al2O3 | 3.5 | 11.3 | 2.7 | 2.0 |
| NiMoW/P(5.0)–Al2O3 | 3.5 | 11.3 | 2.7 | 5.0 |
Для анализа катализаторов набором физико-химических методов было проведено сульфидирование катализаторов в H2/H2S среде (H2/H2S = = 30/70 об.) при температуре 500°C в течение 2 ч. Контроль состава синтезированных катализаторов (Mo, W и Ni) был проведен с помощью EDX800HS рентгено-флуоресцентного анализатора.
Катализаторы анализировали методом ПЭМ ВР на приборе Tecnai G2 20 c LaB6-катодом при ускоряющем напряжении 200 кВ. ПЭМ-снимки получали в светлом поле в условиях недофокусировки без объективной апертуры (фазовый контраст) при увеличении около 200 000. Среднюю длину частиц MoS2 и число слоев в упаковке определяли, принимая в расчет не менее 400 частиц, расположенных на 10–15 различных участках поверхности катализаторов. Используя ПЭМ-изображения, можно получить основные геометрические характеристики активной фазы. Расчет количества центров различной локализации на слоях дисульфида молибдена производили по формулам, приведенным в [26].
Исследование катализаторов методом РФЭС проводили на спектрометре Axis Ultra DLD фирмы Kratos с использованием излучения AlKα (hν = = 1486.6 эВ). Шкала энергий связи (Есв) была предварительно откалибрована по положению пиков остовных уровней Au4f7/2 (84.0 эВ) и Cu2p3/2 (932.67 эВ). Образцы наносили на двухсторонний проводящий скотч. Эффект подзарядки, возникающий в процессе фотоэмиссии электронов, минимизировали с помощью облучения поверхности образца медленными электронами с помощью специального источника (flood gun). Для калибровки использовалась линия C1s (284.8 эВ) от углерода, присутствующего на поверхности катализатора. Шаг по энергии – 1 эВ для обзорного спектра, 0.1 эВ для отдельных линий C1s, Al2p, Ni2p, S2p, Mo3d и др.
Методом РФЭС был определен также состав частиц на поверхности катализаторов. С помощью программы CasaXPS (Version 2.3.16) выполнена деконволюция РФЭ-спектров (вычитание фона с использованием метода Ширли, аппроксимация линий функциями Гаусса (30%) и Лоренца (70%)) для расчета содержания Ni-, Мо- и W-атомных групп. Для каждого металла фотоэлектронный спектр был разложен на главный пик и сателлиты, энергия связи, ширина пика на половине высоте и относительная площадь которых математически связаны с соответствующими характеристиками главного пика. Для всех сульфидных образцов катализаторов было установлено наличие определенных атомных групп: Ni–O, Ni–S, Ni–Mo–W–S, Mo–O, Mo–S–O, Mo–S, W–O, W–S–O, W–S.
Разложение спектров позволяет определить относительное содержание атомных групп по формуле:
Были рассчитаны величины относительных содержаний для всех определенных атомных групп (для всех сульфидных катализаторов). Например, относительное содержание Ni в атомной группе Ni–Mo–W–S фазы вычисляли по формуле:
Проведено определение каталитической активности синтезированных образцов на микропроточной лабораторной установке при следующих параметрах: Т = 275, 300°С, об. скорость подачи сырья 60 ч–1, P = 3.0 МПа, H2/cырье = 300/1. В качестве модельных смесей использованы: 1) дибензотиофен (ДБТ) (0.3 мас. %) в толуоле (МС-1); 2) ДБТ (0.3 мас . %), нафталин (1.5 мас. %), хинолин (0.5 мас. %) (МС-2). Содержание модельных соединений (нафталина, ДБТ) и продуктов реакции определяли методом ГЖХ на хроматографе Кристалл-5000 (детектор ПИД, колонка из плавленного кварца с привитой фазой ZB-1; размеры колонки 30–0.00025 м; газ-носитель гелий).
Константа скорости псевдо-первого порядка ГДС ДБТ была рассчитана по выражению: $k_{{{\text{Г Д С }}}}^{{{\text{Д Б Т }}}} = W{\text{ln}}\frac{{{{C}_{0}}}}{C},\quad$где W – объемная скорость подачи сырья, ч–1, С0 – концентрация ДБТ в сырье, С – концентрация ДБТ в продуктах реакции, мас. %.
Константа скорости псевдо-первого порядка ГИД нафталина была рассчитана по выражению: ${{k}_{{{\text{Г И Д }}}}} = W{\text{ln}}\frac{{{{C}_{0}}}}{C}$, где W – объемная скорость подачи сырья, ч–1, С0 – концентрация нафталина в сырье, С – концентрация нафталина в продуктах реакции, мас. %.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
На рис. 1 представлены ПЭМ-изображения пяти сульфидных катализаторов – NiMoW/P(0)–Al2O3, NiMoW/P(0.5)–Al2O3, NiMoW/P(1.0)–Al2O3, NiMoW/P(2.0)–Al2O3, NiMoW/P(5.0)–Al2O3.
Рис. 1.
ПЭМ-изображения сульфидных (а) NiMoW/P(0)–Al2O3-, (б) NiMoW/P(0.5)–Al2O3-, (в) NiMoW/P(1.0)–Al2O3-, (г) NiMoW/P(2.0)–Al2O3-, (д) NiMoW/P(5.0)–Al2O3-катализаторов.

Методом ПЭМ ВР определена длина плит NiMoWS активной фазы, которая изменяется от 3.9 до 4.3 нм, среднее число слоев в ассоциатах NiMoWS возрастает с 1.9 до 2.6 при увеличении содержания P2O5 до 5.0 мас. % (табл. 2). Доли атомов Mo(W) в реберных и угловых центрах для различных образцов близки между собой, за исключением доли атомов Mo(W) в угловых центрах образца с содержанием фосфора в носителе 5.0 мас. %.
Таблица 2.
Морфологические характеристики активной фазы NiPMoW/Al2O3-катализаторов
| P2O5, мас. % | Средняя длина плит, нм | Среднее число упаковок | $f_{{\text{e}}}^{*}$ | $f_{{\text{c}}}^{*}$ | fe/$f_{{\text{c}}}^{*}$ |
|---|---|---|---|---|---|
| 0.0 | 3.9 | 1.9 | 24.5 | 5.3 | 4.4 |
| 0.5 | 3.9 | 2.2 | 24.9 | 5.5 | 4.8 |
| 1.0 | 3.9 | 1.9 | 24.7 | 5.4 | 3.5 |
| 2.0 | 4.0 | 2.5 | 24.4 | 5.2 | 3.3 |
| 5.0 | 4.3 | 2.6 | 23.0 | 4.4 | 5.9 |
Относительные концентрации атомных групп Ni, Mo и W, рассчитанные на основе деконволюции РФЭ-спектров сульфидных NiMoW/P–Al2O3-катализаторов представлены в табл. 3.
Таблица 3.
Содержания различных Ni-, Mo- и W-атомных групп на поверхности сульфидных NiMoW/P-Al2O3-катализаторов
| P2O5, мас. % | Ni, мас. % | Mo, мас. % | W, мас. % | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Ni–Mo–W–S | Ni–S | Ni–O | Mo–S | Mo–S–O | Mo–O | W–S | W–S–O | W–O | |
| 0.0 | 36 | 46 | 18 | 57 | 26 | 17 | 23 | 3 | 74 |
| 0.5 | 22 | 61 | 17 | 50 | 29 | 21 | 21 | 4 | 75 |
| 1.0 | 27 | 51 | 22 | 55 | 28 | 17 | 24 | 4 | 72 |
| 2.0 | 18 | 54 | 28 | 75 | 3 | 22 | 17 | 2 | 81 |
| 5.0 | 18 | 58 | 24 | 59 | 19 | 22 | 26 | 3 | 71 |
Соотношения атомных групп Mo (Mo–S, Mo–O) значительно отклоняются для образца с 0.5 мас. % P2O5 – наблюдается снижение доли атомных групп Mo–S до 50 мас. %, а для образца с 2.0 мас. % наблюдается повышение доли атомных групп Mo–S до 75 мас. %.
Изменение каталитической активности образцов в реакции гидрогенолиза дибензотиофена и гидрирования нафталина представлено на рис. 2 и 3. Для реакций гидрогенолиза ДБТ при температуре 275°C значения констант скоростей находятся в пределах 16.4–27.8 ч–1 для различных катализаторов, 300°C – 36.1–75.5 ч–1. Для реакции гидрогенолиза ДБТ в присутствии хинолина и нафталина (содержание в модельной смеси ДБТ (0.3 мас. %), нафталин (1.5 мас. %), хинолин (0.5 мас. %) при температуре 275°C для различных образцов kГДС в пределах 11.7–30.9 ч–1, 300°C – 11.5–42.9 ч–1. Для гидрирования нафталина kГИД изменяется от 0.79 до 1.83 при температуре 275°C и от 0.85 до 3.08 при температуре 300°C.
Рис. 2.
Зависимость$\quad$ между $k_{{{\text{Г Д С }}}}^{{{\text{Д Б Т }}}}$ и содержанием P2O5 для NiMoW/P–Al2O3-катализаторов: 1 – 275°C, МС-1; 2 – 300°C, МС-1; 3 – 275°C, МС-2; 4 – 300°C, МС‑2.
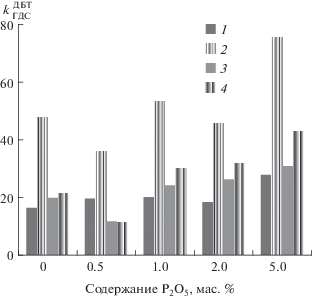
Рис. 3.
Зависимость между kГИД нафталина и содержанием P2O5 для NiMoW/P-Al2O3-катализаторов: 1 – 275°C, МС-2; 2 – 300°C, МС-2.

Изменение констант скорости ГДС для модифицированных образцов носит экстремальный характер. Минимум активности проявляет образец с содержанием фосфора 0.5 мас. %. Аналогичный характер зависимости наблюдается и для константы гидрирования. При этом образец с содержанием P2O5 2.0 мас. % имеет локальный минимум относительно образцов, содержащих 1.0 и 5.0 мас. % P2O5 как в случае активности в реакции ГДС, так и в случае активности в реакции ГИД. На рис. 4 представлена зависимость константы ГИД от константы ГДС. Для представленных точек построены линии тренда, удовлетворительно описывающиеся линейной функцией с доверительными коэффициентами R2 = 0.9904 и R2 = = 0.9886.
Рис. 4.
Зависимость между kГИД нафталина и $k_{{{\text{Г Д С }}}}^{{{\text{Д Б Т }}}}$ для NiMoW/P–Al2O3-катализаторов: 1– 275°C; 2– 300°C.
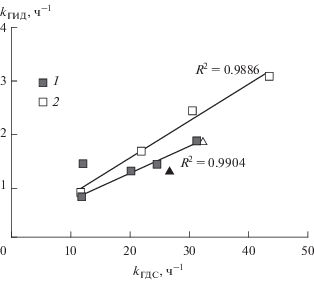
Полученные зависимости свидетельствуют о том, что реакция ГДС и реакция ГИД с высокой степенью вероятности протекают на одних и тех же каталитически активных центрах, что не согласуется с результатами, полученными в работе [27]. При этом следует отметить, что соотношение констант ГИД и ГДС для образца, содержащего 2.0 мас. % P2O5, полученные как для температуры 275°C, так и для температуры 300°C (на рис. 4 обозначены треугольными маркерами), отклоняются от линейной зависимости. Как было указано выше, для этого же образца существуют локальные минимумы по ГДС и ГИД. Морфология активной фазы при этом не имеет существенных отличий (табл. 2).
Анализ данных, полученных методом РФЭС, позволяет заключить, что образец с содержанием P2O5 2.0 мас. % имеет существенно большую концентрацию Ni, Mo и W в составе атомной группы Ni–Mo–W–S. Этот факт может свидетельствовать как об оптимальном соотношении предшественников активной фазы, так и об их восстанавливаемости при заданном соотношении в выбранных условиях сульфидирования. Более детальное понимание причин отличия каталитических свойств для данного образца требует дополнительных исследований.
Авторы выражают глубокую благодарность: к.х.н., н.с. Можаеву А.В. (СамГТУ, Самара) за проведенное сульфидирование образцов: к.ф.-м.н., с.н.с. Маслакову К.И. (МГУ им. М.В. Ломоносова, Москва) и д.х.н., в.н.с. Никульшину П.А. (СамГТУ, Самара) за проведенные запись и обработку РФЭС-спектров.
Работа выполнена при финансовой поддержке Правительства Российской Федерации. Постановление № 220 от 9 апреля 2010 г. Грант № 14.Z50.31.0038 от 20.02.2017.
Список литературы
Usman U., Takaki M., Kubota T., Okamoto Y. // Appl. Catal. 2005. V. 286. P. 148.
Li G., Li W., Zhang M., Tao K. // Appl. Catal. 2004. V. 273. P. 233.
Saih Y., Segawa K. // Catal. Today. 2003. V. 86. P. 61.
Damyanova S., Dimitrov L., Petrov L., Grange P. // A-ppl. Surf. Sci. 2003. V. 214. P. 68.
Ferdous D., Dalai A.K., Adjaye J., Kotlyar L. // Appl. Catal. 2005. V. 294. P. 80.
Ferdous D., Dalai A.K., Adjaye J. // J. Mol. Catal. 2005. V. 234. P. 169.
Huirache-Acuna R., Pawelec B., Rivera-Munoz E., Nava R., Espino J., Fierro J.L.G. // Appl. Catal. 2009. V. 92. P. 168.
Liu Ch., Yu Y., Zhao H. // Fuel Proc. Tech. 2004. V. 86. P. 449.
Usman U., Yamamoto T., Kubota T., OkamotoY. // Appl. Catal. 2007. V. 328. P. 219.
Zepeda T.A., Pawelec B., Obeso-Estrella R., Díaz de Léon J.N., Fuentes S., Alonso-N´ u˜nez G., Fierro J.L.G. // Appl. Catal. 2016. V. 180. P. 569.
Chen W., Maugé F., van Gestel J., Nie H., Li D., Long X. // J. of Catalysis. 2013. V. 304. P. 47.
Rashidi F., Sasaki T., Rashidi A.M., Kharat A.N., Jozani K.J. // J. of Catalysis. 2013. V. 299. P. 321.
Marchal Ch., Uzio D., Merdrignac I., Barre L., Geantet C. // Appl. Catal. 2012. V. 411. P. 35.
Guo X., Song M., Zhao X., Zhao L. // J. of Fuel Chemistry and Technology. 2016. V. 44. P. 1326.
Ferdous D., Dalai A.K., Adjaye J. // Appl. Catal. 2004. V. 260. P.153.
Ding L., Zhang Z., Zheng Y., Ring Z., Chen J. // Appl. Catal. 2006. V. 301. P. 241.
Kunisada N., Choi K.-H., Korai Y., Mochida I., Nakano K. // Appl. Catal. 2005. V. 279. P. 235.
Maity S.K., Ancheyta J., Rana M.S., Rayo P. // Catalysis Today. 2005. V. 109. P. 42.
Jirátová K., Spojakin A., Kaluža L., Palcheva R., Balabánová J., Tyuliev G. // Chinese J. of Catalysis. 2016. V. 37. P. 258.
Rayo P., Ramírez J., Torres-Mancera P., Marroquin G., Maity K., Ancheyta J. // Fuel. 2012. V. 100. P. 34.
Liu C., Yu Y., Zhao H. // Fuel Processing Technology. 2004. V. 86. P. 449.
Sun M., Nicosia D., Prins R. // Catalysis Today. 2003. V. 86. P. 173.
Topsoe H., Clausen B.S., Massoth F.E. // Science and Technology. 1996. V. 11. P. 310.
Pleshakova N.A., Tyshchenko V.A., Tomina N.N., Pimerzin A.A. // Petrol. Chemistry. 2008. V. 48. P. 346.
Томина Н.Н., Антонов С.А., Максимов Н.М., Самсонов М.В., Пимерзин А.А. // Нефтехимия. 2015. Т. 55. № 5. С. 434 [ Petrol. Chemistry. 2015. V. 55. P. 578].
Mingfeng L., Huifeng L., Feng J., Chu Y., Nie H. // Catalysis Today. 2010. V. 149. P. 35.
Сальников В.А, Никульшин П.А., Пимерзин А.А. // Нефтехимия. 2013. Т. 53. № 4. С. 267 [Petrol. Chemistry. 2013. V. 53. P. 233].
Дополнительные материалы отсутствуют.