

Нефтехимия, 2019, T. 59, № 3, стр. 256-260
Энергии активации образования метана и углекислого газа на кобальтовых катализаторах процесса Фишера–Тропша, нанесенных на углеродные нанотрубки
С. А. Черняк 1, *, А. А. Бурцев 1, С. В. Савилов 1, 2, В. В. Лунин 1, 2
1 Московский государственный университет имени М.В. Ломоносова, Химический факультет
Москва, Россия
2 Институт нефтехимического синтеза им. А.В. Топчиева РАН
Москва, Россия
* E-mail: chernyak.msu@gmail.com
Поступила в редакцию 15.11.2018
После доработки 01.12.2018
Принята к публикации 10.12.2018
Аннотация
В работе проведена оценка эффективных энергий активации реакций метанирования СО и конверсии водяного газа в процессе Фишера–Тропша в присутствии кобальтовых катализаторов, нанесенных на окисленные и неокисленные углеродные нанотрубки. Установлено, что энергия метанирования СО не зависит от степени окисления носителя и варьируется в узком диапазоне от 135 до 153 кДж/моль, в то время как значения этого параметра для реакции конверсии водяного газа повышаются при окислении носителя с 40 до 201 кДж/моль в связи с изменением механизма протекания процесса.
Более полувека нас с С.Н. Хаджиевым связывала дружба и любовь к науке. О науке он мог говорить в любое время суток. Настоящий выпуск журнала посвящен памяти выдающегося ученого, великого сына своего народа, государственного человека. Наш долг – служить науке и России так же достойно и преданно, как прожил свою жизнь академик РАН С.Н. Хаджиев.
В.В. Лунин
Процесс Фишера–Тропша (ПФТ) представляет собой конверсию синтез-газа в углеводороды (УВ) и реализуется в промышленности с использованием железных или кобальтовых катализаторов [1]. В результате ПФТ образуется смесь углеводородов С1–С35+, которую в дальнейшем преобразуют в дизельную, керосиновую или бензиновую фракции путем гидрокрекинга [2]. Кобальтовые катализаторы отличаются от железных более высокой устойчивостью к дезактивации и способностью к производству длинных УВ [3]. Основной реакцией, происходящей в ПФТ на кобальтовых катализаторах, является образование линейных парафинов:
(1)
$n{\text{CO }} + 2n{{{\text{H}}}_{2}}\quad \to \left( {--{\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} --} \right)n + n{{{\text{H}}}_{2}}{\text{O}}.$При этом существуют также побочные процессы, к которым можно отнести метанирование СО (2), реакцию конверсии водяного газа (КВГ) (3) и диспропорционирование СО (4):
(2)
${\text{CO}} + 3{{{\text{H}}}_{2}} \to {\text{C}}{{{\text{H}}}_{4}} + {{{\text{H}}}_{2}}{\text{O}},$(3)
${\text{CO }} + {{{\text{H}}}_{2}}{\text{О }} \to {\text{C}}{{{\text{O}}}_{2}} + {{{\text{H}}}_{2}},$Основные индикаторы протекания процессов 2–4 – метан и углекислый газ. Предложено два возможных механизма реакции КВГ: регенеративный механизм (окислительно-восстановительный), при котором поверхность окисляется адсорбированной водой и затем восстанавливается газообразным или адсорбированным СО до СО2 [4, 5], а также ассоциативный, который включает в себя диссоциативную адсорбцию воды на поверхности катализатора с образованием реакционноспособных гидроксильных групп. Последние, находясь вблизи адсорбированной молекулы СО, формируют промежуточное соединение, подобное формиату, которое распадается на углекислый газ и водород [6]. Энергия активации реакции КВГ зависит от металла и носителя, а также от наличия/отсутствия промотора и может составлять, по разным данным, от 40 до 130 кДж/моль [4, 7]. Для катализаторов на основе оксидов церия за счет подвижного кислорода решетки осуществляется окислительно-восстановительный механизм [8], тогда как на других катализаторах возможно существование механизмов обоих типов [5]. Процесс диспропорционирования СО в случае железа, кобальта и никеля может протекать через образование карбидов (в случае железа) или через механизм растворения–осаждения [9], но при температурах ПФТ данная побочная реакция возможна, в основном, только в случае железа, которое образует стабильные карбиды при низких температурах. Это и приводит к сильной дезактивации железных катализаторов за счет зауглероживания их поверхности [3].
Механизм метанирования СО на кобальтовом катализаторе подробно исследован в работе [10]. Установлено, что в ПФТ активация СО протекает через диссоциацию с участием водорода: образуется промежуточная форма CH2O*, которая разлагается до ${\text{CH}}_{x}^{*}$, после чего происходит гидрирование фрагмента CHx до метана.
Кобальтовые катализаторы ПФТ, как правило, представляют собой систему кобальт–носитель. В качестве подложки используют оксидные материалы, такие как оксиды алюминия, кремния или титана [1, 11], а также цеолиты [12]. Вместе с тем, в последнее десятилетие активно развивается направление исследований структурированных углеродных носителей для кобальтовых катализаторов ПФТ, которые обладают более высокими химической инертностью, тепло- и электропроводностью, что позволяет синтезировать активные теплопроводящие системы без введения дорогостоящих структурных промоторов [13–15]. Среди них можно выделить углеродные нанотрубки (УНТ), единственный структурированный углеродный наноматериал, производимый по всему миру в промышленных масштабах [16]. При этом показано, что для равномерного распределения кобальта по поверхности УНТ, а также для повышения стабильности такого катализатора к спеканию, необходима окислительная обработка носителя [17]. В данной работе изучено влияние такой модификации поверхности УНТ на энергии активации реакций метанирования и образования СО2.
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Многостенные УНТ синтезировали пиролизом паров гексана над порошковым катализатором Со-Мо/MgO в кварцевом трубчатом реакторе при температуре 750°С, а затем отмывали концентрированным раствором соляной кислоты и окисляли кипячением в концентрированной азотной кислоте в течение 1, 3 и 15 ч [18–20]. Образец отмытых неокисленных УНТ обозначали как “УНТч”, а окисленных – “УНТx”, где х – время окисления в ч. Кобальтовые катализаторы готовили пропиткой носителя спиртовым раствором нитрата кобальта. После этого растворитель упаривали одновременно с ультразвуковой обработкой в УЗ-ванне (“Ферропласт-Медикал”, Россия) при 60°С. Полученные материалы сушили в сушильном шкафу при 130°С в течение 16 ч. Расчетное массовое содержание кобальта в катализаторах составляло 15% (далее – Co/Sup, где Sup – обозначение соответствующего носителя).
Каталитические испытания проводили в вертикальном проточном кварцевом реакторе диаметром 10 мм, помещенном в трубчатую печь по методике, описанной ранее [20]. Нанесенный нитрат кобальта разлагали при нагревании до 400°С в атмосфере азота (30 мл/мин). Катализатор подвергали предварительному восстановлению в токе водорода (скорость потока 40 мл/мин) при 400°С в течение 4 ч. Гидрирование CO проводили при атмосферном давлении и температурах 190, 205 и 220°С, ступенчато поднимая температуру после достижения стационарного состояния в течение как минимум 20 ч при каждой температуре и при соотношении газов H2 : CO : N2 = 2 : 1 : 0.7, которое задавали с помощью формирователя газовых потоков фирмы “Мета-Хром” (Россия). Азот использовали в качестве внутреннего стандарта. Суммарная скорость потока составляла 2 л/(гкат ч), масса катализатора – 2 г. Продукты реакции исследовали на газовом хроматографе “Agilent Technologies 6890N” (США). Разделение углеводородов осуществляли на капиллярной колонке DB-Petro и пламенно-ионизационном детекторе. Определение CO2, CO и CH4 проводили на катарометре и колонке HayeSep c наполнителем Po-rapak R.
Методом просвечивающей электронной микроскопии (ПЭМ) на приборе “JEOL 2100F” (“JEOL Ltd.”, Япония) с ускоряющим напряжением 200 кВ исследовали структуру катализаторов. Предварительно катализаторы пассивировали при комнатной температуре после окончания каталитической реакции газовой смесью 1%О2 в Ar. Удельную площадь поверхности определяли методом низкотемпературной сорбции азота на сорбтометре “AUTOSORB-1C/MS/TPR” (“Quantachrome”, США) по модели БЭТ. Дегазацию образцов осуществляли при температуре 250°С в течение 3 ч. Рентгеновскую фотоэлектронную спектроскопию (РФЭС) использовали для определения состава поверхности УНТ. Спектры регистрировали на спектрометре Axis Ultra DLD (“Kratos Analytical”, Великобритания) с использованием монохроматического AlKα излучения (1486.6 эВ). Обзорные РФЭС спектры получали при энергии пропускания анализатора равной 160 эВ и шаге 1 эВ. Рентгеновский дифракционный анализ (РФА) проводили с использованием автоматического порошкового дифрактометра STADI-P (“STOE & Cie GmbH”, Германия) с СuKα (λ = 1.54060 Å) излучением в геометрии Брегга–Брентано. Размер частиц рассчитывали по уравнению Шерера. Перед проведением экспериментов катализаторы предварительно подвергали прокаливанию в токе азота (30 мл/мин) при 300°С.
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Содержание кислорода и удельная поверхность носителей представлены в табл. 1. Окисление материала привело к небольшому росту поверхности, а также к значительному увеличению содержания кислорода.
Таблица 1.
Характеристики носителей: атомное содержание поверхностного кислорода, определенное методом РФЭС, и удельная площадь поверхности (Sуд)
| Носитель | Содержание кислорода, ат. % (РФЭС) | Sуд, м2/г |
|---|---|---|
| УНТч | 0.3 | 192 |
| УНТ1 | 6.5 | 225 |
| УНТ3 | 8.0 | 233 |
| УНТ15 | 8.8 | 232 |
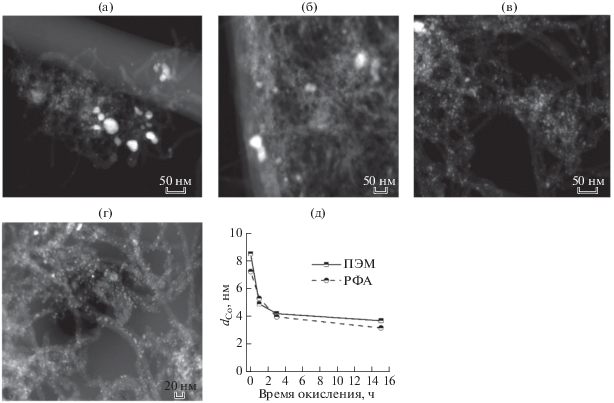
По данным ПЭМ, размер частиц кобальта в исследованных катализаторах различался (рис. 1): наличие крупных (50 нм) кристаллитов кобальта наряду с мелкими (4–10 нм) в образце Со/УНТч обусловлено отсутствием центров кристаллизации металла и малым количеством поверхностных дефектов, которые способствуют стабилизации частиц [17, 21]. Окисление поверхности носителя привело к снижению среднего диаметра частиц кобальта с 7–9 до 3–4 нм по данным ПЭМ и РФА (рис. 1д).
Рис. 1.
Микрофотографии ПЭМ катализаторов Со/УНТч (а), Со/УНТ1 (б), Со/УНТ3 (в), Со/УНТ15 (г); средний размер частиц кобальта, определeнный методами ПЭМ и РФА, в зависимости от времени обработки носителя азотной кислотой (д).
Данные по конверсии СО, селективности катализаторов по метану и СО2 при разных температурах представлены в табл. 2. Заметно, что конверсия СО и селективность по метану на всех катализаторах росла с повышением температуры синтеза, что соответствует литературным данным [22, 23], а наибольшие значения этих параметров зафиксированы на катализаторах СоУНТ3 и СоУНТ15. В этих образцах наблюдался наименьший размер частиц кобальта, что соответствует высокой дисперсии металла и, следовательно, высокой активности, вместе с тем, это также приводит к повышению концентрации водорода на активных центрах и к увеличению выхода метана [24, 25].
Таблица 2.
Конверсия СО (XСО), мольная селективность (S) и выход продукта в мольных процентах от исходного СО (Y = XCO × S/100) при разной температуре ПФТ. Результаты, полученные при 190°С, взяты из работы [17]. P = 1 атм, GHSV = 2 л/гкат/ч
| Катализатор | Т, °С | XСО, % | ${{S}_{{{\text{C}}{{{\text{H}}}_{4}}}}},$ % | ${{S}_{{{\text{C}}{{{\text{O}}}_{2}}}}},$ % | ${{Y}_{{{\text{C}}{{{\text{H}}}_{4}}}}},$% | ${{Y}_{{{\text{C}}{{{\text{O}}}_{2}}}}},$% |
|---|---|---|---|---|---|---|
| CoУНТч | 190 | 3 | 9 | 6.4 | 0.27 | 0.19 |
| 205 | 4 | 19 | 6.3 | 0.76 | 0.25 | |
| 220 | 9 | 25 | 4.6 | 2.25 | 0.41 | |
| CoУНТ1 | 190 | 10 | 14 | 3.9 | 1.40 | 0.39 |
| 205 | 18 | 31 | 4.0 | 5.58 | 0.72 | |
| 220 | 30 | 53 | 7.9 | 15.90 | 2.37 | |
| CoУНТ3 | 190 | 12 | 23 | 6.7 | 2.76 | 0.80 |
| 205 | 23 | 41 | 7.9 | 9.43 | 1.82 | |
| 220 | 40 | 63 | 17.0 | 25.20 | 6.80 | |
| CoУНТ15 | 190 | 14 | 23 | 3.3 | 3.22 | 0.46 |
| 205 | 26 | 45 | 6.2 | 11.70 | 1.61 | |
| 220 | 48 | 63 | 23.1 | 30.24 | 11.09 |
Для оценки эффективных энергий активации метанирования и выделения СО2 использовали значения выхода соответствующего продукта в мольных процентах от исходного количества введенного СО (Y, табл. 1), полученное при трех разных температурах. Аррениусовские зависимости строили в координатах ln(Y) – 1/T, по углу наклона определяли значения Ea. Результаты представлены на рис. 2. Энергия активации метанирования не зависела от структуры носителя и составляла ~135–153 кДж/моль. Полученные значения близки к литературным данным, полученным для кобальтового катализатора, нанесенного на оксид алюминия (113 ± 17 кДж/моль [26]) и на диоксид кремния (77–158 кДж/моль, [27]) и несколько ниже, чем для катализаторов Со/С (167–183 кДж/моль, [27]). Таким образом, можно заключить, что окисление носителя и связанное с ним изменение размера частиц металла, не влияет на энергию активации метанирования CO.
Рис. 2.
Результаты оценки эффективной энергии активации процессов метанирования и образования СО2 в ПФТ на кобальтовых катализаторах, нанесенных на УНТ.
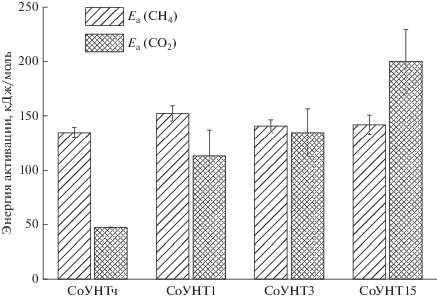
В отличие от метанирования, процесс образования СО2 сильно зависел от структуры катализатора. Учитывая то, что на кобальте, в отличие от железа, с выделением СО2 происходит преимущественно реакция КВГ, можно сделать допущение о том, что рассчитанная эффективная энергия соответствует именно этой реакции. Значения Ea растут при окислении носителя. Это, по-видимому, свидетельствует о смене механизма протекания КВГ при переходе от катализатора, нанесенного на неокисленные УНТ к образцу на окисленных УНТ. Ea для образца СоУНТч (~48 кДж/моль) близко к данным, полученным для широкого круга катализаторов низкотемпературного режима КВГ, тогда как остальные значения (114–201 кДж/моль) ближе к результатам для высокотемпературного режима (при температурах более 230°С) [6]. При этом известно, что низкотемпературный синтез протекает, в основном, по ассоциативному механизму, описываемому моделью Ленгмюра–Хиншельвуда, тогда как высокотемпературный – по окислительно-восстановительному [5, 6]. Полученные результаты можно объяснить высокой интенсивностью ПФТ в случае катализаторов на окисленных УНТ, которая может приводить к возникновению локальных перегревов и направлять процесс по окислительно-восстановительному механизму.
Из приведенных данных видно, что в случае УНТ15 выход СО2 при низких температурах (190 и 205°С) ниже, чем в случае УНТ3, тогда как повышение температуры до 220°С резко увеличивает это значение для УНТ15. Такая разница в выходе может быть обусловлена диффузионным ограничением, ввиду нахождения части кобальтовых частиц внутри каналов УНТ, так и облегчением протекания окислительно-восстановительных процессов на поверхности металла при повышенной температуре.
В некоторых работах утверждается, что активация воды осуществляется на носителе, тогда как СО – на металле [6]. Можно было бы предположить, что снижение размера частиц металла влияет на энергию реакции КВГ за счет повышения количества поверхностных атомов кобальта, находящихся вблизи носителя. В работе [4] показано, что дисперсия металла не влияет на кинетику этого процесса. В то же время, в этой же работе обсуждается разница в энергиях активации реакции КВГ в случае перехода с оксидного носителя (Al2O3, SiO2) платинового катализатора к углеродному (активированный уголь). Отмечено, что при одинаковых значениях Ea для оксидных систем, энергия для системы с углеродным носителем повышается. Авторы связывают это с более сложным и медленным механизмом активации воды на углероде, в отличие от полярных оксидных материалов. Применительно к настоящей работе это может означать, что в случае более дефектного носителя УНТ15 активация воды происходит медленнее и лимитирует скорость реакции.
Таким образом, можно заключить, что энергия активации, а, следовательно, и механизм процесса метанирования СО не зависит от степени окисления носителя УНТ, в то время как в случае КВГ изменение структуры носителя и размера частиц металла сильно влияет на механизм реакции.
Авторы выражают благодарность сотрудникам химического факультета МГУ к.х.н. К.И. Маслакову, к.х.н. А.В. Егорову и к.х.н. Е.В. Сусловой за помощь в осуществлении исследования. Работа выполнена при финансовой поддержке Российского фонда фундаментальных исследований (проект № 18-33-00390 мол_а) с использованием оборудования, приобретенного за счет средств Программы развития Московского университета.
Список литературы
van de Loosdrecht J., Botes F.G., Ciobica I.M., Ferreira A., Gibson P., Moodley D.J., Saib A.M., Visagie J.L., Weststrate C.J., Niemantsverdriet J.W. // Comprehensive Inorganic Chemistry II (2nd Edition). Ch. 7.20. (Reedijk J. and Poeppelmeier K., Editors). Elsevier, 2013. P. 525.
Eliseev O.L., Savost’yanov A.P., Sulima S.I., Lapidus A.L. // Mendeleev Commun. 2018. V. 28. № 4. P. 345.
Khodakov A.Y., Chu W., Fongarland P. // Chem. Rev. 2007. V. 107. № 5. P. 1692.
Spencer M.S. // Catal. Lett. 1995. V. 32. № 1. P. 9.
Mendes D., Chibante V., Mendes A., Madeira L.M. // Ind. Eng. Chem. Res. 2010. V. 49. № 22. P. 11269.
Smirniotis P., Gunugunuri K. Water Gas Shift Reaction: Research Developments and Applications. Elsevier Science, 2015. 280 p.
Grenoble D.C., Estadt M.M., Ollis D.F. // J. Catal. 1981. V. 67. № 1. P. 90.
Li Y., Fu Q., Flytzani-Stephanopoulos M. // Appl. Catal. B. 2000. V. 27. № 3. P. 179.
Boehm H.P. // Carbon. 1973. V. 11. № 6. P. 583.
Yang J., Qi Y., Zhu J., Zhu Y.-A., Chen D., Holmen A. // J. Catal. 2013. V. 308. P. 37.
Savost’yanov A.P., Narochnyi G.B., Yakovenko R.E., Mitchenko S.A., Zubkov I.N. // Petrol. Chemistry. 2018. V. 58. № 1. P. 76.
Asalieva E.Y., Kul’chakovskaya E.V., Sineva L.V., Mordkovich V.Z., Bulychev B.M. // Petrol. Chemistry. 2016. V. 56. № 3. P. 275.
Xiong H., Jewell L.L., Coville N.J. // ACS Catalysis. 2015. V. 5. № 4. P. 2640.
Fu T., Li Z. // Chem. Eng. Sci. 2015. V. 135. P. 3.
Kulikova M.V., Zemtsov L.M., Sagitov S.A., Efimov M.N., Krylova A.Y., Karpacheva G.P., Khadzhiev S.N. // Solid Fuel Chemistry. 2014. V. 48. № 2. P. 105.
Yan Y., Miao J., Yang Z., Xiao F.X., Yang H.B., Liu B., Yang Y. // Chem. Soc. Rev. 2015. V. 44. № 10. P. 3295.
Chernyak S.A., Suslova E.V., Ivanov A.S., Egorov A.V., Maslakov K.I., Savilov S.V., Lunin V.V. // Appl. Catal. A. 2016. V. 523. P. 221.
Savilov S.V., Chernyak S.A., Paslova M.S., Ivanov A.S., Egorova T.B., Maslakov K.I., Chernavskii P.A., Lu L., Lunin V.V. // ACS Appl. Mater. Interfaces. 2018. V. 10. № 24. P. 20983.
Chernyak S.A., Ivanov A.S., Strokova N.E., Maslakov K.I., Savilov S.V., Lunin V.V. // J. Phys. Chem. C. 2016. V. 120. № 31. P. 17465.
Черняк С.А., Селяев Г.Е., Суслова Е.В., Егоров А.В., Маслаков К.И., Харланов А.Н., Савилов С.В., Лунин В.В. // Кинетика и катализ. 2016. V. 57. № 5. P. 645.
Karimi A., Nasernejad B., Rashidi A.M., Tavasoli A., Pourkhalil M. // Fuel. 2014. V. 117B. P. 1045.
Chernyak S.A., Suslova E.V., Egorov A.V., Lu L., Savilov S.V., Lunin V.V. // Fuel Process. Technol. 2015. V. 140. P. 267.
Zaman M., Khodadi A., Mortazavi Y. // Fuel Process. Technol. 2009. V. 90. № 10. P. 1214.
den Breejen J.P., Radstake P.B., Bezemer G.L., Bitter J.H., Frøseth V., Holmen A., Jong K.P.d. // J. Am. Chem. Soc. 2009. V. 131. № 20. P. 7197.
Xiong H., Motchelaho M.A.M., Moyo M., Jewell L.L., Coville N.J. // J. Catal. 2011. V. 278. № 1. P. 26.
Vannice M.A. // J. Catal. 1975. V. 37. № 3. P. 462.
Reuel R.C., Bartholomew C.H. // J. Catal. 1984. V. 85. № 1. P. 78.
Дополнительные материалы отсутствуют.