

Нефтехимия, 2019, T. 59, № 3, стр. 282-288
Особенности образования изопрена из изоамилового спирта в микроканалах конвертера, модифицированного наноразмерными каталитическими Fe–Cr-содержащими системами
А. С. Федотов 1, Д. О. Антонов 1, В. И. Уваров 2, М. В. Цодиков 1, P. Sébastien 3, S. Heyte 3, F. Dumeignil 3
1 Институт нефтехимического синтеза им. А.В. Топчиева РАН
Москва, Россия
2 Институт структурной макрокинетики и проблем материаловедения РАН
Московская область, Черноголовка, Россия
3 Univ. Lille, CNRS, Centrale Lille, ENSCL, Univ. Artois
UMR 8181 – UCCS – Unité de Catalyse et Chimie du Solide, Lille, France
Поступила в редакцию 12.11.2018
После доработки 01.12.2018
Принята к публикации 10.12.2018
Аннотация
Разработан двухстадийный способ получения изопрена из изоамилового спирта путем последовательного осуществления стадий дегидратации изоамилового спирта и дегидрирования образующихся изоамиленов с использованием пористых керамических конвертеров, полученных самораспространяющимся высокотемпературным синтезом, в порах которых золь-гель-методом синтезированы системы Fe–Cr, содержащие осажденные частицы железа и хрома субнанометрового масштаба. Показано, что биметаллическая каталитическая система, полученная раздельным нанесением Fe и Cr, обладает наиболее высокой активностью в ряду изученных в реакции дегидрирования изоамиленов, на котором при 24-х кратном разбавлении водой и температуре 600оС выход изопрена составляет до ~10%, селективность ~23%, а производительность ~0.13 лизопрен/(ч гакт.комп.). Конверсия изоамиленовой фракции при этом составляет 46%.
ВВЕДЕНИЕ
Изопрен (2-метил-1,3-бутадиен) – один из основных мономеров для получения искусственных каучуков, синтезу которых посвящено множество работ, лежащих в основе тонких химических технологий. Несмотря на богатую историю разработки способов получения этого мономера, проблема развития высокоселективных превращений остается нерешенной до настоящего времени. Главная сложность при осуществлении данных процессов заключается в их высокой эндотермичности, что обуславливает необходимость применения высоких температур внутри реактора, в результате чего происходит интенсификация побочных реакций, приводящих к быстрому закоксовыванию катализаторов. В этой связи, в ведущих научных школах мира ведется поиск альтернативных подходов к разработке экологически приемлемых и эффективных процессов получения изопрена [1].
Изопрен является вторым по важности после 1,3-бутадиена мономером с мировым объемом производства, превышающим 1 млн т/г [2]. Технологические методы синтеза изопрена аналогичны бутадиеновым. Наиболее распространенный вариант получения изопрена — извлечение из С5-фракций пиролиза нефти, содержащей обычно 15–20% изопрена. Эта фракция образуется в качестве побочного продукта при получении этилена и пропилена, причем в весьма небольших количествах, не более 14–23% от выхода этилена.
Важно отметить, что современные процессы получения изопрена являются многостадийными и включают такие стадии, как нефтепереработка, органический синтез, разделение углеводородных смесей и др. Использование же в качестве сырья ископаемых топлив увеличивает экологическую нагрузку на места его производства [1]. Таким образом, все предложенные до сих пор способы получения изопрена являются дорогими.
С точки зрения “зеленой химии” для получения изопрена гораздо проще и дешевле утилизировать малоценные побочные продукты спиртового брожения, например, изоамиловый спирт [3], последовательно осуществляя процессы его дегидратации и дегидрирования образующихся изоамиленов в изопрен.
Мировое производство биоэтанола, получаемого в процессе переработки растительного сырья для использования в качестве биотоплива, составляет около 97 млрд л (или 77 млрд т) [4]. При этом обычное содержание различных примесей, образующихся в процессе брожения, составляет около 0.5 мас. % от выхода этилового спирта. Основным компонентом этих продуктов является изоамиловый спирт, концентрация которого в бражке, в зависимости от вида исходного сырья, может достигать 80 мас. % и более [5], что в глобальных производственных объемах на текущий момент превышает 300 млн т/г.
Таким образом, использование изоамилового спирта в процессах получения синтетических каучуков может стать очередным важным этапом развития экологически безопасных химических производств, базирующихся на возобновляемом сырье. Одним из перспективных способов решения данной задачи представляется использование мембранно-каталитической технологии, позволяющей уменьшить общую энергетику процесса путем осуществления реакционной стадии в каналах каталитического конвертера, по сравнению с традиционным реактором со стационарным слоем насыпного катализатора. Эффективность заявленного авторами метода подтверждена ранее проведенными исследованиями по осуществлению процессов углекислотного, парового и смешанного риформинга метана, этанола, продуктов ферментации и диметилового эфира [6, 7].
ЭКСПЕРИМЕНТАЛЬНАЯ ЧАСТЬ
Синтез пористого керамического носителя. Для высокоселективного получения изоамиленовой фракции из изоамилового спирта впервые методом СВС синтезирован пористый каталитический материал на основе γ-Al2O3. Синтез осуществляли на основе геля псевдобемитной структуры AlOOH с использованием одностороннего прессования при давлении 30–90 МПа с последующим спеканием при температуре 750°C в атмосфере воздуха в течение 1 ч. Величина открытых пор синтезированного каталитического материала на основе γ-Al2O3, определенная по методу пузырька, составила 0.5–0.8 мкм.
В качестве исходного сырья для синтеза пористой подложки для осуществления дегидрирования изоамиленовой фракции, использовали порошок α-Al2O3 крупной фракции “Электрокорунд” белый, с размером частиц 100 мкм, производства ООО “Литпром”.
С целью повышения устойчивости к высокой температуре для синтеза конвертера в исходный порошок Al2O3 вводили порошкообразные добавки эвтектического состава оксида магния производства завода “Красный химик” ГОСТ 4526-75 и карбида кремния. Для повышения механической прочности был применен синтез карбида кремния в режиме самораспространяющегося высокотемпературного синтеза (СВС) с восстановительной стадией по нижеприведенной реакции (1) с последующим выщелачиванием в растворе соляной кислоты (2):
(1)
${\text{Si}}{{{\text{O}}}_{2}} + {\text{Mg}} + {\text{C}} \to {\text{SiC}} + {\text{MgO}}\quad{\kern 1pt} ,$(2)
${\text{MgO}} + {\text{ }}2{\text{HCl}} \to {\text{MgC}}{{{\text{l}}}_{2}} + {{{\text{H}}}_{{\text{2}}}}{\text{O}}{\text{.}}$Для придания материалу прочности, к готовому порошку добавляли кремнийорганические соединения, образующие при обжиге активный SiO2, связывающий частицы Al2O3. Данные порошки частично взаимодействуют между собой, превращаясь в муллиты, индиалит (Mg2Al4Si5O18) и шпинель (MgAl2O4). Смешение порошков осуществляли в шаровой мельнице с использованием сферических гранул Al2O3 в течение 1 ч. Далее проводили одностороннее прессование готовой смеси под давлением 30–90 МПа и ее спекание при температуре 1300–1450°C в атмосфере воздуха. При температурах спекания порошков образовывалась жидкая фаза эвтектического состава, содержащая оксид магния и карбид кремния в виде клиноэнстатита, смачивающая частицы Al2O3, образуя прочный пористый каркас конвертера.
Пористый керамический каталитический алюминий-оксидный носитель представляет собой трубку, у которой один конец заглушен для обеспечения принудительной диффузии газов через рабочую поверхность цилиндра от наружной стенки к внутренней, а на другом находится прижимная гайка для герметичной стыковки образца с реактором через графитовую прокладку (рис. 1).
Рис. 1.
Внешний вид носителя (форма, изготовленная из α-Al2O3 является носителем, после модификации которого мы называем его конвертером, ибо сам оксид алюминия в изучаемых реакциях неактивен и является только подложкой для наносимых катализаторов), синтезированного из α-Al2O3. Размеры: общая длина 137 мм; длина рабочей зоны 120 мм; внешний диаметр трубки 25 мм; толщина стенки 7 мм.

Рентгенодифрактометрический анализ алюминий-оксидного носителя показал присутствие в нем фазы α-Al2O3, шпинели MgAl2O4 и смешанного оксида гексагональной структуры Mg2Al4Si5O4.
Величину открытых пор алюминий-оксидного носителя оценивали по модели капиллярного вытеснения на основе формулы Лапласа для “модели цилиндрических пор”, непосредственное измерения величины открытых пор осуществляли с помощью ртутного порометра и методом “пузырька”. Диаметр открытых пор образца, определенный данными методами, составил ~1–3 мкм. Пористость образцов, измеренная методом гидростатического взвешивания, составила более 50%.
Модификация носителя железо–хромовыми каталитическими компонентами. Каталитическую систему формировали на внутренней поверхности пор носителя методом молекулярного наслаивания коллоидного раствора предшественников с целью уменьшения вклада побочных реакций полимеризации и коксообразования, имеющих место при дегидрировании изоамиленов.
С этой целью на первой стадии получали покрытие на основе γ-Al2O3, проводя нанесение изопропилата алюминия [Al(O-изоPr)3] путем прокачки его золя. Прокачку золя осуществляли с внешней стороны трубки через поры α-Al2O3 носителя с помощью водоструйного насоса, с последующей сушкой и термической обработкой. Затем формировали покрытия К2О и CeO2 пропиткой конвертера водным раствором К2СО3, и азотнокислого церия. Осаждение оксидов калия (1+) и церия (4+), а также γ-Al2O3 осуществляли с целью уменьшения вклада побочных реакций полимеризации и коксообразования, имеющих место при дегидрировании изоамиленов.
Раздельное нанесение активных железо–хром-содержащих компонентов на модифицированный носитель проводили из толуольных растворов соответствующих ацетилацетоных комплексов Fe(acac)3 и Cr(acac)3. На следующей стадии формировали дисперсные частицы оксидов Fe(3+) и Cr(3+) путем прокаливания пропитанного образца в муфельной печи в токе воздуха при различных температурах: 500°C – 2 ч, 600°C – 2 ч, 800°C – 1 ч. Количество нанесенных компонентов определяли по привесу образца после стадий термической обработки. Их содержание на поверхности альфа-алюминийоксидного конвертера представлено в табл. 1.
Таблица 1.
Состав каталитического конвертера дегидрирования изоамиленовой фракции
| Содержание компонентов, мас. % | ||||
|---|---|---|---|---|
| нанесенные компоненты | керамический носитель | |||
| K2O | СeO2 | (Fe,Cr)2O3 (1 : 1) | γ-Al2O3 | α-Al2O3 |
| 0.02 | 0.02 | 0.73 | 3.13 | 96.11 |
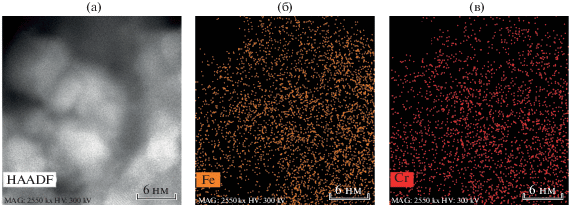
Данные просвечивающей электронной микроскопии с энергодисперсионным анализом Fe–Cr-содержащего конвертера показали равномерное распределение осажденных частиц железа и хрома субнанометрового масштаба на поверхности α-Al2O3-носителя (рис. 2а–в).
Рис. 2.
Данные ПЭМ конвертера состава α-Al2O3 + γ-Al2O3 + K,Ce,Cr (5%) + Fe (5%) – с раздельным нанесением активных компонентов, полученные на приборе TEM FEI TITAN Themis 300: (а) фотомикрография сканируемой поверхности носителя; (б) распределение частиц железа по сканируемой поверхности; (в) распределение частиц хрома по сканируемой поверхности.
Предварительно, с целью определения оптимального состава активных компонентов конвертера, по той же методике были изготовлены модельные образцы моно- (Mn) и би- (Fe–Cr, Fe–Mn) металлических гранулированных катализаторов, совместным или раздельным нанесением активных компонентов на промышленные гранулы γ-Al2O3.
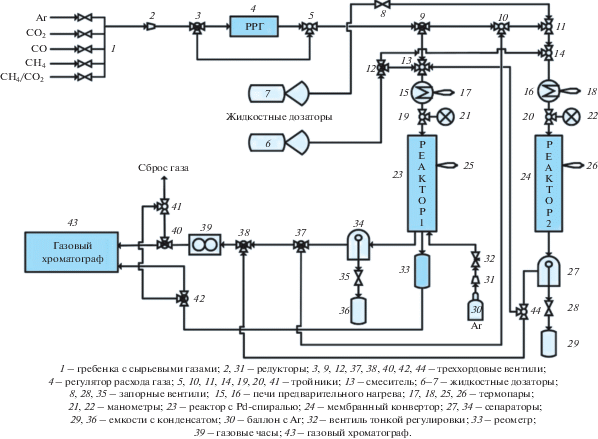
Мембранно-каталитическая установка и гибридный мембранно-каталитический реактор. Превращение изоамилового спирта в изопрен осуществляли с использованием разработанной оригинальной мембранно-каталитической установки (рис. 3), последовательно проводя стадию дегидратации изоамилового спирта в изоамилены в проточном реакторе (1) с их последующим дегидрированием в изопрен на пористом каталитическом конвертере в проточном реакторе (2). Установка снабжена аналитическим оборудованием для on-line определения состава газообразных и жидких продуктов методом газо-жидкостной хроматографии.
Дегидратацию изоамилового спирта в изоамиленовую фракцию осуществляли на гранулах промышленного γ-Al2O3 и гранулированном пористом каталитическом материале, приготовленным методом СВС на основе γ-Al2O3, при следующих одинаковых условиях: mкат = 20 г; Ar/изо-AmOH = 2.5; объемная скорость подачи в реактор паро-газовой смеси воды и аргона, деленная на объем катализатора Q (пар−газ) = 216 ч–1; Т = = 350°С; Робщ = 1 атм.
Дегидрирование полученной изоамиленовой фракции в изопрен осуществляли на гранулированном каталитическом конвертере при следующих параметрах процесса: Н2О/изоамилены = х, х12, х24, х36 (где х – кратность мольного отношения воды к изоамиленам); Q (пар–газ) = = 197–2198 ч–1; Т = 600°С; Робщ = 1 атм.
Методика анализа продуктов реакции. Анализ продуктов осуществляли методами газо-жидкостной хроматографии и хромато-масс-спектрометрии. Содержание водорода, оксидов углерода и метана в продуктах реакции определяли on-line методом газовой хроматографии на хроматографе КристалЛюкс 4000М с использованием в качестве детектора катарометра, газа-носителя – аргона высокой чистоты с расходом 10 мл/мин и адсорбционной насадочной колонкой. Наполнитель колонок – активированный уголь марки СКТ, который перед загрузкой в колонку подвергали обработке паром с целью снижения адсорбционного объема микропор (длина колонки 1 м, диаметр 3 мм, размер частиц 0.2–0.3 мм). Температура колонки, детектора и испарителя установили равной 120°С. Концентрации газов определяли по калибровочным кривым с использованием программного обеспечения NetChrom v2.1; малые концентрации СО определяли с помощью откалиброванного ИК-спектрометра Riken Keiki Model RI-550A.
Углеводородные газы С1–С5 идентифицировали на хроматографе КристалЛюкс 4000М с использованием пламенно-ионизационного детектора (ПИД) и газа-носителя – гелия.
Жидкие органические продукты реакции в органической фазе определяли методом ГЖХ на хроматографе Varian 3600, ПИД, капиллярная колонка: Хромтэк SE-30, 25 м × 0.25 мм, Df = = 0.33 мкм, температурный режим: 50°С (5 мин), 10°C/мин, 280°С, Тинж = 250°С, Ринж = 1 бар, деление потока 1/200, газ-носитель – гелий. Внутренние стандарты: трифторметилбензол (для алкан-олефиновой фракции), н-октан (для спиртовой смеси). Остаточное содержание органических продуктов в водной фазе находили с помощью ГХ-МС по соотношению интегральных сигналов методом абсолютной градуировки.
Методика расчетов. Конверсия изоамилового спирта:
Конверсия изоамиленов:
Выход изопрена на поданные изоамилены:
Селективность по изопрену (выход на превращенные изоамилены):
Селективность по изопрену (выход на превращенный изоамиловый спирт):
Производительность по изопрену, л/(ч гакт. комп.) (активными компонентами синтезированной каталитической системы являются оксиды Fe3+ и Cr3+):
РЕЗУЛЬТАТЫ И ИХ ОБСУЖДЕНИЕ
Получение изопрена из изоамилового спирта. Получение изопрена из изоамилового спирта последовательно осуществляли в соответствии со следующей двухстадийной схемой превращений:
(1)
$\begin{gathered} {{({\text{C}}{{{\text{H}}}_{3}})}_{2}}{\text{CH}}{\kern 1pt} --{\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} --{\kern 1pt} {\text{C}}{{{\text{H}}}_{2}}{\kern 1pt} --{\kern 1pt} {\text{OH}} \to \\ \to \,\,[{{({\text{С }}{{{\text{Н }}}_{3}})}_{2}}{\text{С Н }}{\kern 1pt} --{\kern 1pt} {\text{С Н }}{\kern 1pt} = {\kern 1pt} {\text{С }}{{{\text{Н }}}_{2}} + \\ {\text{ + }}\,\,{\text{С }}{{{\text{Н }}}_{3}}{\kern 1pt} --{\kern 1pt} {\text{С }}{{{\text{Н }}}_{2}}{\kern 1pt} --{\kern 1pt} {\text{С }}\left( {{\text{С }}{{{\text{Н }}}_{3}}} \right){\kern 1pt} = {\kern 1pt} {\text{С }}{{{\text{Н }}}_{2}} + {\text{ }} \\ {\text{ + }}\,\,{\text{С }}{{{\text{Н }}}_{3}}{\kern 1pt} --{\kern 1pt} {\text{С }}\left( {{\text{С }}{{{\text{Н }}}_{{\text{3}}}}} \right){\kern 1pt} = {\kern 1pt} {\text{С Н }}{\kern 1pt} --{\kern 1pt} {\text{С }}{{{\text{Н }}}_{3}}]{\text{ }} + {{{\text{Н }}}_{{\text{2}}}}{\text{О ,}} \\ \end{gathered} $(2)
$\begin{gathered} \left[ {{\text{И з о а м и л е н ы }}} \right] \to {\text{С }}{{{\text{Н }}}_{2}}{\kern 1pt} = {\kern 1pt} {\text{С }}\left( {{\text{С }}{{{\text{Н }}}_{3}}} \right){\kern 1pt} --{\text{С Н }}{\kern 1pt} = {\kern 1pt} {\text{С }}{{{\text{Н }}}_{2}} + {\text{ }}{{{\text{Н }}}_{2}}\quad \\ \Delta Н = 125{\text{ к Д ж /м о л ь }}{\text{.}} \\ \end{gathered} $Дегидратация изоамилового спирта, получение изоамиленовой фракции (стадия 1). Обнаружено, что на промышленных гранулах γ-Al2O3 100%-ная конверсия изоамилового спирта достигается при температуре 350°С, в то время как на образце γ-Al2O3, приготовленном методом СВС, температура исчерпывающей конверсии ниже на 50°С, при этом селективность по изоамиленам составляет более 98%. Оставшиеся 2% приходятся на предельные и непредельные углеводороды С2–С5 и водород. Изомерный состав изоамиленов для обоих катализаторов примерно одинаков и представлен 3-метил-1-бутиленом (~75%), 2-метил-2-бутиленом (~11%) и 2-метил-1-бутиленом (~12%), что соотносится с результатами, полученными на многокомпонентном промышленном катализаторе Dow Type B (Ca–Ni–PO4) [8, 9]. Отмечено, что на образце γ-Al2O3, приготовленнoм методом СВС, с повышением температуры до 350°С содержание 3-метил-1-бутилена и 2-метил-2-бутилена несколько снижается (в пределах 5%), в то время как выход 2-метил-1-бутилена соответственно увеличивается.
Важно отметить, что в известном из литературы процессе получения изоамиленов из изоамилового спирта на катализаторе 4%Cr2O3 ⋅ Al2O3 [9, 10] температура исчерпывающей дегидратации составляет 600°С, что на 300 градусов выше, чем в результате, полученном нами.
Дегидрирование изоамиленовой фракции, получение изопрена (стадия 2). Как видно из табл. 2 наибольшей активностью в реакции дегидрирования полученной изоамиленовой фракции при температуре 600°С обладает гранулированная модельная система состава γ-Al2O3 + K, Ce, Cr (5%) + + Fe (5%) (с раздельным нанесением активных компонентов), на которой селективность по изопрену достигала около 13%, что соотносится с результатом, полученным в одностадийном высокотемпературном процессе дегидрирования С5-фракции на многокомпонентных промышленных катализаторах 1707 и Dow Type B [8, 9, 11, 12]. При этом выход изопрена при 24-х кратном разбавлении субстрата водой достигал ~5%, а при 36-и кратном ~8%; производительность при этом соответственно составила 0.06 и 0.10 лизопрен/(ч гакт. комп.).
Таблица 2.
Дегидрирование изоамиленовой фракции в изопрен на модельных гранулированных катализаторах и на синтезированном конвертере (600°С)
| Состав катализатора | H2O/изо-Am, х-раз |
Xизоамилены, % | Хизопрен, % | Sизопрен, % | ρизопрен, л/(ч гакт.комп) | ${{W}_{{{{{\text{H}}}_{2}}}}},$ л/ч |
|---|---|---|---|---|---|---|
| Гранулы | ||||||
| γ-Al2O3 +K, Ce, Cr (5%) + Fe (5%) – раздельное нанесение |
0 | 89 | 1 | 1 | 0.02 | 2.99 |
| 12 | 81 | 2 | 3 | 0.03 | 3.90 | |
| 24 | 74 | 5 | 6 | 0.06 | 4.88 | |
| 36 | 61 | 8 | 13 | 0.10 | 3.63 | |
| γ-Al2O3 +K, Ce, Mn (5%) + Fe (5%) – совместное нанесение |
0 | 94 | 0 | 0 | 0.00 | 4.03 |
| 12 | 65 | 4 | 6 | 0.05 | 2.75 | |
| 24 | 47 | 4 | 8 | 0.05 | 1.94 | |
| 36 | 41 | 4 | 10 | 0.05 | 2.02 | |
| γ-Al2O3 +K, Ce, Mn (5%) + Fe (5%) – раздельное нанесение |
0 | 81 | 3 | 4 | 0.04 | 1.33 |
| 12 | 68 | 6 | 9 | 0.08 | 2.51 | |
| 24 | 36 | 6 | 17 | 0.08 | 0.38 | |
| 36 | 18 | 7 | 37 | 0.09 | 0.30 | |
| γ-Al2O3 +K, Ce, Mn (10%) |
0 | 89 | 1 | 1 | 0.02 | 3.33 |
| 12 | 86 | 3 | 3 | 0.03 | 4.89 | |
| 24 | 69 | 5 | 8 | 0.07 | 3.64 | |
| 36 | 56 | 6 | 10 | 0.07 | 2.30 | |
| Конвертер | ||||||
| α-Al2O3 + γ-Al2O3 +K, Ce, Cr (5%) + Fe (5%) – раздельное нанесение |
12 | 84 | 4 | 5 | 0.05 | 1.08 |
| 24 | 46 | 10 | 23 | 0.13 | 0.74 | |
| 36 | 43 | 9 | 21 | 0.12 | 0.60 | |
Выход изопрена на модифицированном пористом керамическом каталитическом конвертере состава γ-Al2O3 + K, Ce, Cr (5%) + Fe (5%) (с раздельным нанесением активных компонентов) при 24-х кратном разбавлении водой и температуре 600°С возрос до ~10%, селективность ~23%, а производительность ~0.13 лизопрен/(ч гакт.комп.). Конверсия изоамиленовой фракции при этом составила 46%. Таким образом, применение модифицированного пористого керамического каталитического конвертера позволило значительно увеличить основные показатели по сравнению с гранулированным катализатором (табл. 2, 3).
Таблица 3.
Содержание газообразных компонентов продуктовой смеси дегидрирования изоамиленовой фракции (мол. %) на Fe–Cr-содержащем конвертере в зависимости от избытка воды (600°С)
| Компонент продуктовой смеси, мол. % | H2O/изо-Am, х-раз | ||
|---|---|---|---|
| 12 | 24 | 36 | |
| Водород | 45.7 | 36.9 | 34.1 |
| Мономеры | |||
| Изопрен | 2.0 | 6.4 | 6.3 |
| Непрореагировавшие изоамилены | 8.2 | 33.0 | 39.5 |
| Бутилены | 7.3 | 5.0 | 4.1 |
| Пропилен | 3.2 | 1.8 | 1.5 |
| Этилен | 4.0 | 2.4 | 1.9 |
| Алканы | |||
| Пропан | 0 | 0.1 | 0 |
| Этан | 2.0 | 0.3 | 0.3 |
| Метан | 22.2 | 7.7 | 6.4 |
| Оксиды углерода | |||
| Диоксид углерода | 3.6 | 5.5 | 5.1 |
| Монооксид углерода | 1.7 | 0.7 | 0.7 |
| Конверсия изоамиленов, мол. % | 84 | 46 | 43 |
В табл. 3 представлены данные по составу газообразных продуктов реакции дегидрирования изоамиленовой фракции на Fe–Cr-содержащем конвертере при 600°С в зависимости от мольного соотношения вода–изоамилены, откуда видно, что в продуктах дегидрирования помимо изопрена содержатся и другие ценные мономеры, образующиеся в побочных реакциях крекинга, а также алканы, оксиды углерода и водород. Высокое содержание водорода, вероятно, является результатом протекания реакций риформинга углеводородов и образования коксовых отложений. Установлено, что за 5 ч эксперимента около 6 мол. % углерода от поданной изоамиленовой фракции пошло на коксообразование.
Из данных табл. 3 следует, что оптимальными условиями для получения изопрена является 24-х кратный избыток воды, при котором достигается максимальное содержание изопрена при пониженном содержании побочных продуктов реакции. Важно отметить, что в каталитических микроканалах конвертера существенно медленнее развиваются процессы коксообразования по сравнению с традиционным реактором с гранулированным слоем катализатора, на котором регенерация проводится каждые 8–15 мин.
ВЫВОДЫ
Выход и селективность в образовании изопрена, полученного путем дегидрирования изоамиленов на конвертере, превосходит аналогичные показатели, полученные на гранулированном катализаторе того же состава и на известных промышленных катализаторах.
Способ двухстадийного получения изопрена с использованием каталитического пористого конвертера обладает более высокой стабильностью к зауглероживанию по сравнению с промышленными процессами дегидрирования, в которых стадия регенерации проводится каждые 8–15 мин.
Из полученных результатов следует, что предлагаемый способ, позволяет эффективно осуществлять самосогласованные реакции дегидратации и дегидрирования изоамилового спирта с целью получения важного мономера-изопрена, что существенным образом расширяет возможности современных процессов “зеленой химии”.
Работа поддержана Российским научным фондом (грант № 17-13-01270).
Список литературы
Arpe H.-J., Hawkins S. // Industrial Organic Chemistry. 5th Edition. ISBN: 2010. https://www.wiley.com/ en-us/Industrial+Organic+Chemistry%2C+5th+Edition- p-9783527320028.
https://www.ihs.com/products/chemical-technology-pep-reviews-bio-based-isoprene-2011.html.
Кондаков И.Л. Синтетический каучук его гомологи и аналоги. Императорский Юрьевский ун-т. Юрьев: тип. Маттисена, 1912.
https://www.ethanolrfa.org/wp-content/uploads/2016/ 02/Ethanol-Industry-Outlook-2016.pdf.
Стабников В.Н. Перегонка и ректификация этилового спирта. 2-е издание. М.: Изд-во “Пищевая промышленность”, 1969.
Чистяков А.В., Жарова П.А., Цодиков М.В., Николаев А.С., Кротова И.В., Эзжеленко Д.И. // Кинетика и катализ. 2016. Т. 57. № 6. С. 807.
Fedotov A.S., Antonov, D.O., Bukhtenko O.V., Uvarov V.I., Kriventsov V.V., Tsodikov M.V. // International J. of Hydrogen Energy. 2017. V. 42. № 38. P. 24131.
Gose J. // J. Soc. Org. Synth. Chem. Japan. 1963. V. 21. P. 508.
Oda H. // J. Chem. Soc. Japan. 1967. V. 70. P. 1325.
Nauch O. // Patent Germ. № 264902. 2014.
Friedman B.S. // Patent US № 2853535. 2016.
Огородников С.К., Идлис Г.С. Производство изопрена. Л.: Химия, 1973. 243 c.
Дополнительные материалы отсутствуют.